

Abstract
Background
Ischemic preconditioning delays the onset of electrical uncoupling and prevents loss of the primary ventricular gap junction protein connexin43 (Cx43) from gap junctions during subsequent ischemia.
Methods
To test the hypothesis that these effects are mediated by protein kinase C epsilon (PKCε), we studied isolated Langendorff-perfused hearts from mice with homozygous germline deletion of PKCε (PKCε-KO). Cx43 phosphorylation and distribution were measured by quantitative immunoblotting and confocal microscopy. Changes in electrical coupling were monitored using the 4-electrode technique to measure whole-tissue resistivity.
Results
The amount of Cx43 located in gap junctions, measured by confocal microscopy under basal conditions, was significantly greater in PKCε-KO hearts compared to wildtype but total Cx43 content measured by immunoblotting was not different. These unanticipated results indicate that PKCε regulates subcellular distribution of Cx43 under normal conditions. Preconditioning prevented loss of Cx43 from gap junctions during ischemia in wildtype but not PKCε-KO hearts. Specific activation of PKCε, but not PKCδ, also prevented ischemia-induced loss of Cx43 from gap junctions. Preconditioning delayed the onset of uncoupling in wildtype but hastened uncoupling in PKCε-KO hearts. Cx43 phosphorylation at the PKC site Ser368 increased 5-fold after ischemia in wildtype hearts and, surprisingly, by nearly 10-fold in PKCε-KO hearts. Preconditioning prevented phosphorylation of Cx43 in gap junction plaques at Ser368 in wildtype but not PKCε-KO hearts.
Conclusion
Taken together, these results indicate that PKCε plays a critical role in preconditioning to preserve Cx43 signal in gap junctions and delay electrical uncoupling during ischemia.
Keywords: preconditioning, gap junctions, connexin43, coupling, protein kinase C
Introduction
Ischemic preconditioning, defined as brief intervals of ischemia/reperfusion preceding sustained ischemia, reduces infarct size1-3 and prevents reperfusion arrhythmias.4,5 While the antiarrhythmic mechanism of preconditioning remains unknown, previous studies from our laboratory and others have shown that preconditioning delays electrical uncoupling of the myocardium during ischemia.6-8 Preconditioning also prevents ischemia-induced changes in the distribution7 and phosphorylation7,9,10 of connexin43 (Cx43), the primary ventricular gap junction protein. Cx43 is a critical target in ischemia and preconditioning, as evidenced by the fact that mice deficient in Cx43 show greater incidence of arrhythmias after coronary occlusion11 and loss of preconditioning-induced cardioprotection,12 a finding also observed in myocytes isolated from Cx43-deficient hearts subjected to simulated ischemia/reperfusion.13
Protein kinase C (PKC) is a superfamily of serine/threonine protein kinases implicated in preconditioning.14-16 Specifically, the Ca2+-independent PKC epsilon isoform (PKCε) is thought to play a critical role in preconditioning, as salutary effects of preconditioning on infarct size are absent in mice lacking PKCε.17,18 Conversely, specific activation of PKCε before sustained ischemia mimics preconditioning19,20 and prevents reperfusion arrhythmias.20,21 We have previously shown that non-selective inhibition of PKC by chelerythrine or calphostin C blocks the ability of preconditioning to delay uncoupling and prevent Cx43 redistribution,7 suggesting an important role for PKC in mediating the effects of preconditioning on gap junctions. Furthermore, PKC phosphorylates the intracellular C-terminal domain of Cx43 at Ser36822 and Ser262,23 and these reactions are probably catalyzed at least partially by PKCε, which co-immunoprecipitates with Cx43.24,25 Phosphorylation at Ser368 has been shown to alter channel selectivity26 and gap junction assembly,27 although the effects on cell communication in the intact heart are unclear. Accordingly, the aim of the present study was to determine whether PKCε is responsible for mediating changes in Cx43 distribution and phosphorylation and electrical coupling at gap junctions during ischemic preconditioning. We found that in genetically engineered mice lacking PKCε, preconditioning fails to preserve Cx43 in gap junctions and cellular uncoupling is actually accelerated during sustained ischemia. These results identify PKCε as a potential target for regulating Cx43 distribution and electrical coupling during sustained ischemia.
Materials and Methods
Animals
Mice homozygous for a germline deletion of the gene encoding PKCε (PKCε-KO) were originally produced by Khasar et al.28 F1 generation C57BL/6J and 129SvJae heterozygous progeny (generous gift from Dr. Robert O. Messing) were intercrossed to generate F2 generation hybrid C57Bl/6Jx129SvJae wildtype and PKCε-KO littermates. Previous studies have shown these mice lack responsiveness to preconditioning in the absence of any gross or functional abnormality at baseline compared to wildtype littermates.18 All studies were performed in adult animals 12-20 weeks of age. Experimental protocols were approved by the Animal Studies Committee at Washington University School of Medicine.
Isolated heart perfusion
Hearts of anesthetized adult mice were excised rapidly, transferred to a Langendorff apparatus and perfused via aortic cannula with oxygenated Krebs-Henseleit buffer containing (in mmol/L): NaCl 118.3, KCl 2.7, MgSO4 1.0, KH2PO4 1.4, NaHCO3 29.0, CaCl2 3.4 and glucose 10, with insulin 70 mU/L and BSA 0.4% at 37° C. Flow was adjusted to achieve a retrograde perfusion pressure of 40-50 mmHg. All hearts were initially perfused with oxygenated buffer during a 10-min stabilization period. A subset of hearts was then subjected to a preconditioning protocol consisting of 3 cycles of 3 min of global no-flow ischemia followed by 5 min of normal perfusion before undergoing 30 min of global ischemia, as described previously.7 Other hearts were not subjected to preconditioning but were instead perfused with normoxic buffer for 24 min before undergoing 30 min of global ischemia.
In separate studies, hearts were perfused for 10 min with normoxic buffer containing peptide activators (1 μM) of PKCε19 (KAE1-1, KAI Pharmaceuticals) or PKCδ29 (KAD1-1, KAI Pharmaceuticals) or a non-specific control peptide (C-1, KAI Pharmaceuticals) before undergoing 30 min of global ischemia.
Antibodies
Antibodies used in this study included a rabbit polyclonal antibody (Zymed) directed against epitopes in the C-terminus of rat Cx43 (immunoblotting, 1:5000 dilution); a mouse monoclonal anti-Cx43 antibody (Chemicon MAB3068) (immunohistochemistry and confocal microscopy, 1:400 dilution); a rabbit polyclonal anti-phospho-Cx43 antibody directed against Ser368 (Cell Signaling) (immunoblotting, 1:500 dilution; immunostaining,1:100 dilution); rabbit polyclonal anti-phospho-Cx43 antibodies directed against Ser262, Ser279/282, Ser255 or Tyr265 (Santa Cruz) (immunoblotting); a monoclonal anti-PKCε antibody (BD Biosciences) (immunoblotting, 1:250 dilution); a polyclonal anti-PKCδ antibody (Santa Cruz) (immunoblotting, 1:750 dilution); a monoclonal anti-GAPDH antibody (RDI) (immunoblotting, 1:5000 dilution), and a polyclonal anti-actin antibody (Santa Cruz) (immunoblotting, 1:1000 dilution).
Preparation and quantification of immunoblots
Hearts were removed from the perfusion apparatus and trimmed of atria and great vessels. Apical and basal portions of each ventricle were frozen separately for subsequent analysis. Apical samples were prepared for Cx43 immunoblotting and analyzed as described previously.30 Cx43 band densities were divided by their respective GAPDH density values and then normalized to the same control sample on each gel. A subset of immunoblots was probed for actin to verify that 30 min. of global ischemia does not alter GAPDH levels (data not shown). Immunoblot analysis of PKC isoforms in subcellular fractions was performed on the basal portions as described previously.31 Briefly, samples were pulverized in homogenization buffer (20 mM Tris-HCl (pH 7.4), 2mM EDTA, 0.5 mM EGTA, 100 nM aprotinin, 1 mM benzamidine, 1 μM leupeptin, 1 μM pepstatin, 1 mM PMSF) and centrifuged (100,000g for 45 min, 4° C). The supernatant was saved as the cytosolic fraction and the pellet was resuspended in homogenization buffer and saved as the membrane fraction.
Quantitative confocal immunofluorescence microscopy
Hearts were removed from the perfusion apparatus, fixed in 10% neutral buffered formalin, embedded in paraffin and sectioned for immunohistochemistry and quantitative confocal microscopy as described previously.32,33 The amount of Cx43 signal at intercellular junctions was quantified as described previously32,33 and expressed as a proportion of total tissue area. The amount of phospho-Cx43(Ser368) at junctions was also quantified and expressed as a fraction of total Cx43 signal.
Measurement of whole-tissue resistance
Electrical uncoupling during ischemia was monitored in isolated mouse hearts by measuring changes in whole tissue resistance using the 4-electrode method.7,34-37 Once perfusion with normoxic buffer had been initiated in excised hearts, 4 Teflon-coated silver wire electrodes (0.0015 inch coated diameter) were passed through the anterior surface of the left ventricle in a linear arrangement oriented parallel to the long axis of epicardial fibers. The tip of each wire contained a bead of epoxy to prevent the wire from pulling through the epicardial surface. The Teflon insulation had been removed at a point ∼50μm below the bead to permit measurements at a consistent intramyocardial position below the epicardial surface. The outer two electrodes, each separated from its adjacent inner electrode by a distance of 1.0 mm, were connected to a current source, and the inner 2 electrodes, separated from each other by a distance of 1.0 mm, were connected to a voltage amplifier. A subthreshold alternating current (1000Hz; peak-to-peak amplitude 19μA) was delivered across the outer two electrodes while the voltage drop across the inner two electrodes was recorded. Tissue resistance (rt), a measure of extracellular (ro) and intracellular (ri) resistances arranged in parallel (1/rt = 1/ro + 1/ri), was measured at l min intervals throughout the experiment. Data were normalized to control values obtained during normoxic perfusion to permit comparisons between hearts.
Statistical analysis
All data are expressed as mean ± SEM. Differences between groups were analyzed with ANOVA and Fisher's protected least significant difference test. A value of p<0.05 was considered statistically significant.
Results
Altered Cx43 distribution in PKCε-null ventricular myocytes
To determine whether the absence of PKCε affects Cx43 expression or distribution under basal conditions, hearts from wildtype and PKCε-KO animals were analyzed by confocal microscopy and immunoblotting using a polyclonal anti-Cx43 antibody that detects both phosphorylated and nonphosphorylated Cx43 isoforms.32,33,38 Under basal conditions, in the absence of ischemia, the amount of Cx43 immunoreactive signal in gap junctions was 72% greater in PKCε-KO compared to wildtype hearts (p<0.001) (Figure 1A). However, immunoblotting showed no significant difference in the amount of Cx43 in ventricular tissue lysates from wildtype and PKCε-KO hearts (Figure 1B). These unanticipated results indicate that in the absence of PKCε, there is a shift of Cx43 from junctional to non-junctional pools.
Figure 1.
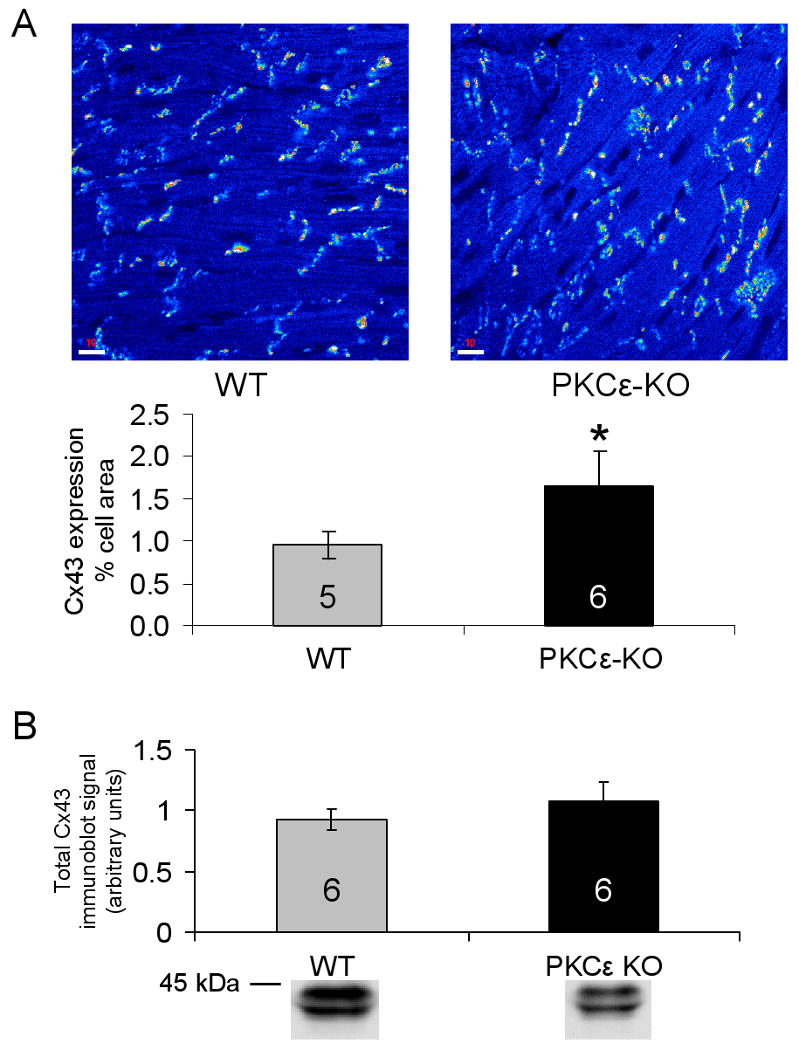
(A) Top: Representative confocal microscopy images showing Cx43 signal at cell-cell junctions in control wildtype (WT) and PKCε-KO hearts. Bottom: Quantitative confocal microscopy measurements showing amount of Cx43 signal as a percent of total tissue area in WT and PKCε-KO hearts; n for each group indicated in bar. *p<0.001 vs WT. (B) Densitometric measurements from immunoblots of total tissue Cx43 in WT and PKCε-KO hearts under basal conditions. Representative immunoblots are shown below each measurement; n for each group indicated in bar.
Regulation by PKCε of Cx43 distribution during ischemia and preconditioning
To elucidate the role of PKCε in mediating changes in Cx43 distribution during ischemia, the amount of Cx43 signal in gap junctions was measured by confocal microscopy in wildtype and PKCε-KO hearts subjected to ischemia with or without prior preconditioning. As previously observed in studies of rat hearts,7,37 the amount of Cx43 signal in gap junctions was reduced by nearly 80% after 30 min of global ischemia in non-preconditioned wildtype hearts (Figure 2). PKCε-KO hearts showed a comparable degree of Cx43 loss from gap junctions during ischemia (Figure 2). Preconditioning resulted in marked preservation of Cx43 signal in junctions in wildtype mouse hearts, a finding also previously observed in rat hearts,7,38 but preconditioning did not prevent loss of Cx43 from gap junctions in PKCε-KO hearts (Figure 2). Immunoblotting studies revealed that ischemia with or without prior preconditioning did not significantly change total Cx43 protein levels in wildtype or PKCε-KO hearts compared to respective controls (Figure 3). Taken together, these results indicate that preconditioning prevents redistribution of Cx43 during ischemia by a PKCε-dependent mechanism.
Figure 2.
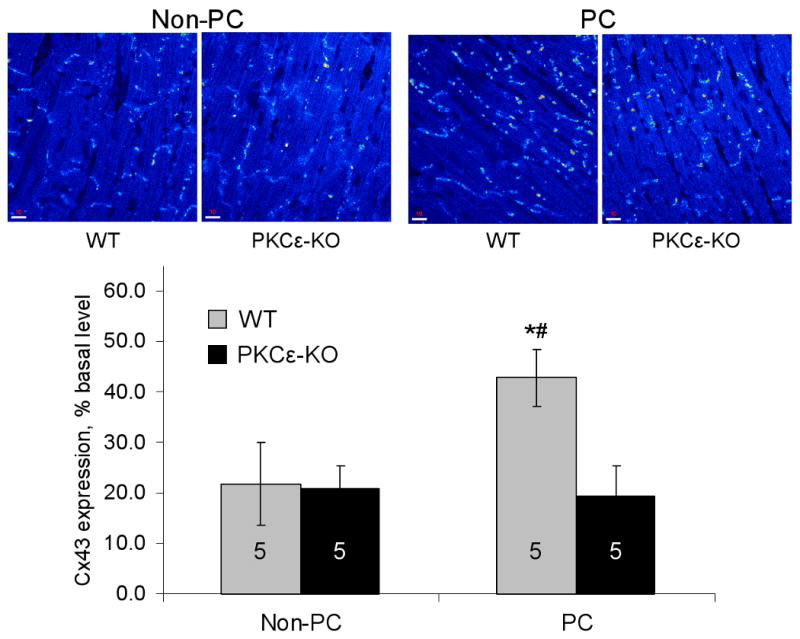
Top: Representative confocal microscopy images showing Cx43 signal at cell-cell junctions in wildtype (WT) and PKCε-KO hearts subjected to 30 min ischemia without (Non-PC) or with preconditioning (PC). Bottom: Quantitative confocal microscopy measurements showing amount of Cx43 signal as a percent of total tissue area. Values are expressed as percent of basal values to account for differences in basal Cx43 signal between WT and PKCε-KO animals; n for each group indicated in bar. *p<0.001 vs. WT non-PC; #p<0.001 vs. PKCε-KO PC.
Figure 3.
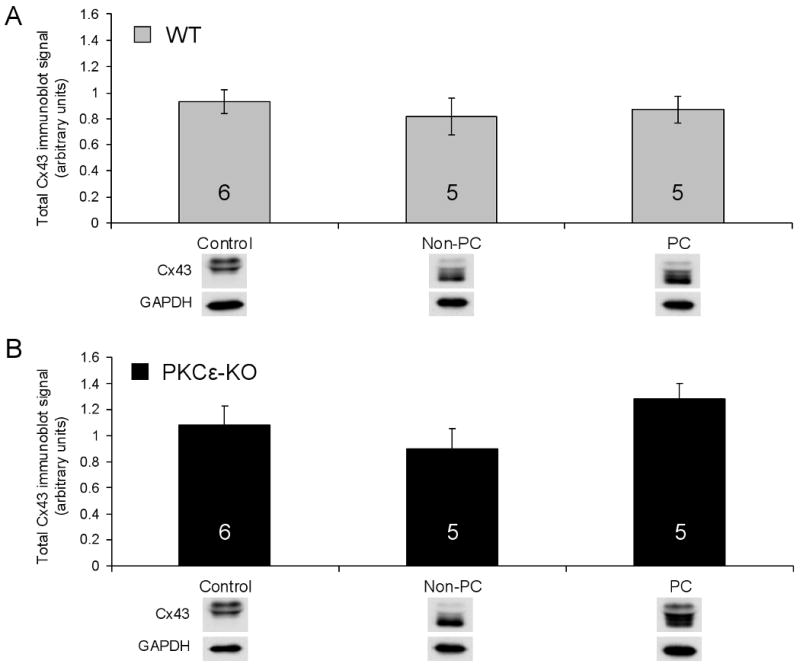
(A) Densitometric measurements from immunoblots of total tissue Cx43 in control, non-PC, and PC hearts from WT animals. (B) Densitometric measurements from immunoblots of total tissue Cx43 in control, non-PC, and PC hearts from PKCε-KO animals. Representative immunoblots are shown below each measurement; n for each group indicated in bar.
To further strengthen the conclusion that activation of PKCε preserves Cx43 signal in gap junctions during ischemia, we used PKC isoform-specific peptide activators20,30 to activate PKC in wildtype hearts independent of preconditioning. We then subjected hearts to ischemia and measured Cx43 in gap junctions by confocal microscopy. As shown in Figure 4, Cx43 signal in junctions following ischemia was >3-fold greater in hearts pretreated with a specific activator of PKCε compared to a non-specific control peptide. In contrast, activation of PKCδ was ineffective in preserving Cx43 levels during ischemia (Figure 4). These results provide independent evidence that activation of PKCε during preconditioning is both necessary and sufficient to preserve Cx43 signal in gap junctions during subsequent ischemia.
Figure 4.

Top: Representative confocal microscopy images showing Cx43 signal at cell-cell junctions in WT hearts perfused for 10 min with a PKCε activator, PKCδ activator, or control peptide before undergoing 30 min of global ischemia. Bottom: Quantitative confocal microscopy measurements showing amount of Cx43 signal as a percent of total tissue area in hearts treated with control peptide (CC), PKCε activator or PKCδ activator; n=3 for each group. *p=0.013 vs. control; #p=0.006 vs. PKCδ activator.
Effects of PKCε on Cx43 phosphorylation during ischemia and preconditioning
In previous immunoblotting studies in rat hearts, we observed that ischemia caused a marked decrease in phosphorylated forms of Cx43 and accumulation of apparently dephosphorylated Cx43.7,37 We also observed that preconditioning limited this shift. In the present study, however, preconditioning did not appear to limit accumulation of more rapidly migrating forms of Cx43 despite the fact that it preserved Cx43 signal in gap junctions in wildtype hearts (Figure 3). Thus, as reported previously,7 there appears to be no mechanistic link between loss of phosphorylated Cx43 and retention of Cx43 in junctions. Furthermore, it is likely that shifts in Cx43 phosphorylation apparent on Western blots reflect not only dephosphorylation but also phosphorylation at new sites in response to ischemia and/or preconditioning, and that phosphorylation at critical residues may underlie the salutary effects of preconditioning on electrical coupling during ischemia. Accordingly, we used phospho-specific anti-Cx43 antibodies to analyze changes in Cx43 at sites known to be phosphorylated in response to various kinases activated by ischemia and/or preconditioning. Densitometric analysis of immunoblots showed that phosphorylation of Cx43 at Ser368 (phospho-Cx43(Ser368)), a known PKC phosphorylation site,22 increased >5-fold after 30 min of ischemia in both non-preconditioned and preconditioned wildtype hearts (Figure 5A). Surprisingly, both non-preconditioned and preconditioned PKCε -KO hearts showed an even greater increase in phospho-Cx43(Ser368) after 30 min of ischemia, indicating that another kinase activated by preconditioning can promote phosphorylation of Cx43 at Ser368. There were no apparent differences between wildtype and PKCε-KO hearts in Cx43 phosphorylation at the PKC phosphorylation site Ser262 (Figure 5B), the MAPK sites Ser255 and Ser 279/282 (Figure 5C), or the Src kinase site Tyr265 (Figure 5D).
Figure 5.

Densitometric measurements from immunoblots of Cx43 phosphorylated at (A) Ser368, (B) Ser 262, (C) Ser255 and Ser279/282, and (D) Tyr265 in control, non-PC and PC hearts from WT and PKCε-KO animals; n for each group indicated in bar. #p<0.03 vs. WT control; *p<0.001 vs. PKCε-KO control; †p<0.02 vs. WT non-PC; §p=0.003 vs. WT PC. Representative immunoblots are shown below each measurement.
To determine whether preconditioning altered Ser368 phosphorylation of Cx43 located within gap junctions, hearts from wildtype and PKCε-KO animals were analyzed by confocal immunofluorescence microscopy using an antibody specific for phospho-Cx43(Ser368). The amount of Cx43 phosphorylated at Ser368 was measured by confocal microscopy and expressed as a proportion of the amount of total Cx43 in gap junctions, which was measured in adjacent sections from the same samples using the polyclonal anti-Cx43 antibody that recognizes all Cx43 isoforms. Under control (non-ischemic) conditions, only a limited amount of phospho-Cx43(Ser368) signal was present in junctions in both wildtype and PKCε-KO hearts (Figure 6). However, the effects of ischemia and preconditioning on phospho-Cx43(Ser 368) in gap junctions differed markedly in the two genotypes. In wildtype hearts, ischemia without prior preconditioning caused a marked increase in the amount of phospho-Cx43(Ser368) in junctions concomitant with electrical uncoupling, whereas preconditioning dramatically reduced accumulation of phospho-Cx43(Ser368) in junctions during subsequent ischemia (Figure 6). In PKCε-KO hearts, ischemia without preconditioning also caused an increase in phospho-Cx43(Ser368) in gap junctions, but preconditioning did not diminish its accumulation and, in fact, led to a marked increase in phospho-Cx43(Ser368) in junctions (Figure 6). Thus, preconditioning not only preserves Cx43 in gap junctions but also reduces the amount of phospho-Cx43(Ser368) accumulating in junctions during ischemia. Both of these processes depend on actions (direct or indirect) of PKCε.
Figure 6.

(A) Representative confocal microscopy images showing amount of Cx43 phosphorylated at Ser368 in cell-cell junctions. (B) Quantitative confocal microscopy measurements showing amount of phosphorylated at Ser368 in cell-cell junctions expressed as a proportion of total Cx43 in junctions; n for each group indicated in bar. *p=0.005 vs. WT control; #p=0.015 vs. WT PC; §p=0.049 vs. PKCε-KO control.
Activation of PKCδ during ischemia and preconditioning in PKCε-null hearts
To determine whether activation of PKCδ could contribute to the increase in phospho-Cx43(Ser368) seen in PKCε-KO hearts after ischemia and preconditioning, we used immunoblotting to measure the relative proportions of PKCε and PKCδ in cytosolic and membrane fractions prepared from ventricular lysates of wildtype and PKCε-KO ventricles (Figure 7). In wildtype hearts under control (non-ischemic) conditions, both PKCε and PKCδ resided primarily in the cytosolic fraction (Figure 7A). Ischemia with or without prior preconditioning led to translocation of PKCε but not PKCδ to the membrane fraction in wildtype hearts, consistent with selective activation of PKCε. In contrast, ischemia in PKCε-KO hearts resulted in marked translocation of PKCδ from the cytosolic to membrane fractions (Figure 7B). Thus, PKCε, but not PKCδ, is activated during ischemia and preconditioning in wildtype hearts whereas PKCδ only is activated in PKCε-KO hearts.
Figure 7.
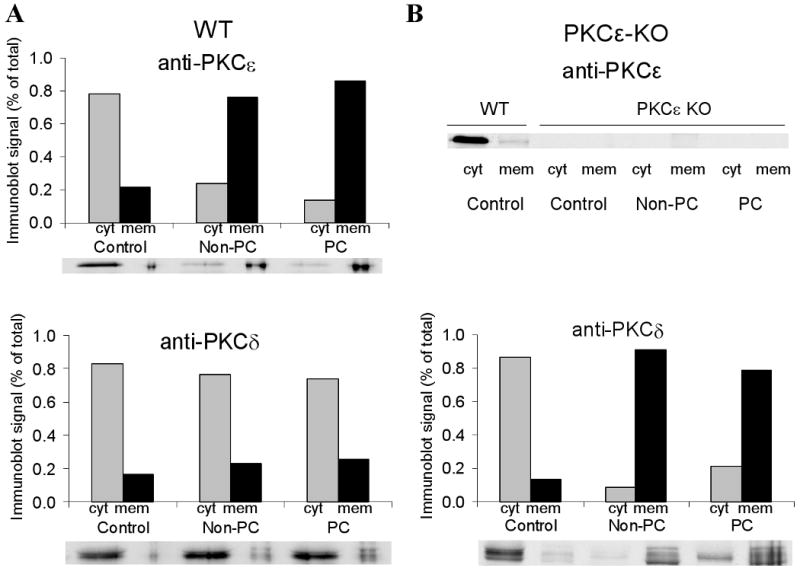
Densitometric measurements from immunoblots of PKCε and PKCδ in cytosolic (cyt) and membrane (mem) fractions of whole ventricular lysates. Immunoblots are shown below each group. Each sample corresponds to two pooled hearts.
Effects of PKCε on electrical uncoupling during ischemia and preconditioning
To determine whether PKCε plays a role in the ability of preconditioning to delay the onset of electrical uncoupling, we used the 4-electrode technique to monitor changes in whole tissue resistance (rt) in wildtype and PKCε-KO hearts. It is well known that rt increases in characteristic phases during ischemia and that the sustained rise in rt reflects electrical uncoupling at gap junctions.35,36 As shown in our previous studies in rat hearts,7 preconditioning delayed the onset of uncoupling following 30 min of ischemia in wildtype mouse hearts (from 22.1±0.9 min in non-preconditioned hearts (n=7) to 25.3±0.7 min in preconditioned hearts (n=8), p<0.02; see Figure 8 for representative traces). Interestingly, uncoupling after ischemia occurred significantly later in non-preconditioned PKCε-KO hearts than in non-preconditioned wildtype hearts (27.7±1.3 (n=6) vs. 22.1±0.9 min, respectively; p<0.001). This difference is presumably related to the greater amount of Cx43 in gap junctions under basal conditions in PKCε-KO hearts (see Figure 1). However, in contrast to wildtype hearts in which preconditioning delayed uncoupling during ischemia, preconditioning accelerated the onset of uncoupling in PKCε-KO hearts (from 27.7±1.3 to 24.2±0.8 (n=6) min; p=0.019; see Figure 8 for representative traces).
Figure 8.
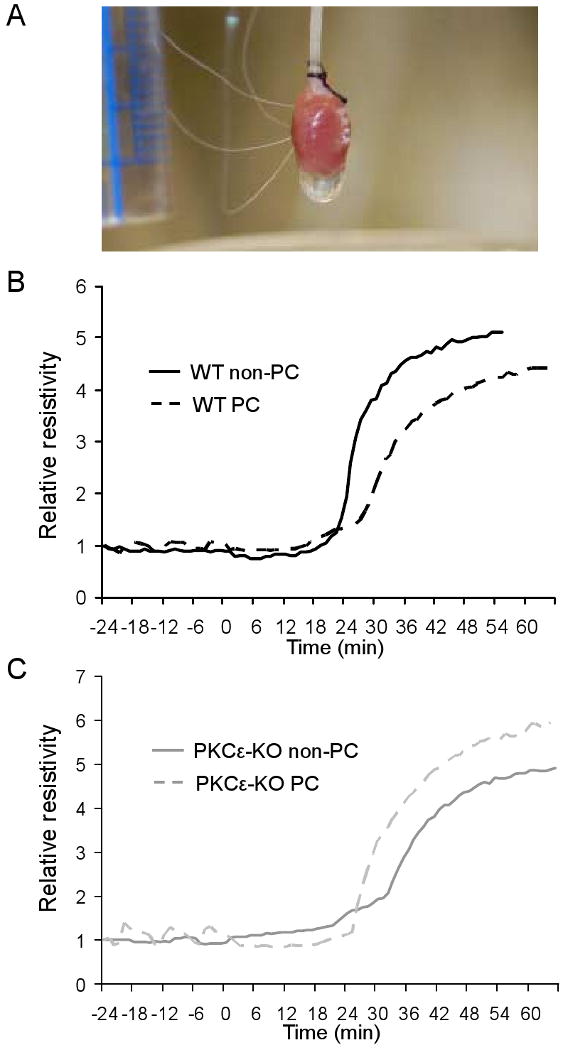
(A) Photograph showing alignment of four electrodes in perfused mouse heart for measurement of whole-tissue resistivity. (B) Representative whole-tissue resistivity curves measured in non-PC and PC hearts from WT animals. Resistivity measurements have been normalized to allow comparisons between experiments. (C) Representative whole-tissue resistivity curves measured in non-PC and PC hearts from PKCε-KO animals.
Discussion
The results of this study indicate that PKCε is necessary for preconditioning to prevent translocation of Cx43 out of gap junctions and to delay electrical uncoupling during subsequent sustained ischemia. Whereas previous studies had implicated a PKC family member as a mediator of preconditioning,7 the use of mice with targeted deletion of PKCε allowed definitive identification of the specific PKC isoform responsible for the palliative effects of preconditioning on electrical coupling. Our findings also show that the timing of PKCε activation is important in mediating the effects of ischemic preconditioning on electrical uncoupling in response to ischemia. Whereas translocation of PKCε from cytosol to membrane fractions occurs during ischemia independent of prior preconditioning, only preconditioned hearts retained Cx43 in gap junctions and exhibited delayed uncoupling, and both of these effects were absent in PKCε-KO hearts. Thus, it appears that activation of PKCε must occur early during injury to produce salutary effects of preconditioning. This conclusion is supported by independent evidence that activation of PKCε by specific peptides prior to ischemia mimicked the effects of preconditioning on Cx43 distribution.
We also identified an unexpected role for PKCε in regulating the amount of Cx43 in gap junctions under normal, basal conditions. Cx43 immunoreactive signal located at sites of cell contacts was significantly greater in PKCε-KO hearts than in wildtype hearts despite the fact that total amounts of Cx43 were the same. While one must be cautious in equating increased immunoreactive signal with a corresponding increase in coupling, these observations suggest that PKCε regulates the subcellular distribution of Cx43 such that when PKCε is eliminated, a greater proportion of total cellular Cx43 resides within the junctional pool. Increased basal levels of Cx43 in gap junctions may also explain why the onset of electrical uncoupling was delayed during ischemia (without prior preconditioning) in PKCε-KO compared to wildtype hearts. Further studies will be required to elucidate mechanisms by which PKCε regulates the amount of Cx43 in gap junctions under basal conditions.
This study provides new insights into potential mechanisms by which PKCε mediates changes in electrical coupling in the heart subjected to ischemia and preconditioning. One mechanism appears to involve regulation of changes in Cx43 subcellular distribution by PKCε, supported by the observations that: 1) PKCε-KO hearts contain more Cx43 in gap junctions and uncouple significantly later than wildtype hearts; and 2) preconditioning prevents loss of Cx43 from gap junctions and delays uncoupling in wildtype but not PKCε-KO hearts. However, the observation that preconditioned and non-preconditioned PKCε-KO hearts show the same level of Cx43 in gap junctions but uncoupling occurs significantly faster in preconditioned hearts suggests an additional mechanism, perhaps involving phosphorylation of Cx43 in gap junction plaques at Ser368. Even though preconditioned PKCε-KO hearts exhibited the same amount of Cx43 in gap junctions as non-preconditioned hearts, significantly more of that Cx43 was phosphorylated at Ser368. In view of evidence linking phosphorylation at Ser368 with changes in Cx43 channel function,26,27 it is plausible to suggest that preconditioning delays uncoupling by limiting Cx43 phosphorylation at Ser368 and, furthermore, that PKCε affects electrical coupling by regulating both the amount and phosphorylation state of Cx43 in gap junctions.
The fact ischemia induced marked Cx43 phosphorylation at Ser368 in PKCε-KO hearts leaves little doubt that another PKC isoform can phosphorylate Cx43 at Ser368. Although we did not systematically investigate all PKC isoforms, one obvious candidate is PKCδ which we found was markedly translocated from cytosolic to membrane fractions during ischemia in PKCε-KO but not wildtype hearts. These observations are consistent with previous studies showing enhanced PKCδ expression and activation in PKCε-KO mice subjected to aortic constriction and transient ischemia.18,39 It is known that PKC isoforms are complexly interdependent40 such that removal of one isoform may affect activation of another.39 Further study is required to determine whether multiple PKC isoforms can phosphorylate Cx43 and whether cross-regulation of PKC isoforms affects Cx43 phosphorylation in the setting of myocardial ischemia.
Phosphorylation of Cx43 at Ser368 is unlikely to be solely responsible for the changes in Cx43 distribution during ischemia, considering the lack of correlation between this phosphorylation event and the amount of Cx43 in gap junctions. Our results also rule out phosphorylation at Ser262 as a possible mechanism. Thus, PKCε may regulate the subcellular distribution of Cx43 indirectly rather than through direct phosphorylation. One potential pathway involves mitochondrial KATP channels which are believed to operate downstream of PKCε.41,42 This hypothesis is consistent with our previous observations that the KATP channel agonist diazoxide prevented translocation of Cx43 from gap junctions during ischemia.7 Another possible pathway involves translocation of Cx43 to the inner mitochondrial membrane, where it is believed to play a role in diazoxide-induced preconditioning, although the role in ischemic preconditioning is unclear.43 PKCε overexpression also modulates expression of an inducible form of the chaperone protein, heat shock protein 70 (Hsp70), in rat neonatal cardiomyocytes44 raising the possibility that PKCε regulates the formation of a Cx43-Hsp90-Hsp70 complex for translocation to the inner mitochondrial membrane.43
It is important to note that the significance of Cx43 phosphorylation at Ser368 in the heart remains controversial as several groups have reported conflicting findings.26,45,46 Our finding that Cx43 phosphorylation at Ser368 increases after 30 min of global ischemia is consistent with a recent study, also in the mouse, from Ek-Vitorin et al.26 However, Matsushita et al. and Axelsen et al. report decreased Cx43 phosphorylation at Ser368 in the rat heart subjected to hypoxic perfusion45 or global ischemia.46 Of course, changes in electrical coupling during ischemia could involve changes in Cx43 phosphorylation at PKC-independent residues. To address this possibility, we probed Cx43 phosphorylation state at four other C-terminal residues (Ser262, Ser255, Ser279/282, and Tyr265) and observed no differences in any group of hearts compared to wild-type control. However, Lampe et al.46 have recently reported dephosphorylation of Cx43 at casein kinase 1 sites Ser325/328/330 coinciding with lateralization of Cx43 during ischemia. Interestingly, dephosphorylation of Cx43 during hypoxia has also been linked to decreased availability of ATP.47 In any event, more detailed information about changes in Cx43 phosphorylation state at particular residues is needed to fully understand molecular mechanisms of Cx43 redistribution and electrical uncoupling during ischemia.
Spatially heterogeneous electrical uncoupling of the myocardium during acute ischemia produces regions of slow conduction that predisposes the heart to arrhythmias.48 Pharmacological targeting of gap junctions to improve intercellular communication is emerging as a potentially promising anti-arrhythmic strategy.49,50 Likewise, recent studies have shown that PKCε activation is anti-arrhythmic in the setting of ischemia-reperfusion.20,21 The results of the present study support the notion that PKCε should be added to the growing list of protein kinases in a drug target class with growing promise for treatment of cardiac arrhythmia.
Acknowledgments
We thank Evelyn Kanter and Erin Gribben for expert technical assistance.
Sources of Support: This work was supported by NIH grants HL50598, HL066350 and HL084979, and by a grant from the Barnes Foundation.
Footnotes
Conflicts of interest: None for any of the authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Y, Downey JM. Ischemic preconditioning protects against infarction in rat heart. Am J Physiol Heart Circ Physiol. 1992;263:H1107–1112. doi: 10.1152/ajpheart.1992.263.4.H1107. [DOI] [PubMed] [Google Scholar]
- 2.Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- 3.Schott RJ, Rohmann S, Braun ER, Schaper W. Ischemic preconditioning reduces infarct size in swine myocardium. Circ Res. 1990;66:1133–1142. doi: 10.1161/01.res.66.4.1133. [DOI] [PubMed] [Google Scholar]
- 4.Wu ZK, Iivainen T, Pehkonen E, Laurikka J, Tarkka MR. Ischemic preconditioning suppresses ventricular tachyarrhythmias after myocardial revascularization. Circulation. 2002;106:3091–3096. doi: 10.1161/01.cir.0000041430.32233.5b. [DOI] [PubMed] [Google Scholar]
- 5.Hagar JM, Hale SL, Kloner RA. Effect of preconditioning ischemia on reperfusion arrhythmias after coronary artery occlusion and reperfusion in the rat. Circ Res. 1991;68:61–68. doi: 10.1161/01.res.68.1.61. [DOI] [PubMed] [Google Scholar]
- 6.Tan H, Mazon P, Verberne H, Sleeswijk M, Coronel R, Opthof T, Janse MJ. Ischaemic preconditioning delays ischemia induced cellular electrical uncoupling in rabbit myocardium by activation of ATP-sensitive potassium channels. Cardiovasc Res. 1993;27:644–651. doi: 10.1093/cvr/27.4.644. [DOI] [PubMed] [Google Scholar]
- 7.Jain SK, Schuessler RB, Saffitz JE. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ Res. 2003;92:1138–1144. doi: 10.1161/01.RES.0000074883.66422.C5. [DOI] [PubMed] [Google Scholar]
- 8.Cinca J, Warren M, Carreno A, Tresanchez M, Armadans L, Gomez P, Soler-Soler J. Changes in myocardial electrical impedance induced by coronary artery occlusion in pigs with and without preconditioning: correlation with local ST-segment potential and ventricular arrhythmias. Circulation. 1997;96:3079–3086. doi: 10.1161/01.cir.96.9.3079. [DOI] [PubMed] [Google Scholar]
- 9.Miura T, Ohnuma Y, Kuno A, Tanno M, Ichikawa Y, Nakamura Y, Yano T, Miki T, Sakamoto J, Shimamoto K. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;286:H214–221. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- 10.Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. Faseb J. 2003;17:1355–1357. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- 11.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 12.Schwanke U, Konietzka I, Duschin A, Li X, Schulz R, Heusch G. No ischemic preconditioning in heterozygous connexin43-deficient mice. Am J Physiol Heart Circ Physiol. 2002;283:H1740–1742. doi: 10.1152/ajpheart.00442.2002. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Heinzel FR, Boengler K, Schulz R, Heusch G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol. 2004;36:161–163. doi: 10.1016/j.yjmcc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Ytrehus K, Downey JM. Evidence that translocation of protein kinase C is a key event during ischemic preconditioning of rabbit myocardium. J Mol Cell Cardiol. 1994;26:661–668. doi: 10.1006/jmcc.1994.1078. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell MB, Meng X, Ao L, Brown JM, Harken AH, Banerjee A. Preconditioning of isolated rat heart is mediated by protein kinase C. Circ Res. 1995;76:73–81. doi: 10.1161/01.res.76.1.73. [DOI] [PubMed] [Google Scholar]
- 16.Speechly-Dick ME, Mocanu MM, Yellon DM. Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res. 1994;75:586–590. doi: 10.1161/01.res.75.3.586. [DOI] [PubMed] [Google Scholar]
- 17.Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, Marber MS. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res. 2002;55:672–680. doi: 10.1016/s0008-6363(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 18.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase Cε. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 19.Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc Natl Acad Sci USA. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 21.Yue Y, Qu Y, Boutjdir M. Protective role of protein kinase C epsilon activation in ischemia-reperfusion arrhythmia. Biochem Biophys Res Commun. 2006;349:432–438. doi: 10.1016/j.bbrc.2006.08.068. [DOI] [PubMed] [Google Scholar]
- 22.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 24.Doble BW, Ping P, Kardami E. The ε subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ Res. 2000;86:293–301. doi: 10.1161/01.res.86.3.293. [DOI] [PubMed] [Google Scholar]
- 25.Bowling N, Huang X, Sandusky GE, Fouts RL, Mintze K, Esterman M, Allen PD, Maddi R, McCall E, Vlahos CJ. Protein kinase C-α and -ε modulate connexin-43 phosphorylation in human heart. J Mol Cell Cardiol. 2001;33:789–798. doi: 10.1006/jmcc.2000.1349. [DOI] [PubMed] [Google Scholar]
- 26.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res. 2006;98:1498–1505. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- 28.Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, 2nd, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of δPKC and εPKC. Proc Natl Acad Sci USA. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson CM, Kanter EM, Green KG, Laing JG, Betsuyaku T, Beyer EC, Steinberg TH, Saffitz JE, Yamada KA. Redistribution of connexin45 in gap junctions of connexin43-deficient hearts. Cardiovasc Res. 2002;53:921–935. doi: 10.1016/s0008-6363(01)00522-3. [DOI] [PubMed] [Google Scholar]
- 31.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms ε and η in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 32.Kwong KF, Schuessler RB, Green KG, Laing JG, Beyer EC, Boineau JP, Saffitz JE. Differential expression of gap junction proteins in the canine sinus node. Circ Res. 1998;82:604–612. doi: 10.1161/01.res.82.5.604. [DOI] [PubMed] [Google Scholar]
- 33.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol Heart Circ Physiol. 2000;278:H1662–1670. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 34.Plonsey R, Barr R. The four-electrode resistivity technique as applied to cardiac muscle. IEEE Trans Biomed Eng. 1982;29:541–546. doi: 10.1109/tbme.1982.324927. [DOI] [PubMed] [Google Scholar]
- 35.Kleber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res. 1987;61:271–279. doi: 10.1161/01.res.61.2.271. [DOI] [PubMed] [Google Scholar]
- 36.Smith WTt, Fleet WF, Johnson TA, Engle CL, Cascio WE. The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Circulation. 1995;92:3051–3060. doi: 10.1161/01.cir.92.10.3051. [DOI] [PubMed] [Google Scholar]
- 37.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 38.Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- 39.Klein G, Schaefer A, Hilfiker-Kleiner D, Oppermann D, Shukla P, Quint A, Podewski E, Hilfiker A, Schroder F, Leitges M, Drexler H. Increased collagen deposition and diastolic dysfunction but preserved myocardial hypertrophy after pressure overload in mice lacking PKCepsilon. Circ Res. 2005;96:748–755. doi: 10.1161/01.RES.0000161999.86198.1e. [DOI] [PubMed] [Google Scholar]
- 40.Rybin VO, Sabri A, Short J, Braz JC, Molkentin JD, Steinberg SF. Cross-regulation of novel protein kinase C (PKC) isoform function in cardiomyocytes. Role of PKC epsilon in activation loop phosphorylations and PKC delta in hydrophobic motif phosphorylations. J Biol Chem. 2003;278:14555–14564. doi: 10.1074/jbc.M212644200. [DOI] [PubMed] [Google Scholar]
- 41.Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A, Shimamoto K. Opening of mitochondrial KATP channel occurs downstream of PKC-ε activation in the mechanism of preconditioning. Am J Physiol Heart Circ Physiol. 2002;283:H440–447. doi: 10.1152/ajpheart.00434.2001. [DOI] [PubMed] [Google Scholar]
- 42.Hassouna A, Matata BM, Galinanes M. PKC-epsilon is upstream and PKC-alpha is downstream of mitoKATP channels in the signal transduction pathway of ischemic preconditioning of human myocardium. Am J Physiol Cell Physiol. 2004;287:C1418–1425. doi: 10.1152/ajpcell.00144.2004. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, Konietzka I, Miro E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99:93–101. doi: 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]
- 44.Coaxum SD, Martin JL, Mestril R. Overexpression of heat shock proteins differentially modulates protein kinase C expression in rat neonatal cardiomyocytes. Cell Stress Chaperones. 2003;8:297–302. doi: 10.1379/1466-1268(2003)008<0297:oohspd>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsushita S, Kurihara H, Watanabe M, Okada T, Sakai T, Amano A. Alterations of phosphorylation state of connexin 43 during hypoxia and reoxygenation are associated with cardiac function. J Histochem Cytochem. 2006;54:343–353. doi: 10.1369/jhc.4A6611.2005. [DOI] [PubMed] [Google Scholar]
- 46.Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein-Rathlou NH, Andersen S, Jensen ON, Hennan JK, Kjolbye AL. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123) J Mol Cell Cardiol. 2006;40:790–798. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, Webster KA, Bishopric NH. Reversible Connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004;95:726–733. doi: 10.1161/01.RES.0000144805.11519.1e. [DOI] [PubMed] [Google Scholar]
- 48.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- 49.Hennan JK, Swillo RE, Morgan GA, Keith JC, Jr, Schaub RG, Smith RP, Feldman HS, Haugan K, Kantrowitz J, Wang PJ, Abu-Qare A, Butera J, Larsen BD, Crandall DL. Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J Pharmacol Exp Ther. 2006;317:236–243. doi: 10.1124/jpet.105.096933. [DOI] [PubMed] [Google Scholar]
- 50.Guerra JM, Everett THt, Lee KW, Wilson E, Olgin JE. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation. 2006;114:110–118. doi: 10.1161/CIRCULATIONAHA.105.606251. [DOI] [PubMed] [Google Scholar]