

Abstract
The trans-Golgi network (TGN) is the major sorting station in the secretory pathway of all eukaryotic cells. How the TGN sorts proteins and lipids to generate the enrichment of sphingolipids and sterols at the plasma membrane is poorly understood. To address this fundamental question in membrane trafficking, we devised an immunoisolation procedure for specific recovery of post-Golgi secretory vesicles transporting a transmembrane raft protein from the TGN to the cell surface in the yeast Saccharomyces cerevisiae. Using a novel quantitative shotgun lipidomics approach, we could demonstrate that TGN sorting selectively enriched ergosterol and sphingolipid species in the immunoisolated secretory vesicles. This finding, for the first time, indicates that the TGN exhibits the capacity to sort membrane lipids. Furthermore, the observation that the immunoisolated vesicles exhibited a higher membrane order than the late Golgi membrane, as measured by C-Laurdan spectrophotometry, strongly suggests that lipid rafts play a role in the TGN-sorting machinery.
Introduction
One of the major functions of membrane trafficking in eukaryotic cells is to provide each organelle connected within the system with its functional complement of proteins and lipids (van Meer, 1989; Mellman and Warren, 2000). Biosynthesis of secretory proteins starts in the ER, and the glycosylation of proteins is completed in the Golgi apparatus (Mellman and Warren, 2000). From the TGN, proteins are distributed to the plasma membrane (PM), endosomes, and lysosomes (Griffiths and Simons, 1986; Traub and Kornfeld, 1997; Bard and Malhotra, 2006; Pfeffer, 2007). The lipid composition changes progressively throughout the secretory pathway; the ER displays a relatively low concentration of sterols and sphingolipids, which in turn accumulate toward the PM (Simons and van Meer, 1988; van Meer and Simons, 1988; van Meer, 1989; Zinser and Daum, 1995).
Lipid rafts have been proposed to be involved in the generation of the lipid gradients in the secretory pathway (Simons and Ikonen, 1997). These membrane structures have evolved from controversial detergent-resistant entities to dynamic, nanometer-sized membrane domains formed by sterols, sphingolipids, saturated glycerophospholipids, and proteins (Simons and Vaz, 2004; Hancock, 2006). Lipid raft coalescence can be induced by lipid–protein and protein–protein interactions to form ordered membrane microdomains involved in signal transduction (Simons and Toomre, 2000), virus assembly (Brügger et al., 2006), and membrane trafficking (Schuck and Simons, 2004). Raft involvement in TGN cargo sorting was suggested from work on apical transport in epithelial cells and formed the basis for the raft concept as it was first formulated (Simons and van Meer, 1988; Simons and Ikonen, 1997). However, direct evidence indicating sorting of lipids in membrane trafficking is still lacking, and the function of rafts in the formation of post-Golgi transport carriers remains elusive.
In this work, we aimed to provide answers to the issue of whether the TGN can sort lipids during transport carrier assembly. If raft coalescence plays a functional role in the sorting machinery that forms carrier vesicles transporting raft proteins to the cell surface, these secretory vesicles should be selectively enriched in sterols and sphingolipids. However, testing this hypothesis has so far been hampered by numerous shortcomings of biochemical and analytical methodology available for such experiments. In this study, we used the yeast Saccharomyces cerevisiae as our experimental system. The molecular lipid composition of S. cerevisiae includes ergosterol (in contrast to cholesterol in mammalian cells), three classes of inositol-containing sphingolipids, inositolphosphoceramide (IPC), mannosyl-IPC (MIPC), and mannosyl-di-IPC (M[IP]2C), glycerophospholipids, and glycerolipids (Daum et al., 1998; Holthuis et al., 2001). Similar to epithelial cells, yeast uses at least two distinct cell surface delivery pathways (Simons and Wandinger-Ness, 1990; Wandinger-Ness et al., 1990; Harsay and Bretscher, 1995; Bagnat et al., 2000; Gurunathan et al., 2002; Harsay and Schekman, 2002; Rodriguez-Boulan et al., 2005). The pathway that transports raft proteins from the TGN to the PM is mediated by a population of light density secretory vesicles (LDSVs), which are similar to apical transport carriers in epithelial cells (Harsay and Bretscher, 1995; Bagnat et al., 2000). In a visual genome-wide screen for identifying sorting factors at the TGN, we found that the chimeric raft protein FusMidGFP displayed a trafficking phenotype in mutants with sphingolipid and ergosterol biosynthesis defects (Proszynski et al., 2004, 2005). Thus, we reasoned that the formation of the TGN-derived secretory vesicles could be generated based on a raft clustering mechanism. To purify the vesicles transporting FusMidGFP, we developed an immunoisolation procedure and implemented a shotgun lipidomics approach for quantitative characterization of the lipidome of secretory vesicles and the donor compartment.
Results
Immunoisolation of TGN-derived secretory vesicles
To isolate TGN-derived secretory vesicles in high purity, we devised an immunoisolation procedure using a transmembrane raft protein as bait. We engineered the bait from the chimeric protein FusMidGFP, a type I transmembrane O-glycosylated raft protein which is directly delivered from the TGN to the PM in LDSVs (Fig. 1; Proszynski et al., 2004, 2005).
Figure 1.

Intracellular accumulation of the immunoisolation baits at the restrictive temperature 37°C in sec6-4 cells. (A) FusMidGFPLTLM9, the raft-carrier cargo immunoisolation bait, and InvRFP expressed for 45 min. At the permissive temperature 24°C, FusMidGFPLTLM9 reached the PM, whereas InvRFP was secreted and therefore not visible (for InvRFP secretion, see also Fig. S3 B). At the restrictive temperature 37°C, both proteins accumulated intracellularly. (B) Gap1GFPLTLM9, the TGN/E immunoisolation bait, and InvRFP expressed for 45 min. At the permissive temperature 24°C, InvRFP was secreted and, thus, not visible (Fig. S3 B), and Gap1GFPLTLM9 localized to intracellular compartments. At the restrictive temperature 37°C, both proteins accumulated intracellularly. DIC, differential interference contrast. Bars, 2 µm.
Several experimental factors were important to ensure successful immunoisolation of secretory vesicles. First, we had to localize the bait protein specifically into the TGN-derived transport carriers. Second, the bait protein had to be engineered with a high affinity epitope for immunoisolation. Third, we developed a specific immunoadsorbent with high binding capacity and low unspecific binding. Fourth, we introduced a protease cleavage site into the bait that enabled tobacco etch virus (TEV) protease–specific release of the secretory vesicles from the immunoadsorbent (Aebersold and Mann, 2003).
At the C terminus of the bait protein FusMidGFP, we introduced a high affinity 9× myc (M9) tag. A TEV protease site (T) was inserted between GFP and M9 and flanked by linker regions (L), generating the immunoisolation raft carrier bait FusMidGFPLTLM9, hereafter abbreviated as FusMidp.
To isolate post-Golgi secretory vesicles from living cells, we expressed the bait FusMidp in the temperature-sensitive exocyst mutant sec6-4 (Novick et al., 1980). When expressed at the permissive temperature 24°C, FusMidp reached the cell surface, as observed by fluorescence microscopy (Fig. 1 A). At the restrictive temperature 37°C, FusMidp accumulated intracellularly and did not translocate to the PM, as demonstrated by fluorescence microscopy and biochemistry (Fig. 1 A and Fig. S1 A). Electron microscopy demonstrated that the block of secretion at 37°C led to intracellular accumulation of vesicles (Fig. 2, A and B). By tomography, we observed that the vesicles were spherical with a diameter of ∼100 nm (Fig. 2 C and Video 1).
Figure 2.

Accumulation of secretory vesicles in sec6-4 cells at the restrictive temperature 37°C. (A) Transmission electron micrograph of a budding sec6-4 cell cultured at the permissive temperature 24°C. (B) Transmission electron micrograph of a budding sec6-4 cell cultured for 45 min at the restrictive temperature 37°C. White arrowheads indicate accumulated secretory vesicles with a diameter of ∼100 nm. (C) 3D reconstruction of a budding cell with accumulated secretory vesicles (red) in a tomogram recorded from a 200-nm thick section prepared from the same culture of cells as shown in B; the PM is reconstructed in blue, the vacuole is orange, and other organelles such as Golgi cisternae or endosomal structures are in yellow. V, vacuole. Bars, 1 µm.
For the specific isolation of TGN-derived FusMidp-vesicles, we combined conventional subcellular fractionation with a novel immunoisolation procedure. Based on previous work, we designed an immunoadsorbent composed of cellulose and sheep anti–mouse antibody (Hales and Woodhead, 1980; Wandinger-Ness et al., 1990). This immunoadsorbent showed high binding specificity, quantitative vesicle pickup, and efficient organelle release after TEV protease cleavage.
Secretory vesicles were isolated by cell lysis, differential fractionation, and organelle prepurification by isopycnic sucrose gradient centrifugation followed by immunoisolation (Figs. S2 and S3). Mouse anti-myc antibody was added to the prepurified organelle fraction enriched in the bait FusMidp and incubated with the sheep anti–mouse immunoadsorbent. Vesicles were specifically eluted by addition of TEV protease, producing the expected truncation of the bait (Fig. 3 A).
Figure 3.

Immunoisolation of FusMidp-vesicles and the TGN/E. (A and B) Several organelle markers were monitored throughout the isolation of FusMidp-vesicles (A) and the TGN/E (B), with GFP detecting the respective baits. InvRFP was used as a specific marker for HDSVs. Gas1p is the major GPI-anchored protein in yeast and is an LDSV marker. Pep12p is the late endosome t-SNARE, Dpm1p is an ER protein, and Tlg1p is a late Golgi marker. Input, sucrose gradient fraction 7 (A) and 4 (B); FT, flow through; Bind, material bound to the immunoadsorbent; Wash, supernatant of the last wash; Remain, material not released from the cellulose fibers after TEV cleavage; ELUATE, eluted material released from the immunoadsorbent after TEV cleavage.
Immunodepletion of endosomes reduces organelle cross-contamination of immunoisolated secretory vesicles
To specifically recover FusMidp–secretory vesicles, we implemented the immunodepletion of endosomes by applying a mouse antibody against the endosomal t-SNARE Pep12p (Fig. 3 A). The addition of TEV protease released the bait-labeled vesicles specifically because Pep12p was not cleaved and therefore retained endosomes on the immunoadsorbent. This strategy was essential for the successful purification of the FusMidp-vesicles (Fig. 3 A) and their molecular characterization.
Assessment of the purity of FusMidp-vesicles
To document the relative enrichment of different organelle markers throughout the isolation procedure, we performed Western blot analysis. First, we showed that the bait protein was fully glycosylated (Proszynski et al., 2004) in the immunoisolated vesicles, thus confirming that they were TGN derived (Fig. 3 A). We verified that binding and release of the FusMidp-vesicles were specific because an unspecific mouse antibody did not retain vesicles on the immunoadsorbent (Fig. S3 A, b). Moreover, release of a bait construct lacking the TEV protease site was not possible (Fig. S3 A, c). Importantly, in analyzing whether other potential raft proteins were included in the vesicles, we found that the major glycosyl-phosphatidylinositol (PI [GPI])–anchored protein Gas1p (Mayor and Riezman, 2004) was present in FusMidp-vesicles (Fig. 3 A).
The soluble secreted protein invertase (the RFP fusion protein invertase-RFP [InvRFP]), a marker of an alternative cell surface delivery pathway mediated by heavy density secretory vesicles (HDSVs; Harsay and Bretscher, 1995), was not detectable in the eluate, demonstrating that FusMidp-vesicles are distinct from InvRFP-carriers (Fig. 3 A). We confirmed by Western blotting of the extracellular medium that InvRFP was correctly processed at both temperatures and secreted by sec6-4 cells at 24°C (Fig. S3 B).
Assessing the contamination with ER, we monitored the protein Dpm1p, which was efficiently depleted by subcellular fractionation (in P20; Fig. S1, A and B). Residual amounts of Dpm1p were detected in the sucrose gradient (Fig. S2 A), but Dpm1p was essentially removed from the isolated vesicles (Fig. 3 A). The design of the immunoisolation procedure also depleted Pep12p-containing endosomes (Fig. 3 A). As expected, residual amounts of late Golgi vesicular t-SNARE Tlg1p were present in the isolated FusMidp-vesicles (Fig. 3 A; Holthuis et al., 1998).
In addition, we verified that the sample amounts used for assessment of the vesicle purification gave responses within the dynamic range of the Western blot analysis (unpublished data). We hereby conclude that we have established a robust immunoisolation procedure that, for the first time, allows specific isolation of a pure population of secretory vesicles.
Morphology and integrity of immunoisolated secretory vesicles
To assess whether the immunoisolation procedures preserved the integrity of the secretory vesicles, we first examined the morphology of the FusMidp-vesicles by electron microscopy. We found a homogenous population of spherical vesicles with a diameter of 100 nm (Fig. 4). This result agrees well with the size of the vesicles observed in living sec6-4 cells at 37°C (Fig. 2, B and C). To evaluate the integrity of the isolated vesicles, we performed a floatation experiment as previously described (Harsay and Bretscher, 1995). We bottom loaded the immunoisolated FusMidp-vesicles on a linear Nycodenz gradient and centrifuged to equilibrium. The vesicle marker FusMidp and the GPI-anchored Gas1p floated to the top of the gradient with densities as LDSVs (Fig. S4; Harsay and Bretscher, 1995). In addition, the soluble secretory LDSV marker Bgl2p cofractionated with FusMidp and Gas1p. Thus, we concluded that the immunoisolation procedure produced a homogenous population of secretory vesicles with preserved morphology and integrity.
Figure 4.

Transmission electron micrographs of FusMidp-vesicles in different magnifications. (A) Overview of FusMidp-vesicles in an electron micrograph shows the morphological homogeneity of the FusMidp-vesicle population with a diameter of ∼100 nm. These isolated vesicles have the same morphological characteristics as the structures that accumulate in sec6-4 cells at 37°C; see Fig. 2 (B and C). (B) Transmission electron micrograph of a single FusMidp-vesicle. The 100-nm vesicular structure is formed by a membrane whose electron density pattern shows the integrity of the lipid bilayer. (C) A group of FusMidp-vesicles. The lipid bilayer is visible as in B. Bars: (A) 1,000 nm; (B and C) 100 nm.
Immunoisolation of the TGN/endosome system (TGN/E)
For comparative purposes, we devised an approach to isolate the donor organelle of the FusMidp-vesicles. In S. cerevisiae, the TGN is tightly connected to endosomes. Surprisingly, there is no defined protein that exclusively localizes to the TGN. Known TGN residents cycle between the TGN and endosomes (Pelham, 1999). Gap1p is such a protein that cycles under our experimental conditions within these compartments (Roberg et al., 1997; Gao and Kaiser, 2006) and does not mislocalize after epitope tagging at the C terminus (Fig. 1 B). Thus, we used Gap1GFPLTLM9 as bait for the immunoisolation of the donor compartment TGN/E. We applied the same immunoisolation procedure as for the FusMidp-vesicles but without anti-Pep12p antibody–mediated endosome depletion.
Western blot analysis showed (Fig. 3 B) that the isolated TGN/E contained Gap1GFP, which, after tag cleavage, exhibited the expected shift in electrophoretic mobility. The ER marker Dpm1p could not be detected in the TGN/E. We also confirmed that the TGN/E did not contain detectable amounts of the GPI-anchored raft protein Gas1p (Muniz et al., 2001) and that it was separated from the HDSV marker InvRFP (Fig. 3 B). Isolated TGN/E contained both the late Golgi/early endosome t-SNARE Tlg1p and the late endosome marker Pep12p (Fig. 3, A and B). Given that we recovered and analyzed comparable amounts of FusMidp-vesicles and TGN/E (with total lipid concentrations of 25 pmol/µl and 33 pmol/µl, respectively), we concluded that the immunoisolation protocol served as an efficient tool for the isolation of compositionally distinct secretory vesicles and the donor compartment TGN/E.
Furthermore, electron microscopy demonstrated that immunoisolated TGN/E was comprised of a heterogeneous population of 100–300-nm membrane structures and small vesicles of ∼40–50 nm (unpublished data). Moreover, the FusMidp-vesicles and TGN/E were morphologically distinct from purified yeast PM (Fig. S1 C).
Because the vesicle isolation was performed at 4°C, which can induce phase separation and vesicle pinch-off in simple model membranes (Lipowsky, 1993), we sought to determine whether the immunoisolation of vesicles from cells could be compromised by an artifactual fragmentation of the donor compartment. To do this, we performed a set of control experiments. First, we examined whether there was a difference in the amount of vesicles produced during subcellular fractionation at 4 and 24°C (Fig. S5 A), and, second, we investigated whether subcellular fractionation at 4 and 24°C affected the abundance of secretory cargo within the TGN/E (Fig. S5 B). These data showed that the immunoisolated secretory vesicles were not contaminated with artifactual membrane entities created by purification at 4°C.
Collectively, these results demonstrated that the immunoisolation procedure recovered a specific population of secretory vesicles from living cells, carrying the raft proteins FusMidp and Gas1p, and that the secretory vesicles had a distinct morphology and composition in comparison with their donor compartment TGN/E and the yeast PM.
FusMidp-vesicles are enriched in ergosterol and sphingolipids
To determine whether the TGN exhibits the capacity to selectively sort lipids, we performed a comparative lipidomics study of FusMidp-vesicles and the TGN/E. To do this, we used a novel quantitative shotgun lipidomics platform for absolute quantification (i.e., picomole or mole percentage) of ergosterol, sphingolipid, glycerophospholipid, and glycerolipid species that yields accurate assessment of the stoichiometric relationship between the membrane lipid constituents (Ejsing et al., 2009). Lipid species were quantified by spiking samples with defined amounts of internal lipid standards for each monitored lipid class, which correct for biased lipid recoveries and differential ionization efficiencies in the subsequent steps of lipid extraction and quantitative mass spectrometric analysis (Ejsing et al., 2006a,b). Comparative lipidome analysis of FusMidp-vesicles, the TGN/E, and yeast cell lysates resulted in the absolute quantification of 83 molecular lipid species constituting 12 lipid classes.
The most abundant lipid species in the FusMidp-vesicles was ergosterol, comprising 22.8 mol% of the lipidome (corresponding to 5.7 pmol/µl), followed by 11.1 mol% M(IP)2C 18:0;3/26:0;1 (Fig. 5). The lipidome of the TGN/E contained 8.5 mol% PI 16:0-16:1 as the most abundant lipid species followed by 9.8 mol% ergosterol. In comparison, ergosterol and M(IP)2C 18:0;3/26:0;1 were 2.3- and 2.6-fold enriched, respectively, in the FusMidp-vesicles as compared with the TGN/E. Furthermore, we observed a similar enrichment of IPC 18:0;3/26:0;1 (2.2-fold) and MIPC 18:0;3/26:0;1 (2.4-fold) in the FusMidp-vesicles. Evaluating the lipid class composition (sum of all lipid species constituting the same lipid class), we observed that PI comprised the most abundant class in both FusMidp-vesicles and the TGN/E (Fig. 6). In addition to the enrichment of ergosterol and sphingolipids, the FusMidp-vesicles also showed a 3.2-fold enrichment of phosphatidic acid (PA). FusMidp-vesicles displayed a concomitant 3.7–1.6-fold reduction in phosphatidylserine (PS), phosphatidylethanolamine (PE), DAG, and phosphatidylcholine (PC) as compared with TGN/E. Comparing the lipid composition of FusMidp-vesicles and the TGN/E with that of total yeast cell extracts (Fig. 6), we concluded that the immunoisolation procedure enabled the specific recovery of subcellular organelles having selective differences in molecular lipid composition in addition to the differences in protein composition and morphology.
Figure 5.

Molecular lipid species composition of FusMidp-vesicles and the donor TGN/E. Quantitative lipidomic analysis of FusMidp-vesicles and the TGN/E allowed absolute quantification of 83 molecular lipid species. Lipid composition is shown in mole percentage to demonstrate the stoichiometric relationship between the lipid species (n = 4 independent experiments; mean estimate ± SD). Chemical structures of membrane lipids are illustrated on the right. Lipid species are annotated by their molecular composition (see Materials and methods). Cer, ceramide; PG, phosphatidylglycerol.
Figure 6.

Lipid class composition of FusMidp-vesicles, the TGN/E, and total cell extract. The mole percentage of lipid class was calculated as the sum of the mole percentage of lipid species of the respective lipid class. (n = 4 independent experiments; mean estimate ± SD). Cer, ceramide; PG, phosphatidylglycerol.
The membrane organization of FusMidp-vesicles is more ordered than the TGN/E
Because the lipidome data demonstrated that the FusMidp-vesicles had a composition compatible with raft lipid enrichment (ergosterol and sphingolipids; Simons and Vaz, 2004), we sought to determine whether there was a difference in the membrane structure of the FusMidp-vesicles as compared with the TGN/E. If the enrichment of ergosterol and sphingolipids in FusMidp-vesicles were to reflect a difference in bilayer order caused by raft coalescence, the prediction would be that the bilayer would be more packed than the donor membranes from where the vesicles form (Simons and Vaz, 2004). To measure membrane order, we used a spectrophotometric assay based on fluorescent C-Laurdan (Parasassi et al., 1991; Gaus et al., 2006; Kim et al., 2007). The fluorescence emission spectrum of C-Laurdan is sensitive to membrane order and may be used to calculate a parameter called the general polarization (GP), whose magnitude serves as a relative measure of membrane order. The data demonstrated that the membrane of the FusMidp-vesicles (GP = 0.24 ± 0.01) was more ordered than the TGN/E (GP = 0.09 ± 0.05; Fig. 7). This result indicated that the biogenesis of FusMidp-vesicles involves a modulation of membrane architecture during its formation at the TGN.
Figure 7.

C-Laurdan spectrophotometry reveals higher membrane order for FusMidp-vesicle as compared with the TGN/E. Immunoisolated membranes were stained with C-Laurdan and subsequently analyzed by fluorescence spectrophotometry. The GP is a relative measure for membrane order (see Materials and methods). FusMidp-vesicles (red bar) exhibited a higher GP value than the TGN/E (blue bar; n = 3 independent experiments; mean estimate ± SD).
Discussion
Selective lipid segregation in cargo sorting at the TGN
It is well known that all steps in the biosynthetic and endocytic transport pathways involve protein sorting. Despite numerous genetic screens designed to identify the molecular sorting machinery at the TGN that generates transport carriers targeted to the cell surface, we are still left with an incomplete picture of this fundamental process (Novick et al., 1980; Walch-Solimena and Novick, 1999; Proszynski et al., 2005; Sciorra et al., 2005; Harsay and Schekman, 2007).
One model in the field is that lipid raft clustering contributes to sorting processes at the TGN (Simons and Ikonen, 1997; Mellman and Warren, 2000; Rodriguez-Boulan et al., 2005). The main consequence for the membrane lipid composition of a vesicle that is generated by such a lipid raft–dependent TGN sorting mechanism would be a selective enrichment for sterols and sphingolipids. Until now, rigorous testing of this prediction was hindered by technical caveats in methodology; it was not possible to isolate TGN-derived secretory vesicles with a purity sufficient to enable the characterization of their molecular lipid composition. Furthermore, methodology for assessing the stoichiometric relationship between the membrane lipid constituents was not available.
The main focus of this work was to determine whether the lipid composition of TGN-derived secretory vesicles transporting the raft protein FusMidp would change during formation at the TGN. By engineering a novel immunoisolation procedure, we were able to purify secretory vesicles and late Golgi compartments from living cells. Western blot analysis and electron microscopy demonstrated that the immunoisolated FusMidp-vesicles represent a specific population of secretory vesicles. We compared the molecular lipid composition of FusMidp-vesicles with their donor organelle TGN/E using a novel quantitative shotgun lipidomics platform with unsurpassed analytical sensitivity and specificity (Ejsing et al., 2009). The comparative lipidome analysis demonstrated that FusMidp-vesicles comprised 2.3-fold more ergosterol and sphingolipids as compared with the TGN/E (Fig. 5). These compositional differences between vesicle and donor compartment membranes unequivocally demonstrate that lipid sorting occurs in the TGN. Furthermore, the enrichment of these lipids corroborates the model that the TGN sorting machinery uses a raft-clustering mechanism during the formation of secretory vesicles. In addition to the enrichment of ergosterol and sphingolipids, the lipid analysis also documented differences in other lipid classes between the FusMidp-vesicles and the TGN/E. PA was also elevated in the FusMidp-vesicles (Figs. 5 and 6). This lipid has previously been implicated in post-Golgi secretion through phospholipase D action (Routt et al., 2005). Interestingly, PS, PE, and PC were depleted in FusMidp-vesicles.
The molecular constituents of secretory vesicles suggest a functional role for sphingolipids and sterols in TGN sorting
The results presented in this study corroborate our previous finding that ergosterol and sphingolipids are functionally important for cell surface transport of FusMidp (Proszynski et al., 2005). Several other studies have also implicated ergosterol and sphingolipids in the delivery of other raft-associated proteins, including Gas1p, from the Golgi to the PM in yeast (Bagnat et al., 2000; Eisenkolb et al., 2002; Umebayashi and Nakano, 2003; Gaigg et al., 2005, 2006). Interestingly, sphingolipid mutants identified in the screen produce sphingolipid species with shortened fatty acid moieties (elo3Δ) or species having a dihydrosphingosine (sur2Δ) instead of a phytosphingosine moiety (Haak et al., 1997; Oh et al., 1997; Ejsing et al., 2009). These molecular attributes have been implicated in the formation of liquid-ordered domains by augmenting sphingolipid–sterol interactions by hydrogen bonding (sur2Δ cannot hydroxylate the long chain base at C4; Kuikka et al., 2001) and by coupling the two membrane leaflets in raft domains by interdigitation of the very long chain fatty acid moiety from the luminal leaflet into the underlying cytoplasmic leaflet (elo3Δ synthesizes C22:0 and C24:0 fatty acids instead of C26:0; Veiga et al., 2001). The effects of these seemingly minor modifications of lipid structure on membrane trafficking only emphasize the importance of lipidomic analysis in studies of membrane trafficking.
We note that the lipid composition of transport vesicles that exit from the Golgi toward endosomes and lysosomes remains to be determined. However, from our finding that the TGN/E contains a lower ergosterol and sphingolipid concentration as compared with the FusMidp-vesicles, we predict that the route to the endosomes is less dependent on these lipids. This prediction is corroborated by the absence of ergosterol and sphingolipid mutants in genetic screens analyzing these pathways (Seaman et al., 1997; Bonangelino et al., 2002). Another important open issue is the lipid composition of the vesicles carrying invertase. Unfortunately, the transmembrane receptor for invertase is unknown, and, therefore, we could not address this aspect.
Mechanistic implications
The functional involvement of raft-based sorting in the assembly of transport carriers at the TGN could imply the formation of stabilized liquid-ordered domains from small rafts in the TGN membrane (Simons and Ikonen, 1997; Hancock, 2006). Raft coalescence would be promoted by an increase in sphingolipid and sterol concentration toward the trans side of the Golgi complex. Work from our laboratory has recently demonstrated that raft coalescence can be induced at a physiologically relevant temperature to form domains selectively enriched in raft proteins and lipids (Lingwood et al., 2008). Thus, induction of raft clustering in some similar way could lead to selective segregation of proteins and lipids in the TGN membrane. Interestingly, the incorporation of the GPI-anchored raft protein Gas1p into FusMidp-vesicles is consistent with a raft-clustering mechanism. So is the observation that FusMidp-vesicles exhibited a higher membrane order than the TGN/E, as measured by C-Laurdan spectrophotometry. This assay corroborates the notion that the vesicle bilayer undergoes a structural change, as predicted by a raft clustering mechanism (Simons and Vaz, 2004).
The lipid-facilitated sorting mechanism underlying the generation of the raft carriers at the TGN would lead to an enrichment of sphingolipids and ergosterol at the PM and enable the establishment of the lipid gradient characteristic of the secretory pathway (Hechtberger et al., 1994; Zinser and Daum, 1995; Schneiter et al., 1999; Holthuis et al., 2001). This gradient could be enhanced by retrograde removal of glycerophospholipids by coat protein I (COPI)–mediated transport. Brügger et al. (2000) produced COPI vesicles in vitro from rat liver and COS cell Golgi membranes. Mass spectrometric analysis of COPI vesicles showed a reduction in both cholesterol and sphingomyelin content. If similar segregation of ergosterol and sphingolipids also occurred during yeast COPI vesicle biogenesis, the selective transport of sterols and sphingolipids anterogradely and the recycling of phosphatidylcholine retrogradely could be a basic element in the processes responsible for Golgi sorting functions.
Recently, the cytoplasmic protein complex termed exomer was shown to be important for TGN to PM transport in yeast (Wang et al., 2006). Additionally, peripheral membrane proteins such as BAR (BIN/amphiphysin/RVS) domain–containing proteins could facilitate the budding process at the TGN (Itoh and De Camilli, 2006). But what are missing are the factors responsible for raft clustering in the TGN. We postulate that luminal or cytosolic proteins are required for induction of raft coalescence. Candidates could be Chs5p, Kes1p, or Rvs161p, which have been previously identified in the screen for sorting factors (Proszynski et al., 2005).
Lipid species–dependent phase segregation in biological membranes contributes to cargo sorting and membrane sculpting
Immiscibility of two liquid phases in membrane bilayers can promote membrane bending (Lipowsky, 1993). Recent work on shiga toxin has demonstrated that when this pentameric protein binds the glycosphingolipid Gb3 on the cell surface, it creates tubular membrane invaginations that internalize the toxin into cells (Romer et al., 2007). This process is energy independent and seems to be driven by a phase segregation process. Similarly, polyoma virus binds to the cell surface ganglioside GM1, and this multimeric binding leads to domain-induced budding into the cell (Lipowsky, 1993; Damm et al., 2005; Ewers et al., 2005). Thus, a new principle is emerging in membrane trafficking that uses lipid–lipid and lipid–protein interactions driving protein and lipid sorting during transport carrier formation. The novel strategy presented in this work will make it possible to isolate and characterize other sorting stations and carriers in the secretory pathway of living cells and thereby contribute to unraveling the molecular mechanisms of this complex distribution machinery.
Materials and methods
Yeast strain and culture media
The yeast strain KSY302 used in this study expressed the temperature-sensitive allele sec6-4 of the exocyst subunit SEC6 (provided by P. Novick, Yale University School of Medicine, New Haven, CT). The genotype of KSY302 is sec6-4; leu2-3,112; ura3-52. KSY302 was cultured in synthetic complete media containing yeast nitrogen base (Difco), CSM-URA-LEU (BIO101), and 2% raffinose (Sigma-Aldrich). Construct expression was induced by adding 2% galactose (Merck).
Plasmids
To produce the vesicle bait FUSMIDGFPLTLM9 (plasmid p147), LTLM9 was amplified by PCR and integrated by homologous recombination 3′ of FUSMIDGFP into HindIII-linearized plasmid TPQ55 (based on pRS416; Proszynski et al., 2004) by recombination in yeast. The TGN/E bait, GAP1GFPLTLM9 (plasmid p165), was engineered by recombining LTLM9 3′ of GAP1GFP into XhoI-linearized p163 (GAP1GFP; provided by C. Kaiser, Massachusetts Institute of Technology, Cambridge, MA). INVRFP (plasmid p145) was made by restriction digestion of a construct carrying InvRFP using SacI and HindIII and ligation into pRS415. The bait without a TEV site, FUSMIDGFPLM9 (plasmid p137), was produced as p147 with a PCR fragment of LM9 not containing the TEV site.
Antibodies
Mouse IgG ChromPure whole molecule (Jackson ImmunoResearch Laboratories), mouse anti–human CD3 (clone OKT3; eBioscience), sheep anti–mouse antibody (Wandinger-Ness et al., 1990), mouse anti-myc antibody (c-Myc [9E10]; Santa Cruz Biotechnology, Inc.), mouse anti-GFP (Santa Cruz Biotechnology, Inc.), rabbit anti-RFP (provided by M. Zerial, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany), rabbit anti-Gas1p (provided by H. Riezman, University of Geneva, Geneva, Switzerland), mouse anti-Pep12p (Invitrogen), mouse anti-Dpm1p (Invitrogen), rabbit anti-Tlg1p (provided by J. Holthuis, Utrecht University, Utrecht, Netherlands; Holthuis et al., 1998), and rabbit anti-Bgl2p (provided by R. Schekman, University of California, Berkeley, Berkeley, CA) were used.
Microscopy
Microscopy was performed with a microscope (BX61; Olympus) and a UPlanSApo 100× NA 1.40 oil immersion objective (Olympus). Pictures were acquired at room temperature in synthetic complete medium with a camera (SPOT; Diagnostic Instruments, Inc.) using MetaMorph software (MDS Analytical Technologies).
Subcellular fractionation and immunoisolation
KSY302 (sec6-4) transformed with the constructs FusMidGFPLTLM9 (p147) and InvRFP (p145) or Gap1GFPLTLM9 (p165) and p145 was cultured at 24°C in 500 ml SC-URA-LEU and 2% raffinose to 1 OD600. For simultaneous construct expression and vesicle accumulation, cells were shifted into 250 ml SC-URA-LEU and 2% raffinose containing 2% galactose and incubated for 45 min at 37°C. To arrest membrane traffic, the medium was supplemented with 10 mM NaN3 and incubated for another 20 min at 37°C.
Cells were pelleted (4 min at 4,000 g and 4°C), resuspended in 3 ml of lysis buffer (800 mM sorbitol, 1 mM EDTA, 10 mM triethanolamine, pH 7.4, CLAP [chymostatin, leupeptin, antipain, and pepstatin], 1 mM PMSF [Roche], and 10 mM NaN3), and disrupted by 0.5-mm zirconia beads (BioSpec Products, Inc.). The supernatant of a centrifugation at 2,000 g (10 min at 4°C) was collected and centrifuged for another 30 min at 20,000 g (4°C), which produced the supernatant S20. S20 was loaded on top of a sucrose gradient (in wt/wt% sucrose in 10 mM triethanolamine, pH 7.4; 2 ml 50%, 2.5 ml 45%, 2.5 ml 40%, 1 ml 35%, and 1 ml 30%) and centrifuged to equilibrium (2.5 h at RCFmax 401,747 g in a vertical gradient rotor [VTi65.1; Beckman Coulter] at 4°C). The gradient was fractionated into 1-ml fractions. Refractive index and protein concentration of fractions were determined. Fraction 7 was used for FusMidp-vesicle isolation, and fraction 4 was used to isolate the TGN/E.
Respective fractions were diluted with 1 vol PBSG (PBS and 0.1 wt/vol% gelatin) For isolation of FusMidp-vesicles, mouse anti-myc and mouse anti-Pep12p antibodies were added (1:1,000 to total protein and 1:2,000 to total protein, respectively), and the solution was rotated for 2 h at 4°C. Next, 600 µl immunoadsorbent (10 mg adsorbent/ml [in PBSG]; the adsorbent was composed of 200 µg of sheep anti–mouse antibody/mg of cellulose) was added for vesicle binding, and the solution was rotated overnight at 4°C.
The immunoadsorbent with bound vesicles was pelleted (at 1,500 g for 2 min in an Eppendorf table-top centrifuge with swing-out rotor at 4°C) and washed three times in 1 ml PBSG. For vesicle elution, the cellulose was resuspended in 1 ml PBSG containing 1 mM DTT and 100 µg of TEV protease and incubated for 4 h at 4°C. Then, the immunoadsorbent was pelleted, and FusMidp-vesicles were recovered in the supernatant. For lipidome analysis, the vesicles were pelleted (1 h at RCFmax 186,340 g in a fixed angle rotor [TLA55; Beckman Coulter]) and resuspended in 200 µl of 150 mM NH4HCO3, pH 8.0. The TGN/E was isolated with exactly the same procedure except that Gap1GFPLTLM9 was used as immunoisolation bait, TGN/E input material was sucrose gradient fraction 4, and the immunodepletion with anti-Pep12p antibody was not applied.
Purification of sheep anti–mouse antibody
Sheep anti–mouse antibody was affinity purified using a mouse IgG–coupled N-hydroxy-succinimide column (GE Healthcare) prepared according to the recommendations of the manufacturer. Sheep serum was circulated with a peristaltic pump over the mouse IgG–coupled N-hydroxy-succinimide column. The sheep antibody was eluted by a pH change to pH 2.5 in 100 mM glycine. The eluted antibody was rapidly neutralized to pH 7.5 (using 1 M Tris-Cl, pH 8.8) and dialyzed overnight with 200 mM of borate buffer, pH 8.2.
Production of the immunoadsorbent
The cellulose-based sheep anti–mouse-coupled immunoadsorbent was prepared as previously described (Hales and Woodhead, 1980; Wandinger-Ness et al., 1990). In brief, cellulose powder (microgranular cellular powder CC41; GE Healthcare) was activated to aminocellulose, which was stable for several months. For antibody binding, aminocellulose was modified to diazocellulose, which was immediately coupled to sheep anti–mouse antibody. The immunoadsorbent was stable for up to 6 mo.
PM isolation
PM was prepared as previously described (Serrano, 1988). 250 OD600 units of cells were treated exactly as for isolation of FusMidp-vesicle and TGN/E. The pellet of the 20,000 g spin (P20) was resuspended in 300 µl of 20% glycerol, pH 5, and loaded on top of a 4-ml step gradient (1.3 ml of 55% [wt/wt] sucrose in 10 mM triethanolamine, pH 7.4; and 2.7 ml of 45% [wt/wt] sucrose in 10 mM triethanolamine, pH 7.4). After 5 h of centrifugation (RCFmax 129,481 g, 31 krpm, at 4°C in a swing-out rotor [SW60; Beckman Coulter]), the visible interface was collected with a gradient fractionator (LabConco). The 500-µl interface fraction was diluted twofold with 20% glycerol, pH 5, and membranes were pelleted (1 h at RCFmax 186,340 g in a TLA55 fixed angle rotor). The pellet was resuspended in 300 µl of 20% glycerol and loaded on a second step gradient (55%/45% sucrose) and centrifuged for 16 h (RCFmax 129,481 g, 31 krpm, at 4°C in an SW60 swing-out rotor). The gradient was fractionated into eight 500-µl fractions. The gradient profile was monitored by Western blotting against the PM markers Pma1p and Gas1p and against GFP to detect the immunoisolation baits and against the ER integral membrane protein Dpm1p and the late endosome marker Pep12p.
Floatation of immunoisolated secretory vesicles
FusMidp-vesicles were immunoisolated and sedimented (RCFmax 123,722 g, 38 krpm, for 90 min in a swing-out rotor [TLS55; Beckman Coulter]) on a 40% Nycodenz cushion. The pellet was resuspended and adjusted to 40% Nycodenz and bottom loaded on a 4-ml linear Nycodenz gradient prepared in an SW60 centrifugation tube. The gradient was centrifuged for 16 h (RCFmax 121,262 g, 30 krpm, at 4°C in an SW60 swing-out rotor). The gradient profile was monitored by Western blotting for FusMidGFP, Gas1p, and Bgl2p.
Mass spectrometric lipid analysis
FusMidp-vesicles, TGN/E, and total cell extracts were subjected to quantitative shotgun lipidomic analysis (Ejsing et al., 2009). In short, samples were mixed with 20 µl of internal lipid standard mixture, providing a total spike of 15 pmol DAG 17:0-17:0, 29 pmol PA 17:0-14:1, 59 pmol PE 17:0-14:1, 15 pmol phosphatidylglycerol 17:0-14:1, 49 pmol PS 17:0-14:1, 58 pmol PC 17:0-14:1, 62 pmol PI 17:0-17:0 (provided by C. Thiele, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany), 15 pmol ceramide 18:0;3/18:0;0, 23 pmol IPC 18:0;2/26:0;0, 20 pmol MIPC 18:0;2/26:0;0, 30 pmol M(IP)2C 18:0;2/26:0;0, and 160 pmol stigmasta-5,7,22-trienol. Samples were subsequently subjected to lipid extraction executed at 4°C (Ejsing et al., 2009). The lower organic phases were isolated and evaporated in a vacuum exicator at 4°C. Lipid extracts were dissolved in 100 µl chloroform/methanol (1:2; vol/vol) and subjected to quantitative lipid analysis on both a hybrid i quadrupole time of flight mass spectrometer (QSTAR Pulsar; MDS Analytical Technologies) and an LTQ Orbitrap instrument (Thermo Fisher Scientific) equipped with the robotic nanoflow ion source TriVersa NanoMate (Advion Biosciences, Inc.). DAG, PA, PS, PE, PI, and phosphatidylglycerol species were quantified by negative ion mode multiple precursor ion scanning analysis (Ejsing et al., 2006a); PC and ceramide species were quantified by consecutive positive ion mode precursor ion scanning m/z 184.1 and multiple reaction monitoring analysis, respectively. IPC, MIPC, and M(IP)2C species were quantified by negative ion mode flow through mass analysis on an LTQ Orbitrap mass spectrometer; ergosterol was quantified after chemical acetylation by multiple reaction monitoring. Automated processing of acquired mass spectra and identification and quantification of detected molecular lipid species were performed by dedicated softwares (MDS Analytical Technologies; Ejsing et al., 2006a) and ALEX (Analysis of Lipid Experiments) software (Ejsing et al., 2009).
Lipid species were annotated by their molecular composition. Glycerophospholipid and DAG species are annotated as: <lipid class><number of carbon atoms in the first fatty acid moiety>:<number of double bonds in the first fatty acid moiety>-<number of carbon atoms in the second fatty acid moiety>:<number of double bonds in the second fatty acid moiety> (e.g., PI 16:0-18:1). Sphingolipid species are annotated as: <lipid class><number of carbon atoms in the long chain base moiety>:<number of double bonds in the long chain base moiety>;<number of hydroxyl groups in the long chain base moiety>/<number of carbon atoms in the fatty acid moiety>:<number of double bonds in the fatty acid moiety>;<number of hydroxyl groups in the fatty acid moiety>. For example, IPC 18:0;3/26:0;1; is an IPC species containing a C18 phytosphingosine (having three hydroxyl groups and no double bonds) and a C26:0 amide-linked fatty acid moiety with a hydroxyl group.
C-Laurdan fluorescence spectrophotometry
FusMidp-vesicles and TGN/E were stained for 15 min with 100 nM C-laurdan (provided by B.R. Cho, Korea University, Seoul, Korea). Samples were excited at 385 nm, and emission spectra were recorded from 400 to 530 nm at 23°C on a fluorescence spectrometer (Fluoromax-3; Horiba). Spectra of unstained samples were subtracted from the sample labeled with C-Laurdan. GP values were calculated according to Parasassi et al. (1991):
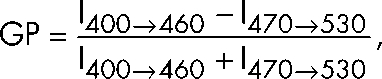 |
where I400→460 and I470→530 stand for the total fluorescence intensity recorded from 400 to 460 nm and from 470 to 530 nm, respectively.
Electron microscopy
Yeast cells were cryoimmobilized using a high pressure freezer (EMPACT2 + RTS; Leica; Manninen et al., 2005). Samples were freeze-substituted in an automatic freeze-substitution machine (Leica) at −90°C in 1% osmium tetroxide plus 0.1% uranyl acetate in anhydrous acetone for 24 h. The temperature was increased 4°C/h (27 h) to 20°C. Samples were washed in acetone and embedded at 24°C in epon/Araldite. 70- and 200-nm sections were cut using a microtome (Ultracut UCT; Leica). Sections were collected on Formvar-coated Cu grids and poststained in 5% uranyl acetate in 50% methanol for 3 min and aqueous lead citrate. Thin sections (70 nm) were viewed on a transmission electron microscope (Tecnai 12; FEI) at 100 kV. 200-nm sections for tomographic imaging were prepared identically; however, a solution of 15-nm gold was applied to both sides of the grids as fiducial markers to assist in the tomographic imaging procedure.
Intermediate voltage electron microscopy and tomography
Regions of interest were located on 200-nm sections on a Tecnai F30 (FEI). Identified targets underwent a preirradiation step with a large spread electron beam to minimize the polymer shrinkage during the tilt series acquisition. The collection of the tomographic data was controlled by the program SerialEM (Mastronarde, 2005). Series of 121 micrographs were acquired by rotating the specimen from ±60° around the y axis in 1° tilt increments at a magnification of 9,400× with an accelerate voltage of 300 kV produced by a field emission gun. Electron micrographs were collected on a cooled charge-coupled device camera (US1000; Gatan). 2,048 × 2,048–pixel images were aligned, and the 3D volume was reconstructed by r-weighted back projection with the IMOD program suite (Mastronarde, 1997). The obtained 3D electron density map is a volume of 1,848 × 1,848 × 206 voxels with anisotropic information. The vesicles and structures were identified in the x–y plane and segmented manually in every 10 planes with 3DMOD (Kremer et al., 1996). The final model was also rendered in 3DMOD.
Conventional chemical fixation of isolated organelles
Membrane pellets of isolated FusMidp-vesicles and the TGN/E were fixed in 2.5% glutaraldehyde in PBS for 1 h, rinsed in PBS, and fixed in osmium tetroxide (1% aqueous solution for 1 h). After washing in distilled water, pellets were refrigerated in a solution of aqueous uranyl acetate (4% for 1 h), dehydrated in an ascending ethanol series (30–100 with 10% concentration steps) two times for 5 min at each concentration, and embedded in epon/Araldite for microtomy. Sections were cut at 30–70 nm and mounted on Formvar-coated Cu grids, poststained, and examined as described in Electron microscopy.
Online supplemental material
Fig. S1 shows the molecular and morphological characterization of the yeast PM. Fig. S2 shows the organelle marker profile of the sucrose gradient used for prepurification of FusMidp-vesicles and the TGN/E. Fig. S3 shows the specificity of the immunoisolation procedure and the secretion of InvRFP. Fig. S4 shows that the immunoisolation procedure preserves the physical integrity of FusMidp-vesicles. Fig. S5 shows that subcellular fractionation at 4°C neither promotes artifactual production of secretory vesicles nor modulates the concentration of secretory cargo in isolated TGN/E. Video 1 shows an animated 3D reconstruction of a tomogram of a sec6-4 cell at the restrictive temperature 37°C. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200901145/DC1.
Acknowledgments
We are grateful to the members of the Simons laboratory for stimulating discussions and Marino Zerial, Vineeth Surendranath, and Madhavi Krishnan for critical reading of the manuscript. We also thank Christoph Thiele for providing synthetic PI 17:0-17:0; Angela Wandinger-Ness, Jean Gruenberg, Ünal Coskun, Aki Manninen, David Drechsel, Regis LeMaitre, and Martine Ruer for supporting the development of the immunoisolation procedure; and Randy Schekman, Peter Novick, Christiane Walch-Solimena, Joost Holthuis, Howard Riezman, Chris Kaiser, Bong Rae Cho, and Marino Zerial for providing indispensable reagents. We thank Eva Duchoslav for advice on Lipid Profiler software, Igor Chernushevich for advice on QqTOF mass spectrometry, and members of the Shevchenko laboratory and Vineeth Surendranath for experimental tips on mass spectrometry and data processing. We also thank Kim Ekroos, Reinaldo Almeida, and Mark Baumert for their advice on TriVersa NanoMate operation and Teresa Dunn for providing the sur2Δscs7Δ double mutant for isolation of sphingolipid standards. Finally, we thank Thomas Doebel and Gunar Fabig for tomogram segmentation.
This work was supported by the Deutsche Forschungsgemeinschaft (grant SFB/TR 13, project D1, to A. Shevchenko and grants SFB/TR 13, project A1, and SP1175 to K. Simons) and a European Union Sixth Framework Program PRISM grant (to K. Simons).
Footnotes
Abbreviations used in this paper: COPI, coat protein I; GP, general polarization; GPI, glycosyl-PI; HDSV, heavy density secretory vesicle; InvRFP, invertase-RFP; IPC, inositolphosphoceramide; LDSV, light density secretory vesicle; M(IP)2C, mannosyl-di-IPC; MIPC, mannosyl-IPC; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PM, plasma membrane; PS, phosphatidylserine; TEV, tobacco etch virus; TGN/E, TGN/endosome system.
References
- Aebersold R., Mann M. 2003. Mass spectrometry-based proteomics.Nature. 422:198–207 [DOI] [PubMed] [Google Scholar]
- Bagnat M., Keranen S., Shevchenko A., Shevchenko A., Simons K. 2000. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast.Proc. Natl. Acad. Sci. USA. 97:3254–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F., Malhotra V. 2006. The formation of TGN-to-plasma-membrane transport carriers.Annu. Rev. Cell Dev. Biol. 22:439–455 [DOI] [PubMed] [Google Scholar]
- Bonangelino C.J., Chavez E.M., Bonifacino J.S. 2002. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae.Mol. Biol. Cell. 13:2486–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brügger B., Sandhoff R., Wegehingel S., Gorgas K., Malsam J., Helms J.B., Lehmann W.D., Nickel W., Wieland F.T. 2000. Evidence for segregation of sphingomyelin and cholesterol during formation of COPI-coated vesicles.J. Cell Biol. 151:507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brügger B., Glass B., Haberkant P., Leibrecht I., Wieland F.T., Kräusslich H.G. 2006. The HIV lipidome: a raft with an unusual composition.Proc. Natl. Acad. Sci. USA. 103:2641–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm E.M., Pelkmans L., Kartenbeck J., Mezzacasa A., Kurzchalia T., Helenius A. 2005. Clathrin- and caveolin-1–independent endocytosis: entry of simian virus 40 into cells devoid of caveolae.J. Cell Biol. 168:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., Lees N.D., Bard M., Dickson R. 1998. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae.Yeast. 14:1471–1510 [DOI] [PubMed] [Google Scholar]
- Eisenkolb M., Zenzmaier C., Leitner E., Schneiter R. 2002. A specific structural requirement for ergosterol in long-chain fatty acid synthesis mutants important for maintaining raft domains in yeast.Mol. Biol. Cell. 13:4414–4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejsing C.S., Duchoslav E., Sampaio J., Simons K., Bonner R., Thiele C., Ekroos K., Shevchenko A. 2006a. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning.Anal. Chem. 78:6202–6214 [DOI] [PubMed] [Google Scholar]
- Ejsing C.S., Moehring T., Bahr U., Duchoslav E., Karas M., Simons K., Shevchenko A. 2006b. Collision-induced dissociation pathways of yeast sphingolipids and their molecular profiling in total lipid extracts: a study by quadrupole TOF and linear ion trap-orbitrap mass spectrometry.J. Mass Spectrom. 41:372–389 [DOI] [PubMed] [Google Scholar]
- Ejsing C.S., Sampaio J.L., Surendranath V., Duchoslav E., Ekroos K., Klemm R.W., Simons K., Shevchenko A. 2009. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry.Proc. Natl. Acad. Sci. USA. 106:2136–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers H., Smith A.E., Sbalzarini I.F., Lilie H., Koumoutsakos P., Helenius A. 2005. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes.Proc. Natl. Acad. Sci. USA. 102:15110–15115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaigg B., Timischl B., Corbino L., Schneiter R. 2005. Synthesis of sphingolipids with very long chain fatty acids but not ergosterol is required for routing of newly synthesized plasma membrane ATPase to the cell surface of yeast.J. Biol. Chem. 280:22515–22522 [DOI] [PubMed] [Google Scholar]
- Gaigg B., Toulmay A., Schneiter R. 2006. Very long-chain fatty acid-containing lipids rather than sphingolipids per se are required for raft association and stable surface transport of newly synthesized plasma membrane ATPase in yeast.J. Biol. Chem. 281:34135–34145 [DOI] [PubMed] [Google Scholar]
- Gao M., Kaiser C.A. 2006. A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast.Nat. Cell Biol. 8:657–667 [DOI] [PubMed] [Google Scholar]
- Gaus K., Zech T., Harder T. 2006. Visualizing membrane microdomains by Laurdan 2-photon microscopy.Mol. Membr. Biol. 23:41–48 [DOI] [PubMed] [Google Scholar]
- Griffiths G., Simons K. 1986. The trans Golgi network: sorting at the exit site of the Golgi complex.Science. 234:438–443 [DOI] [PubMed] [Google Scholar]
- Gurunathan S., David D., Gerst J.E. 2002. Dynamin and clathrin are required for the biogenesis of a distinct class of secretory vesicles in yeast.EMBO J. 21:602–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak D., Gable K., Beeler T., Dunn T. 1997. Hydroxylation of Saccharomyces cerevisiae ceramides requires Sur2p and Scs7p.J. Biol. Chem. 272:29704–29710 [DOI] [PubMed] [Google Scholar]
- Hales C.N., Woodhead J.S. 1980. Labeled antibodies and their use in the immunoradiometric assay.Methods Enzymol. 70:334–355 [DOI] [PubMed] [Google Scholar]
- Hancock J.F. 2006. Lipid rafts: contentious only from simplistic standpoints.Nat. Rev. Mol. Cell Biol. 7:456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsay E., Bretscher A. 1995. Parallel secretory pathways to the cell surface in yeast.J. Cell Biol. 131:297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsay E., Schekman R. 2002. A subset of yeast vacuolar protein sorting mutants is blocked in one branch of the exocytic pathway.J. Cell Biol. 156:271–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsay E., Schekman R. 2007. Avl9p, a member of a novel protein superfamily, functions in the late secretory pathway.Mol. Biol. Cell. 18:1203–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hechtberger P., Zinser E., Saf R., Hummel K., Paltauf F., Daum G. 1994. Characterization, quantification and subcellular localization of inositol-containing sphingolipids of the yeast, Saccharomyces cerevisiae.Eur. J. Biochem. 225:641–649 [DOI] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols B.J., Dhruvakumar S., Pelham H.R. 1998. Two syntaxin homologues in the TGN/endosomal system of yeast.EMBO J. 17:113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J.C., Pomorski T., Raggers R.J., Sprong H., Van Meer G. 2001. The organizing potential of sphingolipids in intracellular membrane transport.Physiol. Rev. 81:1689–1723 [DOI] [PubMed] [Google Scholar]
- Itoh T., De Camilli P. 2006. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature.Biochim. Biophys. Acta. 1761:897–912 [DOI] [PubMed] [Google Scholar]
- Kim H.M., Choo H.J., Jung S.Y., Ko Y.G., Park W.H., Jeon S.J., Kim C.H., Joo T., Cho B.R. 2007. A two-photon fluorescent probe for lipid raft imaging: C-laurdan.ChemBioChem. 8:553–559 [DOI] [PubMed] [Google Scholar]
- Kremer J.R., Mastronarde D.N., McIntosh J.R. 1996. Computer visualization of three-dimensional image data using IMOD.J. Struct. Biol. 116:71–76 [DOI] [PubMed] [Google Scholar]
- Kuikka M., Ramstedt B., Ohvo-Rekila H., Tuuf J., Slotte J.P. 2001. Membrane properties of D-erythro-N-acyl sphingomyelins and their corresponding dihydro species.Biophys. J. 80:2327–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D., Ries J., Schwille P., Simons K. 2008. Plasma membranes are poised for activation of raft phase coalescence at physiological temperature.Proc. Natl. Acad. Sci. USA. 105:10005–10010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R. 1993. Domain-induced budding of fluid membranes.Biophys. J. 64:1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manninen A., Verkade P., Le Lay S., Torkko J., Kasper M., Fullekrug J., Simons K. 2005. Caveolin-1 is not essential for biosynthetic apical membrane transport.Mol. Cell. Biol. 25:10087–10096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde D.N. 1997. Dual-axis tomography: an approach with alignment methods that preserve resolution.J. Struct. Biol. 120:343–352 [DOI] [PubMed] [Google Scholar]
- Mastronarde D.N. 2005. Automated electron microscope tomography using robust prediction of specimen movements.J. Struct. Biol. 152:36–51 [DOI] [PubMed] [Google Scholar]
- Mayor S., Riezman H. 2004. Sorting GPI-anchored proteins.Nat. Rev. Mol. Cell Biol. 5:110–120 [DOI] [PubMed] [Google Scholar]
- Mellman I., Warren G. 2000. The road taken: past and future foundations of membrane traffic.Cell. 100:99–112 [DOI] [PubMed] [Google Scholar]
- Muniz M., Morsomme P., Riezman H. 2001. Protein sorting upon exit from the endoplasmic reticulum.Cell. 104:313–320 [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway.Cell. 21:205–215 [DOI] [PubMed] [Google Scholar]
- Oh C.S., Toke D.A., Mandala S., Martin C.E. 1997. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation.J. Biol. Chem. 272:17376–17384 [DOI] [PubMed] [Google Scholar]
- Parasassi T., De Stasio G., Ravagnan G., Rusch R.M., Gratton E. 1991. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence.Biophys. J. 60:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham H.R. 1999. SNAREs and the secretory pathway-lessons from yeast.Exp. Cell Res. 247:1–8 [DOI] [PubMed] [Google Scholar]
- Pfeffer S.R. 2007. Unsolved mysteries in membrane traffic.Annu. Rev. Biochem. 76:629–645 [DOI] [PubMed] [Google Scholar]
- Proszynski T.J., Simons K., Bagnat M. 2004. O-glycosylation as a sorting determinant for cell surface delivery in yeast.Mol. Biol. Cell. 15:1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proszynski T.J., Klemm R.W., Gravert M., Hsu P.P., Gloor Y., Wagner J., Kozak K., Grabner H., Walzer K., Bagnat M., et al. 2005. A genome-wide visual screen reveals a role for sphingolipids and ergosterol in cell surface delivery in yeast.Proc. Natl. Acad. Sci. USA. 102:17981–17986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg K.J., Rowley N., Kaiser C.A. 1997. Physiological regulation of membrane protein sorting late in the secretory pathway of Saccharomyces cerevisiae.J. Cell Biol. 137:1469–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E., Kreitzer G., Musch A. 2005. Organization of vesicular trafficking in epithelia.Nat. Rev. Mol. Cell Biol. 6:233–247 [DOI] [PubMed] [Google Scholar]
- Romer W., Berland L., Chambon V., Gaus K., Windschiegl B., Tenza D., Aly M.R., Fraisier V., Florent J.C., Perrais D., et al. 2007. Shiga toxin induces tubular membrane invaginations for its uptake into cells.Nature. 450:670–675 [DOI] [PubMed] [Google Scholar]
- Routt S.M., Ryan M.M., Tyeryar K., Rizzieri K.E., Mousley C., Roumanie O., Brennwald P.J., Bankaitis V.A. 2005. Nonclassical PITPs activate PLD via the Stt4p PtdIns-4-kinase and modulate function of late stages of exocytosis in vegetative yeast.Traffic. 6:1157–1172 [DOI] [PubMed] [Google Scholar]
- Schneiter R., Brügger B., Sandhoff R., Zellnig G., Leber A., Lampl M., Athenstaedt K., Hrastnik C., Eder S., Daum G., et al. 1999. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane.J. Cell Biol. 146:741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S., Simons K. 2004. Polarized sorting in epithelial cells: raft clustering and the biogenesis of the apical membrane.J. Cell Sci. 117:5955–5964 [DOI] [PubMed] [Google Scholar]
- Sciorra V.A., Audhya A., Parsons A.B., Segev N., Boone C., Emr S.D. 2005. Synthetic genetic array analysis of the PtdIns 4-kinase Pik1p identifies components in a Golgi-specific Ypt31/rab-GTPase signaling pathway.Mol. Biol. Cell. 16:776–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M.N., Marcusson E.G., Cereghino J.L., Emr S.D. 1997. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products.J. Cell Biol. 137:79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano R. 1988. H+-ATPase from plasma membranes of Saccharomyces cerevisiae and Avena sativa roots: purification and reconstitution.Methods Enzymol. 157:533–544 [DOI] [PubMed] [Google Scholar]
- Simons K., van Meer G. 1988. Lipid sorting in epithelial cells.Biochemistry. 27:6197–6202 [DOI] [PubMed] [Google Scholar]
- Simons K., Wandinger-Ness A. 1990. Polarized sorting in epithelia.Cell. 62:207–210 [DOI] [PubMed] [Google Scholar]
- Simons K., Ikonen E. 1997. Functional rafts in cell membranes.Nature. 387:569–572 [DOI] [PubMed] [Google Scholar]
- Simons K., Toomre D. 2000. Lipid rafts and signal transduction.Nat. Rev. Mol. Cell Biol. 1:31–39 [DOI] [PubMed] [Google Scholar]
- Simons K., Vaz W.L. 2004. Model systems, lipid rafts, and cell membranes.Annu. Rev. Biophys. Biomol. Struct. 33:269–295 [DOI] [PubMed] [Google Scholar]
- Traub L.M., Kornfeld S. 1997. The trans-Golgi network: a late secretory sorting station.Curr. Opin. Cell Biol. 9:527–533 [DOI] [PubMed] [Google Scholar]
- Umebayashi K., Nakano A. 2003. Ergosterol is required for targeting of tryptophan permease to the yeast plasma membrane.J. Cell Biol. 161:1117–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G. 1989. Lipid traffic in animal cells.Annu. Rev. Cell Biol. 5:247–275 [DOI] [PubMed] [Google Scholar]
- van Meer G., Simons K. 1988. Lipid polarity and sorting in epithelial cells.J. Cell. Biochem. 36:51–58 [DOI] [PubMed] [Google Scholar]
- Veiga M.P., Arrondo J.L., Goni F.M., Alonso A., Marsh D. 2001. Interaction of cholesterol with sphingomyelin in mixed membranes containing phosphatidylcholine, studied by spin-label ESR and IR spectroscopies. A possible stabilization of gel-phase sphingolipid domains by cholesterol.Biochemistry. 40:2614–2622 [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C., Novick P. 1999. The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi.Nat. Cell Biol. 1:523–525 [DOI] [PubMed] [Google Scholar]
- Wandinger-Ness A., Bennett M.K., Antony C., Simons K. 1990. Distinct transport vesicles mediate the delivery of plasma membrane proteins to the apical and basolateral domains of MDCK cells.J. Cell Biol. 111:987–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.W., Hamamoto S., Orci L., Schekman R. 2006. Exomer: A coat complex for transport of select membrane proteins from the trans-Golgi network to the plasma membrane in yeast.J. Cell Biol. 174:973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser E., Daum G. 1995. Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae.Yeast. 11:493–536 [DOI] [PubMed] [Google Scholar]