

Abstract
Objective
To study the phenotypic characteristics of patients with a novel p.E292K mutation in BEST1.
Methods
Affected individuals underwent ophthalmic examination and testing that included photography, autofluorescence, OCT, and electrophysiological testing. DNA was analyzed for BEST1 mutations.
Results
Five patients (aged 5–59) expressing the p.E292K mutation in BEST1 were ascertained from three families. EOG light-rise was subnormal in all probands and carriers. Carriers had normal fundus examination, mfERG, visual acuity, and were emmetropic or myopic. Only probands had hyperopia and fundus findings typical of Best macular dystrophy. OCT of vitelliform lesions demonstrated RPE elevation without subretinal fluid; atrophic lesions exhibited disruption of the hyper-reflective outer retina-RPE complex. Intense hyperautofluorescence correlated to the vitelliform lesion.
Conclusions
Patients with Glu292Lys variation in BEST1 exhibit intra- and interfamilial phenotypic variability. A disproportionate fraction (26%) of Best-disease-causing mutations occur in exon 8, suggesting that the portion of protein encoded by this exon (amino acids 290–316) may be especially important to bestrophin’s function. Relatively good visual acuity with vitelliform lesions can be explained by preservation of the outer retina demonstrated by OCT.
Clinical relevance
We demonstrate findings that can be seen with novel mutation in this region of BEST1 that carries implications for disease pathogenesis.
Best macular dystrophy (BMD) is an autosomal dominant condition caused by mutations in the BEST1 gene.1, 2 Mutations in this gene result in a defective protein product, bestrophin, which localizes to the basolateral membrane of the retinal pigment epithelium (RPE)3 and has been associated with conductance abnormalities in a family of chloride channels4, 5 and voltage-gated calcium channels.6–8 These alterations may account for the diminished light peak-dark trough ratio (Arden ratio typically ≤ 1.5) of the electro-oculogram (EOG) characteristic of BMD; full-field ERG is typically normal.9, 10
The fundus findings associated with BEST1 mutations are quite varied and include a normal fundus appearance with an abnormal EOG; vitelliform lesions with a ‘sunny side up’ egg yolk appearance in the central macula; a ‘pseudohypopyon’ in which the yellow material gravitates inferiorly in the sub-RPE space; a ‘scrambled egg’ characterized by yellowish subretinal deposits admixed with patches of hyperpigmentation and atrophy of the retinal pigment epithelium; geographic atrophy; nodular subretinal gliosis centered on fixation; and rarely, choroidal neovascularization.11 Visual acuity is often preserved in at least one eye throughout life, with more substantial visual loss occurring when BMD is complicated by choroidal neovascularization12–14 or extensive geographic atrophy.
Newer diagnostic techniques have refined our understanding of the anatomy of the macular lesions seen in patients with Best disease.15–20 For example, one of the first optical coherence tomography (OCT) studies in these patients showed that the vitelliform material may lie between the outer retina and the RPE.21 Increased fluorescence of vitelliform fundus lesions on fundus autofluorescence (AF)22, 23 images may be due to enhanced accumulation of fluorophores such as A2E.18
Although the precise structure of the protein has yet to be elucidated, it has been hypothesized that bestrophin has four transmembrane domains with amino and carboxyl terminals located in the cytoplasm. The majority of reported BEST1 mutations causing BMD have been missense mutations,1, 2, 22, 24–40 and a disproportionate fraction of these mutations occur in exon 8, suggesting that the portion of the protein encoded by this exon may be especially critical to its function. At the time of this writing, the Carver Nonprofit Genetic Testing Laboratory has observed 155 instances of 84 different BEST1 mutations in probands affected with Best disease, and 22 of these mutations (26%) lie within exon 8 (Kinnick and Stone, unpublished data). Additional evidence of the functional importance of this portion of the protein is that exon 8 is highly conserved among the BEST1 orthologs of C.elegans, D. melanogaster, and mice.2 In addition, a functional analysis of an exon 8 variant (Q293H) in human embryonic kidney cells revealed a severe reduction of chloride current that behaved in a dominant-negative manner.27
In the present study, we identified individuals from three apparently unrelated families with a missense mutation in BEST1, causing a change at position 292 of glutamic acid to lysine. Clinical characterization, including AF, OCT, and full-field and multi-focal electroretinography (ERG; ffERG, mfERG) of these individuals, demonstrates highly variable expression between individuals, as well as intrafamilial variability with nonpenetrance of fundus findings in two consecutive generations.
METHODS
Three probands (aged 5–59) from three unrelated families were identified by characteristic fundus findings and abnormal Arden ratio on electro-oculography (EOG). Family members of the 5-year-old male were also examined, and electrophysiology was performed. After informed consent was obtained, blood samples were taken for DNA extraction, and subsequent mutation screening of BEST1 was performed at the John and Marcia Carver Nonprofit Genetic Testing Laboratory in Iowa City, IA. The protocol of the study adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review boards of the institutions involved.
A full medical history was taken and ophthalmic examination was performed. Patients underwent digital color fundus photography. The AF images of the 54-year-old subject were obtained using a TopCon 50EX digital fundus camera equipped with AF filters purchased from OIS similar to a system previously described.41, 42 The OCT images were obtained utilizing the Stratus III (Stratus OCT 4.0.2 software; Zeiss instruments, Dublin, CA). Electrophysiological assessment included ffERG and EOG, using recording methods laid out by ISCEV standards and recommendations for electroretinography43, 44 and electro-oculography. 43 The mfERG was performed in four cases according to guidelines that have been reported elsewhere.41, 45
RESULTS
Mutation analysis revealed a missense variation resulting in a change of glutamic acid to lysine at amino acid position 292 in BEST1 in all probands and obligate carriers. Genotyping of four informative short tandem repeat polymorphisms at the BEST1 locus revealed a distinctly different disease-associated haplotype in family 1, strongly suggesting that the Glu292Lys mutation in that family occurred independently (data not shown). Summaries of the clinical findings for patients in this study are shown in Table 1.
Table 1.
Summary of clinical findings in 3 families of patients with the Glu292Lys BEST1 variation.
| State | Age | Sex | BCVA | Refraction | Irides | Fundus | Ethnicity |
|---|---|---|---|---|---|---|---|
| proband 1 | 5 | M | 20/40 OU |
+5.25 sph OU | Green | vitelliform OU |
Scandinavian |
| Irish | |||||||
| German | |||||||
| French | |||||||
| asymptomatic | 30 | F | 20/20 OU |
emmetropic | Green | normal | Scandinavian |
| mother of proband 1 |
Irish | ||||||
| German | |||||||
| proband 2 | 54 | M | 20/200 OD |
+2.25+075@180 OD |
Brown | atrophic OD |
East Indian |
| 20/20 OS |
+1.75+075@165 OS |
vitelliform OS |
|||||
| proband 3 | 59 | F | 20/70 OD |
+1.25+050@170 OD |
Green | atrophic OU |
Norwegian |
| 20/50 OS |
+1.00+025@010 OS |
Jewish |
PROBAND 1 AND FAMILY MEMBERS
The parents of a 5-year-old male noted intermittent convergent strabismus in the child for 8 weeks. Refractive error of +5.25 diopter sphere (DS) OU resulted in orthophoria with best-corrected visual acuity (BCVA) of 20/40 OU. Irides were green. Anterior segment examination, including anterior chamber depth, was within normal limits. Fundus examination revealed bilateral central vitelliform lesions (Figures 1A and B). The ffERG was normal and EOG revealed Arden ratios of 1.1 OD and 1.4 OS. The OCT showed a discrete elevation of RPE and widening of the hyper-reflective signal from the outer-retina-RPE complex (Figures 1C and D).
Figure 1.
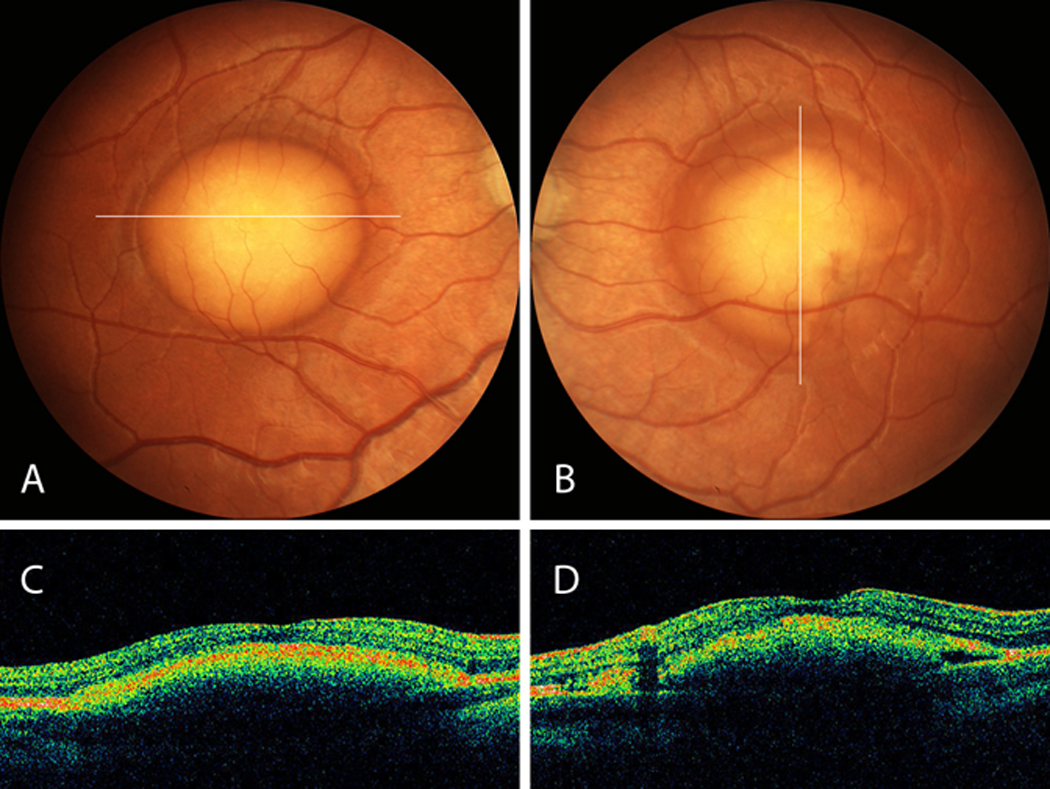
A,B, Fundus of proband 1 shows well-demarcated vitelliform lesions in the central macula. C, D, Corresponding OCT sections shows elevation at the level of the RPE with preservation of the outer retina layer.
The asymptomatic mother, who is of Scandinavian/Irish/German descent, and father, who is of French/German/Norwegian descent, both had VA of 20/20 without correction and normal fundus examination. However, EOG of the 30-year-old mother revealed Arden ratios of 1.2 OD, 1.3 OS. Her ffERG and mfERG were normal. Genetic testing of the mother revealed that she had the same mutation in BEST1 Glu292Lys as her son.
This led to examination and testing of other family members (Figure 2) to further characterize the inheritance pattern. The proband’s maternal grandfather, aged 57, was asymptomatic, with BCVA of 20/20. He had Arden ratios of 1.3 OU, and his ffERG and mfERG were normal. Manifest refraction was −0.75 +1.00@055 OD, −1.00 +1.00@180 OS. Intraocular pressure (IOP) was 14 mm Hg OU. Irides were hazel colored. Anterior chamber depth was normal. Crystalline lens was clear. Posterior segment examination revealed rare RPE changes but no vitelliform lesion or atrophy.
Figure 2.

Pedigree of proband 1 (indicated by shaded box) demonstrates lack of penetrant fundus findings and hyperopia in carriers.
In addition, EOG testing of the above proband and asymptomatic carriers revealed prolonged light-peak slow oscillation, in keeping with previously reported findings in humans46 and animals47 with BMD.
PROBAND 2
The patient is a 54-year-old male of East Indian descent who was referred for reduced vision in the right eye for the past 10 years. Family history was unremarkable for similar vision problems. His BCVA was 20/200 (eccentric) OD and 20/20 OS. Refractive error was +2.25 +0.75@180 OD, +1.75 +0.75@165 OS. His IOP was 12 mm Hg OD and 14 mmHg OS.
Anterior segment exam revealed brown irides and normal anterior chamber depth. Fundus examination revealed a central circumscribed area of atrophy OD and a vitelliform lesion OS (Figures 3A and B). Both eyes exhibited deep, fleck-like changes nasal to the disc and along the temporal arcades. The AF images of the right eye (Figure 3E) showed a central macular hypoautofluorescent lesion that corresponded to the area of atrophy on fundus examination with patches of increased AF, especially in the periphery of the lesion. AF of both eyes allowed better visualization of the fleck-like lesions around the disc and arcades, exhibiting mixed hyper- and hypoautofluorescence that was confluent in many areas. The AF images of the left eye (Figure 3F) showed a central area of homogenously increased autofluorescence corresponding to the vitelliform lesion. OCT imaging (Figure 3C) revealed increased backscatter from the underlying sclera in the region of RPE atrophy with some irregular disruptions in the outer retina-RPE complex at the edges of the atrophic lesion in the right eye; OCT images in the left eye (Figure 3D) showed discrete elevation of the RPE without discontinuity of the hyperreflective outer retina-RPE complex.
Figure 3.
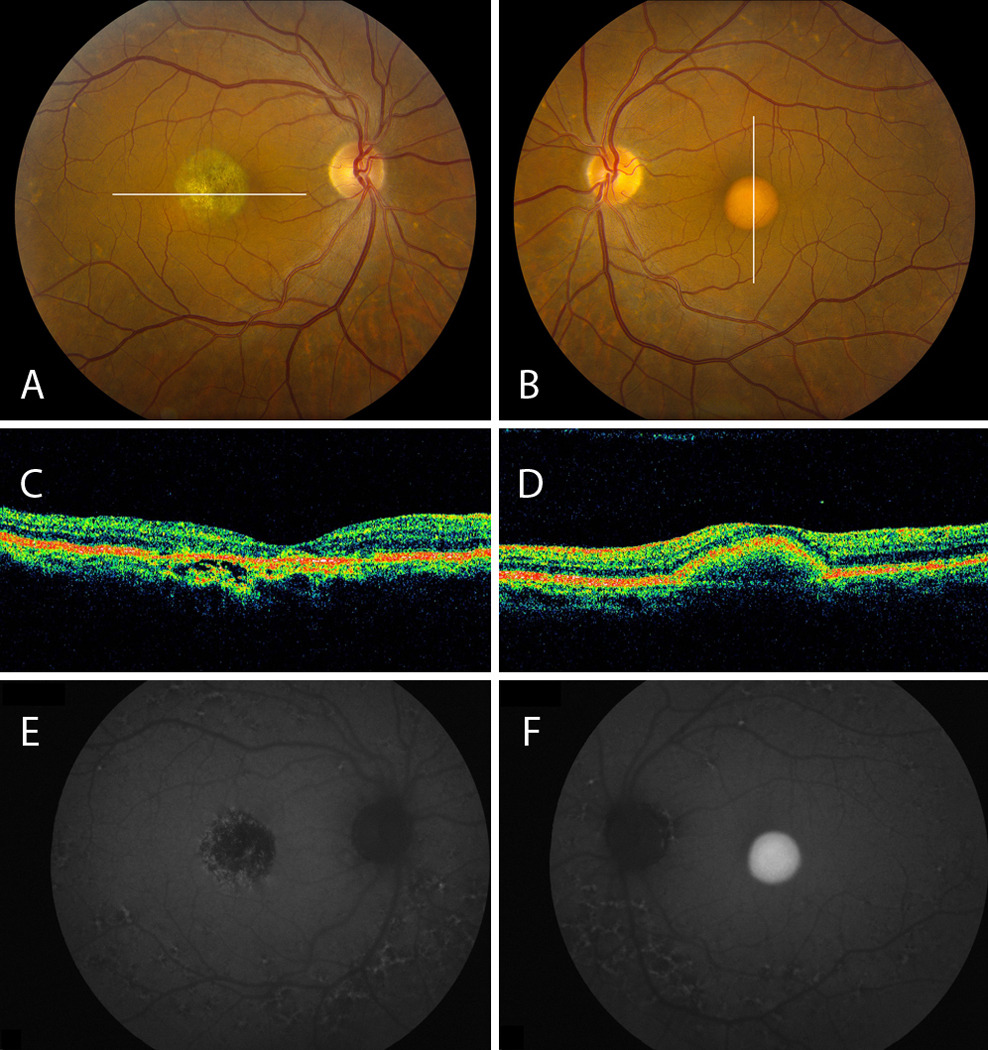
A, B, Proband 2 fundus demonstrates central atrophy OD and a vitelliform lesion OS, respectively. Both exhibit fleck-like changes nasal to the disc and around the arcades. C, OCT OD reflects RPE atrophy with disruption in the outer retina-RPE complex. D, OCT OS shows discrete RPE elevation. E, AF OD demonstrates a central hypoautofluorescent lesion. Fleck-like lesions around the disc and arcades reveal mixed hyper- and hypoautofluorescence in both eyes. F, AF OS shows central hyperautofluorescence corresponding to the vitelliform lesion.
Electrophysiological assessment revealed Arden ratio of 1.5 OD and 1.3 OS. The ffERG was normal. Fixation was not stable enough to permit reliable mfERG recording using a pupil camera in the right eye; but mfERG OS demonstrated reduced responses from the central 1–5 degrees OS (Figure 4) with relative preservation of the response amplitude and timing from the surrounding macula.
Figure 4.
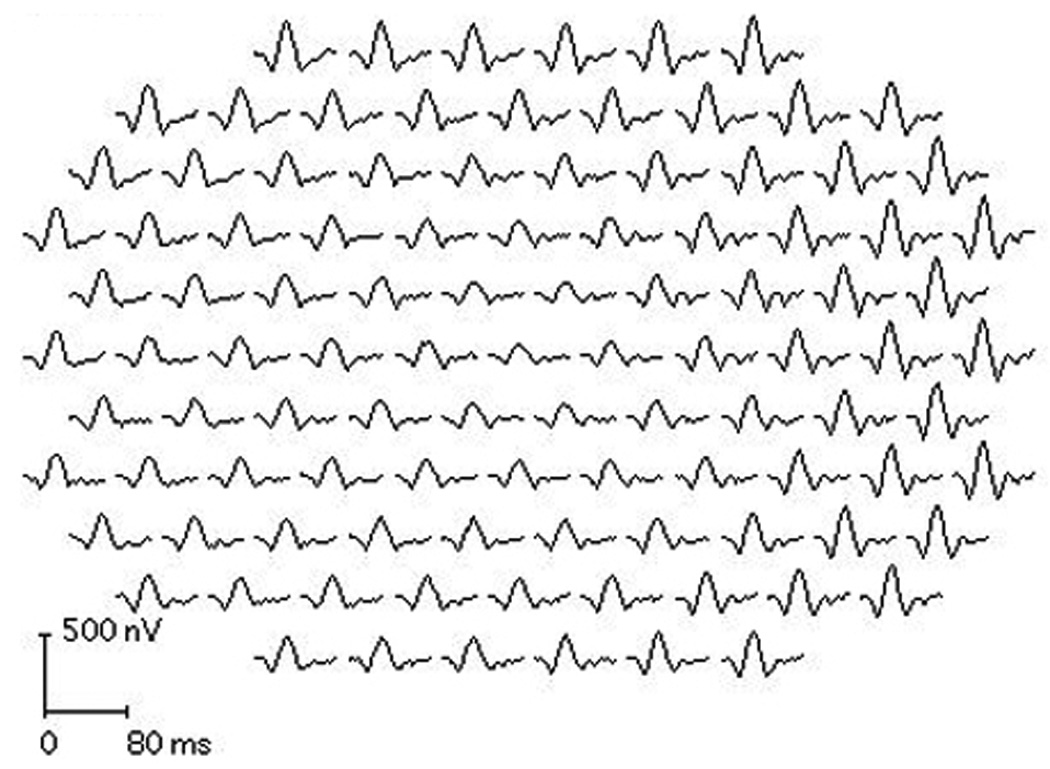
mfERG OS from proband 2 demonstrates reduced responses from the central 1–5 degrees OS with relative preservation of the response amplitude and timing from the surrounding macula.
PROBAND 3
This 59-year-old woman of Norwegian/Jewish descent with green irides was found on routine examination 22 years earlier to have fundus findings suspicious for BMD. Two sisters and a nephew are thought to have BMD, and while her mother had vision problems, no diagnosis had been made at the time of her death. Ocular history was significant for hyperopia and laser peripheral iridotomy OU for occludable angles. Past medical history was significant for well-controlled diabetes mellitus type II, polymyalgia rheumatica, reflux disease, and osteoarthritis.
Visual acuity was 20/70 OD with +1.25 +0.50@170 and 20/50 OS with +1.00 +0.25@010. On biomicroscopy, the laser peripheral iridotomies were patent and there was mild nuclear sclerosis OU. Fundus examination revealed bilateral central atrophy with few drusen in the mid-periphery (Figures 5A and B). Corresponding to the area of atrophy seen on examination, OCT revealed attenuation of the outer retina and hyper-reflectivity of the RPE with increased signal posterior to the RPE (Figures 5C and D).
Figure 5.
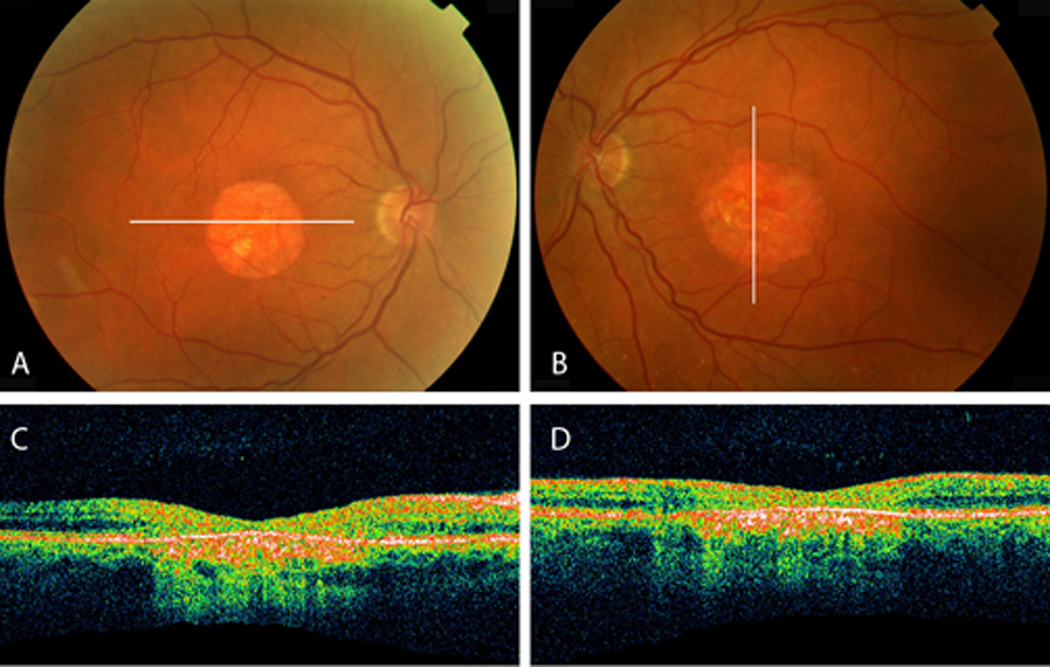
A, B, Fundus of proband 3 reveals bilateral central atrophy with few drusen in the mid-periphery of both eyes. C, D, Optical coherence tomography indicates areas of atrophy seen with increased signal posterior to the retinal pigment epithelium and attenuation of the outer retina.
The Arden ratio was 1.2 OD and 1.1 OS. Her ffERG was normal; mfERG (Figure 6) exhibited attenuation of amplitude and latency delay that was most prominent in the central macula with relative sparing of the peripheral macula.
Figure 6.

mfERG of proband 3 demonstrates attenuation of amplitude and latency delay most prominent in the central macula with relative sparing of the periphery.
DISCUSSION
BEST1, together with BEST2, BEST3, and BEST4, are part of a closely related gene family characterized by several transmembrane spanning domains and an invariant arginine-phenylalanine-proline (RFP) motif. These four human genes are believed to be orthologous to a gene in C.elegans that shares a highly conserved 26-amino acid sequence beginning at position 289.48 Three lines of evidence support the idea that the novel bestrophin variation reported here is disease-causing. First, the glutamic acid residue normally present at position 292 is highly conserved evolutionarily. Second, glutamic acid is negatively charged at neutral pH while the lysine residue found in affected individuals is positively charged. This is the most extreme charge difference possible for a point mutation. Finally, the fact that the mutation was observed in three unrelated families with different bestrophin haplotypes suggests that the variation arose more than once. This makes it very likely that Glu292Lys is the disease-causing variation in the gene and not simply a non-disease-causing polymorphism in linkage disequilibrium with a true disease causing mutation nearby. Mutations are common in the region of the human BEST1 gene encoding amino acids 290–316, suggesting that this portion of the protein is critical to its function.
In the present study, we performed a detailed clinical and electrophysiological evaluation of five subjects from three unrelated families that share a previously undescribed missense mutation in BEST1, a change from glutamic acid to lysine at amino acid position 292. We observed variable expressivity in our cohort of patients. Only the reduced light-peak on the EOG was completely penetrant. Hyperopia was also found in all probands but was distinctly absent in carriers. Though it is tempting to attribute this to reduced axial length from the elevated fundus lesion, hyperopia was also found in our patients with flat, atrophic retinas, as demonstrated on OCT. Moreover, similar degrees of hyperopia were seen in proband 2 despite atrophy in one eye and a vitelliform lesion in the other. Hyperopia is a common finding in patients with BMD22, 49 and is found in other conditions caused by mutations in BEST1such as autosomal dominant vitreoretinochoroidopathy50 and autosomal recessive bestrophinopathy.51 Findings of hyperopia in probands may be related to genetic modifiers such as microphthalmia-associated transcription factor (MITF).52, 53 Interactions between MITF and BEST1 have not been fully explored. While it is unclear whether our probands were hyperopic before the development of lesions, it would be interesting to compare the prevalence of hyperopia in probands and carriers and determine whether hyperopia is a predictive factor for developing vitelliform lesions. Such a finding would have implications for prognosis and counseling of patients and families. The lack of hyperopia among carriers in our study could indicate a favorable prognostic sign in patients with this mutation.
While proband 1 had mildly diminished visual acuity despite the presence of vitelliform lesions, both his mother and his 57-year-old maternal grandfather had relatively normal fundi despite diminished EOG and presence of the Glu292Lys variation. In addition to emmetropia, normal visual acuity, and lack of fundus lesions, both carriers also had normal ffERG and mfERG responses. This appears to be the first demonstration of non-penetrant fundus findings in two consecutive generations with a confirmed BEST1 mutation. Given the early age at which large vitelliform lesions are typically observed, it seems more likely that the variable clinical findings among patients carrying the same BEST1 variation are due to modifying genes rather than to environmental factors.
The results of OCT and AF performed in our patients allow us to speculate on the nature of the vitelliform lesion, for which histopathology in a human eye has not been performed. Models of BEST1 mutations in dogs54 and mice47 are not adequate for this purpose as these animals do not develop typical vitelliform lesions. Human eyes with the ‘scrambled egg’ appearance have abnormally high amounts of lipofuscin.17 Studies in elderly individuals homozygous or heterozygous for mutations in BEST1 have demonstrated modestly elevated levels of A2E compared to controls.18 However, it is unknown whether A2E is a by-product of the latter stages of disease or whether other components of lipofuscin contribute to the hyperautofluorescent appearance in early stages. Intense hyperautofluorescence corresponding to the vitelliform lesion was seen in the left eye of proband 2, but a circumscribed patch of decreased AF corresponded to the atrophic lesion with speckled increased AF. In light of histopathological studies of late BMD,15, 16 this loss of AF is likely due to irregularities in the RPE cell monolayer with secondary loss of photoreceptor outer segment turnover correlating with the poor level of vision in this eye. Relatively preserved visual acuity was seen in probands 1 and 2 with vitelliform lesions causing RPE elevations. Notably, OCT revealed no evidence of serous detachment in our patients, as recently reported in BMD patients studied with spectral-domain OCT.55 This contrasts with other studies of autofluorescent vitelliform lesions that showed fluid between the RPE and outer retina.21, 56 One hypothesis accounting for our findings is that mutated bestrophin in RPE, which likely contains lipofuscin in the vitelliform stage, causes impaired transport of fluid to the choroid, resulting in separation of RPE from choroid. Progression causes defective pumping of subretinal fluid to RPE, resulting in the detachment of the outer retina from RPE that was shown in previous studies of eyes with more advanced disease.
Patients with Glu292Lys variation in BEST1 exhibit intra- and interfamilial phenotypic variability. Thus, it is important for clinicians to realize that the identification of the variation by genetic testing does not always portend the eventual development of macular disease. Our findings support the idea that the portion of the protein consisting of amino acids 290–316 may be critical to the function of bestrophin. Relatively good visual acuity with vitelliform lesions can be explained by preservation of the outer retina demonstrated by OCT.
Acknowledgements
Funded in part by Research to Prevent Blindness (JLD, PJF, and RGW), Foundation Fighting Blindness (JLD, PJF, EMS and RGW), NEI grant EY002162 (JLD), That Man May See, Inc. (JLD), the Bernard A. Newcomb Macular Degeneration Fund (JLD), Hope for Vision (JLD), the Karl Kirchgessner Foundation (JLD), the Curran Fund for Research on Best Disease (RGW), and the Howard Hughes Medical Institute (EMS). The authors thank Karmen Trzupek, Susan Clarke, Jean Andorf and Becky Johnston for their excellent assistance.
References
- 1.Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19(3):241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 2.Marquardt A, Stöhr H, Passmore LA, Krämer F, Rivera A, Weber BH. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease) Hum Mol Genet. 1998;7(9):1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 3.Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2000;97(23):12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci U S A. 2002;99(6):4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsunenari T, Sun H, Williams J, et al. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem. 2003;278(42):41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marmorstein LY, Wu J, McLaughlin P, et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1) J Gen Physiol. 2006;127(5):577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal R, Bakall B, Kinnick T, et al. Expression Of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006;20(1):178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- 8.Yu K, Xiao Q, Cui G, Lee A, Hartzell HC. The best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J Neurosci. 2008;28(22):5660–5670. doi: 10.1523/JNEUROSCI.0065-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutman AF. Electro-oculography in families with vitelliform dystrophy of the fovea. Detection of the carrier state. Arch Ophthalmol. 1969;81(3):305–316. doi: 10.1001/archopht.1969.00990010307001. [DOI] [PubMed] [Google Scholar]
- 10.Krill AE, Morse PA, Potts AM, Klien BA. Hereditary vitelliruptive macular degeneration. Am J Ophthalmol. 1966;61(6):1405–1415. doi: 10.1016/0002-9394(66)90478-8. [DOI] [PubMed] [Google Scholar]
- 11.Miller SA, Bresnick GH, Chandra SR. Choroidal neovascular membrane In Best's vitelliform macular dystrophy. Am J Ophthalmol. 1976;82(2):252–255. doi: 10.1016/0002-9394(76)90428-1. [DOI] [PubMed] [Google Scholar]
- 12.Mohler CW, Fine SL. Long-term evaluation of patients with Best's vitelliform dystrophy. Ophthalmology. 1981;88(7):688–692. doi: 10.1016/s0161-6420(81)34965-3. [DOI] [PubMed] [Google Scholar]
- 13.Fishman GA, Baca W, Alexander KR, Derlacki DJ, Glenn AM, Viana M. Visual acuity in patients with best vitelliform macular dystrophy. Ophthalmology. 1993;100(11):1665–1670. doi: 10.1016/s0161-6420(93)31420-x. [DOI] [PubMed] [Google Scholar]
- 14.Chung MM, Oh KT, Streb LM, Kimura AE, Stone EM. Visual outcome following subretinal hemorrhage in Best disease. Retina. 2001;21(6):575–580. doi: 10.1097/00006982-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Frangieh GT, Green WR, Fine SL. A histopathologic study of Best's macular dystrophy. Arch Ophthalmol. 1982;100(7):1115–1121. doi: 10.1001/archopht.1982.01030040093017. [DOI] [PubMed] [Google Scholar]
- 16.O'Gorman S, Flaherty WA, Fishman GA, Berson EL. Histopathologic findings in Best's vitelliform macular dystrophy. Arch Ophthalmol. 1988;106(9):1261–1268. doi: 10.1001/archopht.1988.01060140421045. [DOI] [PubMed] [Google Scholar]
- 17.Weingeist TA, Kobrin JL, Watzke RC. Histopathology Of Best's macular dystrophy. Arch Ophthalmol. 1982;100(7):1108–1014. doi: 10.1001/archopht.1982.01030040086016. [DOI] [PubMed] [Google Scholar]
- 18.Bakall B, Radu RA, Stanton JB, et al. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2) Exp Eye Res. 2007;85(1):34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Invest Ophthalmol Vis Sci. 2007;48(7):3372–3380. doi: 10.1167/iovs.06-0868. [DOI] [PubMed] [Google Scholar]
- 20.Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Late development of vitelliform lesions and flecks in a patient with best disease: clinicopathologic correlation. Arch Ophthalmol. 2005;123(11):1588–1594. doi: 10.1001/archopht.123.11.1588. [DOI] [PubMed] [Google Scholar]
- 21.Pianta MJ, Aleman TS, Cideciyan AV, et al. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res. 2003;76(2):203–211. doi: 10.1016/s0014-4835(02)00280-4. [DOI] [PubMed] [Google Scholar]
- 22.Renner AB, Tillack H, Kraus H, et al. Late onset is common in best macular dystrophy associated with VMD2 gene mutations. Ophthalmology. 2005;112(4):586–592. doi: 10.1016/j.ophtha.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 23.von Rückmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997;115(5):609–615. doi: 10.1001/archopht.1997.01100150611006. [DOI] [PubMed] [Google Scholar]
- 24.Bakall B, Marknell T, Ingvast S, et al. The mutation spectrum of the bestrophin protein--functional implications. Hum Genet. 1999;104(5):383–389. doi: 10.1007/s004390050972. [DOI] [PubMed] [Google Scholar]
- 25.Caldwell GM, Kakuk LE, Griesinger IB, et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics. 1999;58(1):98–101. doi: 10.1006/geno.1999.5808. [DOI] [PubMed] [Google Scholar]
- 26.Eksandh L, Bakall B, Bauer B, Wadelius C, Andréasson S. Best's vitelliform macular dystrophy caused by a new mutation (Val89Ala) in the VMD2 gene. Ophthalmic Genet. 2001;22(2):107–115. doi: 10.1076/opge.22.2.107.2226. [DOI] [PubMed] [Google Scholar]
- 27.Marchant D, Yu K, Bigot K, et al. New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy. J Med Genet. 2007;44(3):e70. doi: 10.1136/jmg.2006.044511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wabbels B, Preising MN, Kretschmann U, Demmler A, Lorenz B. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2006;244(11):1453–1466. doi: 10.1007/s00417-006-0286-6. [DOI] [PubMed] [Google Scholar]
- 29.Apushkin MA, Fishman GA, Taylor CM, Stone EM. Novel de novo mutation in a patient with Best macular dystrophy. Arch Ophthalmol. 2006;124(6):887–889. doi: 10.1001/archopht.124.6.887. [DOI] [PubMed] [Google Scholar]
- 30.Krämer F, Mohr N, Kellner U, Rudolph G, Weber BH. Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD) Hum Mutat. 2003;22(5):418. doi: 10.1002/humu.9189. [DOI] [PubMed] [Google Scholar]
- 31.Krämer F, White K, Pauleikhoff D, et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8(4):286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Wang G, Dong B, et al. A novel mutation of the VMD2 Gene in a Chinese family with best vitelliform macular dystrophy. Ann Acad Med Singapore. 2006;35(6):408–410. [PubMed] [Google Scholar]
- 33.Marchant D, Gogat K, Boutboul S, et al. Identification of novel VMD2 gene mutations in patients with best vitelliform macular dystrophy. Hum Mutat. 2001;17(3):235. doi: 10.1002/humu.9. [DOI] [PubMed] [Google Scholar]
- 34.Seddon JM, Sharma S, Chong S, Hutchinson A, Allikmets R, Adelman RA. Phenotype and genotype correlations in two best families. Ophthalmology. 2003;110(9):1724–1731. doi: 10.1016/S0161-6420(03)00575-X. [DOI] [PubMed] [Google Scholar]
- 35.Sodi A, Passerini I, Simonelli F, Testa F, Menchini U, Torricelli F. A novel mutation in the VMD2 gene in an italian family with Best maculopathy. J Fr Ophtalmol. 2007;30(6):616–620. doi: 10.1016/s0181-5512(07)89667-7. [DOI] [PubMed] [Google Scholar]
- 36.White K, Marquardt A, Weber BH. VMD2 mutations in vitelliform macular dystrophy (Best disease) and other maculopathies. Hum Mutat. 2000;15(4):301–308. doi: 10.1002/(SICI)1098-1004(200004)15:4<301::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 37.Palomba G, Rozzo C, Angius A, Pierrottet CO, Orzalesi N, Pirastu M. A novel spontaneous missense mutation in VMD2 gene is a cause of a best macular dystrophy sporadic case. Am J Ophthalmol. 2000;129(2):260–262. doi: 10.1016/s0002-9394(99)00327-x. [DOI] [PubMed] [Google Scholar]
- 38.Schatz P, Klar J, Andréasson S, Ponjavic V, Dahl N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006;27(2):51–56. doi: 10.1080/13816810600677990. [DOI] [PubMed] [Google Scholar]
- 39.Lotery AJ, Munier FL, Fishman GA, et al. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41(6):1291–1296. [PubMed] [Google Scholar]
- 40.Marchant D, Gogat K, Dureau P, et al. Use of denaturing HPLC and automated sequencing to screen the VMD2 gene for mutations associated with Best's vitelliform macular dystrophy. Ophthalmic Genet. 2002;23(3):167–174. doi: 10.1076/opge.23.3.167.7880. [DOI] [PubMed] [Google Scholar]
- 41.Duncan JL, Zhang Y, Gandhi J, et al. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48(7):3283–3291. doi: 10.1167/iovs.06-1422. [DOI] [PubMed] [Google Scholar]
- 42.Spaide RF. Fundus autofluorescence and age-related macular degeneration. Ophthalmology. 2003;110(2):392–399. doi: 10.1016/S0161-6420(02)01756-6. [DOI] [PubMed] [Google Scholar]
- 43.Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Arch Ophthalmol. 1993;111(5):601–604. doi: 10.1001/archopht.1993.01090050035023. [DOI] [PubMed] [Google Scholar]
- 44.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 Update). International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol. 1998;97(2):143–156. doi: 10.1023/a:1002016531591. [DOI] [PubMed] [Google Scholar]
- 45.Marmor MF, Hood DC, Keating D, et al. Guidelines for basic multifocal electroretinography (mfERG) Doc Ophthalmol. 2003;106(2):105–115. doi: 10.1023/a:1022591317907. [DOI] [PubMed] [Google Scholar]
- 46.Weleber RG. Fast and slow oscillations of the electro-oculogram in Best's macular dystrophy and retinitis pigmentosa. Arch Ophthalmol. 1989;107(4):530–537. doi: 10.1001/archopht.1989.01070010544028. [DOI] [PubMed] [Google Scholar]
- 47.Marmorstein AD, Stanton JB, Yocom J, et al. A model of best vitelliform macular dystrophy in rats. Invest Ophthalmol Vis Sci. 2004;45(10):3733–3739. doi: 10.1167/iovs.04-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stöhr H, Marquardt A, Nanda I, Schmid M, Weber BH. Three novel human VMD2-like genes are members of the evolutionary highly conserved RFP-TM family. Eur J Hum Genet. 2002;10(4):281–284. doi: 10.1038/sj.ejhg.5200796. [DOI] [PubMed] [Google Scholar]
- 49.Bard LA, Cross HE. Genetic counseling of families with Best macular dystrophy. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1975;79(6):OP865–OP873. [PubMed] [Google Scholar]
- 50.Yardley J, Leroy BP, Hart-Holden N, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest Ophthalmol Vis Sci. 2004;45(10):3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- 51.Burgess R, Millar ID, Leroy BP, et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82(1):19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Esumi N, Kachi S, Campochiaro PA, Zack DJ. VMD2 promoter requires two proximal E-box sites for its activity in vivo and is regulated by the MITF-TFE family. J Biol Chem. 2007;282(3):1838–1850. doi: 10.1074/jbc.M609517200. [DOI] [PubMed] [Google Scholar]
- 53.Esumi N, Oshima Y, Li Y, Campochiaro PA, Zack DJ. Analysis of the VMD2 promoter and implication of E-box binding factors in its regulation. J Biol Chem. 2004;279(18):19064–19073. doi: 10.1074/jbc.M309881200. [DOI] [PubMed] [Google Scholar]
- 54.Guziewicz KE, Zangerl B, Lindauer SJ, et al. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci. 2007;48(5):1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G, Souied EH. High-definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol. 2008;146(4):501–507. doi: 10.1016/j.ajo.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 56.Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmology. 2006;113(8):1392–1400. doi: 10.1016/j.ophtha.2006.03.023. [DOI] [PubMed] [Google Scholar]