

Abstract
The purpose of this study was to use cardiac MRI to define the morphology of the reversibly injured peri-infarct border zone in patients treated with primary percutaneous coronary intervention (PPCI) for acute ST elevation myocardial infarction. In 15 patients, T2-weighted myocardial edema imaging was used to identify the ischemic bed or area at risk (AAR), and late gadolinium enhancement imaging was used to measure infarct size. Images were coregistered, and the boundaries of edema and necrosis were defined using an edge-detection methodology. We observed that infarction always involved the subendocardium but showed variable transmural extension within the AAR. The mean infarct size was 22 ± 19% (range: 8–48%), and the mean AAR was 34 ± 12% (range: 20–57%). The infarcted myocardium was always smaller than the ischemic AAR and involved between 34% and 99% (mean 72 ± 21%) of the ischemic bed primarily due to variation in transmural infarct extension. Although a lateral border zone of potentially viable myocardium was often present, its extent was limited (range: 0–11 mm, mean: 5 ± 4 mm). As a result of this, infarcts occupied the majority (range: 70–100%, mean: 82 ± 13%) of the width of the AAR. The mean fractional wall thickening in the infarcted, peri-infarcted, and remote myocardium was 3.6 ± 16.0%, 40.5 ± 26.4%, and 88.2 ± 39.3%, respectively. These findings demonstrate that myocardial salvage is largely determined by epicardial limitation of the infarct within the ischemic AAR after PPCI. The lateral boundaries of necrosis approximate to the lateral extent of the ischemic bed and systolic wall motion abnormalities extend well beyond the infarct border zone.
Keywords: angioplasty, magnetic resonance imaging, myocardial infarction
primary percutaneous coronary intervention (PPCI) is the preferred strategy for reestablishing myocardial perfusion after acute ST elevation myocardial infarction (STEMI) and leads to an improvement in short- and long-term adverse cardiac events (19, 29). Nevertheless, myocardial salvage after PPCI is frequently modest, and a significant proportion of patients develop transmural necrosis despite epicardial coronary reperfusion (27). A major clinical challenge for improving outcomes after STEMI is not only the timely reestablishment of epicardial coronary artery flow but also the development of adjuvant cardioprotective therapies (42).
After coronary artery occlusion, cardiomyocytes in the hypoperfused vascular territory rapidly become ischemic, leading to loss of function and intracellular edema (18). However, cell death is not simultaneous but is thought to progress as a wavefront starting from the subendocardium with subsequent extension into the midmyocardium and then the subepicardium over a period of several hours (33, 34). Reestablishing reperfusion of the reversibly injured myocardium therefore has the potential for salvaging viable mid- and subepicardial myocytes. Noninvasive assessment of the morphology of the salvaged myocardium in the peri-infact border zone would be of value in the development of interventions designed to limit infarct size.
Cardiac MRI (CMRI) techniques have been used to identify changes in myocardial signal relaxation after ischemia-reperfusion injury and have shown the potential for quantifying myocardial salvage (13). The ischemic myocardium has increased water content due to the accumulation of osmotic load (18) and consequently has a longer T2 relaxation time (8). The myocardium downstream of the coronary artery occlusion may therefore be retrospectively determined for up to a month after subsequent reperfusion (28). This ischemic area at risk (AAR), defined as the region of edema on T2-weighted imaging, coregisters with histopathological findings (12), shows equivalence to radionuclide single photon emission computed tomography (SPECT) measurement of the AAR (11), and includes the reversibly injured myocardium, which is capable of functional recovery (1). When combined with late gadolinium imaging to identify irreversibly damaged necrotic tissue, CMRI provides a potential method for characterizing the peri-infarct border zones. A study (21) of the peri-infarct border zone in postmortem human experiments has revealed that the infarct area is primarily determined by transmural limitation of necrosis as the lateral infarct margins are consistently narrow. Although the AAR appears to be larger than the area of necrosis using CMRI (13), the anatomic relationship between the infarct and the ischemic bed has not been investigated using imaging techniques. Furthermore, the accurate determination of the border zone is complicated by the presence of postreperfusion hemorrhage, which opposes the T2 signal changes due to edema (24, 30), the relatively low intrinsic contrast of the border between normal and edematous myocardium, and also the potential for misregistration between imaging sequences.
The purpose of this study was to compare the size and location of the infarcted myocardium with the anatomic distribution of the ischemic bed using CMRI and image segmentation techniques. These methods would be used to measure the amount of viable myocardium that remained at the lateral and peripheral infarct border zones after PPCI.
METHODS
Study population.
Research Ethics Committee approval was obtained for the study, and all participants gave written informed consent prior to enrollment. Fifteen consecutive patients (13 men and 2 women, mean age: 59 yr) who had undergone PPCI for acute STEMI were prospectively studied. The diagnosis of STEMI required an ST elevation of ≥1 mm in two contiguous ECG leads and chest pain onset within 12 h of presentation. All patients proceeded to PPCI within 24 h of the onset of symptoms. Patients with a prior history of myocardial infarction or contraindications to gadolinium-enhanced CMRI were excluded. Three patients had diabetes mellitus, two patients had arterial hypertension, two patients had hyperlipidaemia, three patients were active smokers, and a further two patients were ex-smokers.
Coronary intervention.
Before PPCI, patients received aspirin and clopidogrel, and intravenous heparin was administered as a bolus with the aim of achieving an activated clotting time of >250 s. Patients underwent left heart catheterisation using either transfemoral or transradial approaches. Selective coronary catheterisation was used to identify and treat the infarct-related artery, and all patients received either a bare metal or drug-eluting stent. Nonculprit atherosclerotic vessels were not treated. Eight patients received a glycoprotein IIb/IIIa inhibitor periprocedurally.
Angiographic analysis.
Determination of the culprit lesion location according to the American Heart Association coronary segment classification was performed by two independent observers (I. Nadra and S. J. Corbett) who were blinded to the CMR results. Flow in the infarct-related artery was graded using the Thrombolysis in Myocardial Infarction (TIMI) Trial criteria (39a) before and after interventions. Epicardial collateral flow was assessed using the Cohen and Rentrop score (35), where 0 = no filling, 1 = filling of side branches of the artery without visualization of the epicardial segment, 2 = partial filling of the epicardial segment via collateral channels, and 3 = complete filling of the epicardial segment of the artery via collateral channels.
Cardiac magnetic resonance examination.
CMRI experiments were performed on a whole body 1.5-T MRI scanner (Achieva, Philips Medical Systems, Best, The Netherlands) using a five-element cardiac phased-array receiver coil. Uniformity correction was performed to eliminate spatial signal variations in the chest (10). A balanced steady-state-free precession cine sequence acquired in the short-axis plane was used to measure left ventricular function [flip angle: 60°, slice thickness: 8 mm, echo time (TE): 1.5 ms, repetition time (TR): 3.0 ms, and acquired voxel size: 1.25 × 1.25 × 8 mm]. All subsequent imaging was obtained in middiastole using the same rate-specific trigger delay. Myocardial edema was imaged in the short-axis plane using a fat-suppressed T2-weighted STIR black blood sequence (flip angle: 90°, slice thickness: 10 mm, TE: 100 ms, TR: two R-R intervals, and acquired voxel size: 1.4 × 2.0 × 8 mm, reconstructed to 0.7 × 0.7 × 8 mm). An intravenous bolus of extracellular contrast medium (Magnevist, Bayer Schering Pharma, Berlin, Germany) was then administered at a dose of 0.2 mmol/kg. After 15 min, a delayed enhancement sequence was used to demonstrate infarct size using an inversion recovery gradient echo sequence (flip angle: 15°, slice thickness: 8 mm, TE: 1.4 ms, TR: 4.3 ms, and acquired voxel size: 1.5 × 1.7 × 8 mm, reconstructed to 0.7 × 0.7 × 8 mm) acquired in the same slice position and trigger delay as the T2-weighted images. Optimum nulling of the remote myocardium was achieved using a Look-Locker technique to set the inversion time delay (23).
Image analysis.
The endo- and epicardial borders of the T2-weighted and late gadolinium images were manually traced to ensure that only pixels within the myocardium were used for subsequent image analysis. Papillary muscles were excluded even when they formed part of the endocardial border. Residual differences in alignment between the T2-weighted and late enhancement sequences were corrected for using a vector-spline algorithm for image registration (4). Boundary detection of the infarct on late enhancement images and the edema on T2-weighted images employed a level-set segmentation algorithm to identify the interfaces between different regions of signal intensity. This method is particularly suited for the detection of nonsharp boundaries due to low contrast or background noise (26).
The regions of late enhancement were quantified using an automated algorithm (Segment 1.8, Medviso, Sweden) with a section-specific signal intensity threshold and level-set boundary detection. In-plane partial volume effects at the infarct boundary were compensated for using subvoxel correction to derive a transmurality index (15). This index was determined for 80 radial cords around the left ventricular myocardium. The volume of nonenhancing microvascular obstruction was separately segmented and included in the infarct volume.
Owing to the lower contrast of the T2-weighted images, a semiautomated method was used for defining the boundary of edema. Two experienced CMRI readers (D. P. O'Regan and S. A. Cook) each defined a point along the edge of the region of edema, and a level-set algorithm (Medical Image Processing, Analysis, and Visualization, National Institutes of Health, Bethesda, MD) was used to generate a single contiguous boundary around the ischemic AAR including regions of low-signal hemorrhage within the area of microvascular obstruction (MVO) (30). The mean value of each observer was used for analysis, and interobserver agreement was assessed using the intraclass correlation coefficient. A transmurality index for the hyperintense edema was calculated for the same 80 radial cords that were used in the late enhancement analysis.
The peripheral border zone was considered to be the mean difference in the transmurality index between the T2 and late enhancement images along each respective radial cord. The lateral border zone was considered as the circumferential distance between the lateral margins of edema and late enhancement and was measured at the midpoint of infarct thickness. The proportion of the edematous myocardium that did not show late enhancement was used to calculate a myocardial salvage index. The endocardial borders of edema and late enhancement on the segmented images were separately measured. The presence of low-signal hemorrhagic foci on the T2 images was also noted.
Volumetric analysis of the cine images was performed using CMRtools (Cardiovascular Imaging Solutions, London, UK) to derive left ventricular mass, end-diastolic volume, end-systolic volume, and ejection fraction. Regional systolic wall thickening was analyzed in three myocardial zones. The extent of delayed enhancement was used to define the infarct zone, a 30° sector either side of the lateral infarct border was used to define the peri-infarct zone, and the remainder of the myocardium that did not demonstrate edema or enhancement was used to define the remote myocardium (7). Wall thickening (WT) was defined as the wall thickness in end systole minus wall thickness in end diastole (WTED). Fractional wall thickening (WTf) was defined as follows:
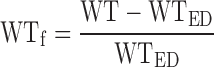 |
Statistical analysis.
Statistical analysis was performed with SPSS version 16 (SPSS, Chicago, IL). All continuous data are reported as means ± SD. The association between variables was assessed by linear least-squares regression. Statistical significance was assumed at P < 0.05.
RESULTS
Patient characteristics.
Patient and infarct characteristics are shown in Table 1. The culprit coronary arteries were as follows: left anterior descending coronary artery, n = 7; right coronary artery, n = 6; circumflex artery, n = 1; and obtuse marginal artery, n = 1. Thirteen subjects had a TIMI flow of 0, and one patient each had TIMI flows of 1 and 3 before PPCI of the culprit coronary artery. After PPCI, 12 patients had a TIMI flow of 3, and two subjects had a TIMI flow of 2. Patients underwent PPCI at a mean of 4.7 ± 3.5 h (range: 1.5–14 h) after symptom onset. The mean interval between PPCI and CMRI examination was 3.2 ± 2.1 days (range: 28 hours–7 days).
Table 1.
Infarct characteristics for each patient
| Patient | Culprit Coronary Artery | Rentrop Score | Pain to Balloon Time, h | PCI to CMR Interval, days | Ischemic AAR, %LV area | Infarct Size, %LV area | Myocardial Salvage, % |
|---|---|---|---|---|---|---|---|
| 1 | RCA | 2 | 4 | 7 | 23 | 20 | 13 |
| 2 | RCA | 2 | 1.5 | 2 | 20 | 8 | 60 |
| 3 | LAD | 0 | 3 | 1 | 29 | 27 | 5 |
| 4 | RCA | 1 | 3 | 4 | 43 | 39 | 9 |
| 5 | OM | 0 | 6 | 3 | 33 | 14 | 59 |
| 6 | RCA | 1 | 3.5 | 3 | 23 | 19 | 18 |
| 7 | LAD | 1 | 8 | 2 | 48 | 48 | 0 |
| 8 | LAD | 0 | 2.5 | 6 | 57 | 38 | 33 |
| 9 | LAD | 1 | 3 | 3 | 40 | 14 | 66 |
| 10 | LCx | 2 | 2.5 | 7 | 25 | 18 | 28 |
| 11 | LAD | 0 | 14 | 3 | 55 | 44 | 20 |
| 12 | RCA | 1 | 2.5 | 4 | 22 | 15 | 34 |
| 13 | LAD | 1 | 10 | 1 | 35 | 26 | 24 |
| 14 | LAD | 0 | 1.5 | 1 | 30 | 24 | 21 |
| 15 | RCA | 2 | 6 | 1 | 28 | 22 | 23 |
PCI, percutaneous coronary intervention; CMR, cardiac magnetic resonance; AAR, area at risk; LV, left ventricle; RCA, right coronary artery; LAD, left anterior descending coronary artery; OM, obtuse marginal coronary artery; LCx, left circumflex artery.
AAR, infarction, and wall motion.
Hyperintense myocardial edema was present in all subjects on T2-weighted images and corresponded to the vascular territory downstream of the occluded coronary artery (Fig. 1). The level-set edge detection tool determined the boundary of myocardial edema with excellent interobserver reliability (intraclass correlation coefficient = 0.93). All subjects showed a contiguous zone of myocardial edema, and no multifocal areas of ischemia were identified. Hypointense foci of hemorrhage in the midmyocardial core of the infarct were present in five transmural infarcts. No subjects had dilated ventricles or evidence of significant valvular incompetence.
Fig. 1.

Cardiac MRI (CMRI) images acquired after primary percutaneous coronary intervention (PPCI; top row) with demarcation of the endocardial (red) and epicardial (green) borders. Analysis of contractile function and transmural extent of signal abnormality was performed along multiple radial cords around the left ventricle (bottom row). A balanced steady-state-free precession (b-SSFP) sequence (left) was used to quantify systolic fractional thickening. A T2-STIR sequence (middle) was used to measure the extent of myocardial edema (yellow), which is a marker for the ischemic area at risk (AAR). A late gadolinium enhancement sequence (right) was used to quantify the extent of infarcted myocardium (yellow).
Regional wall motion abnormalities were present in all subjects in the infarct-related territory. Six subjects demonstrated negative fractional wall thickening in dyskinetic infarcted myocardium owing to paradoxical wall thinning during systole. The mean fractional wall thickening in the infarcted, peri-infarcted, and remote myocardium was 3.6 ± 16.0%, 40.5 ± 26.4%, and 88.2 ± 39.3%, respectively. In the five infarcts with MVO or hemorrhage, the fractional wall thickening in the infarcted zone was significantly reduced compared with those without (−6.1 ± 12.4% vs. 8.3 ± 9.8%, P < 0.05).
All subjects demonstrated late gadolinium enhancement in the same territory as the myocardial edema on T2-weighted images. A single contiguous region of infarct enhancement was present, and no multifocal lesions were present. Late enhancement was always present in the subendocardial region and showed variable transmural extension. The same five subjects that showed midmyocardial hypointense foci on T2-weighted images had corresponding regions of nonenhancement consistent with MVO. The enhancing infarct was confined to the boundary of myocardial edema demonstrated on T2-weighted images.
Endocardial margin of edema.
The subendocardial border of edema on T2-weighted images showed a close correlation to the subendocardial late enhancement (r = 0.94, P < 0.001). In no case was the subendocardium spared from either edema or necrosis despite reperfusion occurring within 2 h. The length of the endocardial border of edema showed a modest correlation with the ischemic AAR (r = 0.34, P < 0.05) but not with the total infarct area (r = 0.15, P = 0.15).
Peripheral and lateral border zones.
The mean infarct size, indicated by late enhancement, was 22 ± 16% with a range of 8–48%. The mean infarct transmurality was 85 ± 20% with nine patients demonstrating transmural infarcts and six patients demonstrating partial thickness infarcts. A peri-infarct border zone was visible in each patient (Fig. 2). The depth of the peripheral infarct border zone from the epicardial surface ranged between 0 and 7 mm (mean: 2 mm). The mean ischemic AAR, indicated by myocardial edema, was 34 ± 12% with a range of 20–57%. Infarct size showed a linear relationship to the ischemic AAR (r = 0.75, P < 0.01; Fig. 3) with a mean myocardial salvage of 27 ± 21%. There was no significant relationship between myocardial salvage and pain to balloon time (r = 0.2). The infarcted myocardium was always smaller than the ischemic AAR and involved between 34% and 99% (mean: 72 ± 21%) of the ischemic bed due to variation in transmural infarct extension. A lateral zone of viable myocardium was present but was limited in extent and ranged from between 0 and 11 mm (mean: 5 ± 4 mm) so that infarcts involved 70–100% (mean: 82 ± 13%) of the width of the AAR. The mean lateral infarct border zone in transmural infarcts was 5 ± 4 mm and that in partial thickness (<75%) infarcts was 5 ± 3 mm. In transmural infarctions, the total lateral infarct border zone represented 12 ± 5% of the ischemic AAR. The width of the lateral border zone showed no relationship to infarct size (r = −0.04, P = 0.4) and was not significantly different in each coronary territory.
Fig. 2.

CMRI images acquired after PPCI to an obtuse marginal artery occlusion. Endo- and epicardial borders are shown in blue. The edges of the ischemic AAR and the infarct have been defined using boundary detection algorithms on late enhancement (left) and T2-weighted sequences (middle), respectively. A coregistered image is shown on the right with its deformation grid. The green area shows the reversibly injured ischemic peri-infarct zone surrounding the infarcted red area. In this partial thickness infarct, myocardial salvage has been largely due to epicardial limitation of the infarct as the lateral peri-infarct border zone is narrow.
Fig. 3.

Correlation between the gadolinium-enhancing infarct area and the ischemic AAR on T2-weighted images (r = 0.75, P < 0.01; expressed as a percentage of the left ventricular area).
DISCUSSION
The major finding of this study is that there is limited potential for myocardial salvage after reperfusion at the lateral infarct border zones compared with the subepicardial peripheral border zone as the lateral infarct margin approximates to the edge of the ischemic bed in both partial thickness and transmural infarcts. These results support ex vivo imaging and histopathological observations of the peri-infarct border zone (21).
In this study, we have shown that the infarcted myocardium is consistently surrounded by a zone of myocardial edema. Infarct size is closely associated to the extent of the ischemic AAR, which depends on the vascular territory of the occluded coronary artery. As in both ex vivo human and canine studies, the ischemic bed size is a major determinant of infarct size (21). After PPCI, the subjects in our study demonstrated that infarct size, as a proportion of the AAR, ranged between 34% and 99% and, therefore, up to 66% of the ischemic vascular bed had been salvaged after reperfusion. In this series of patients, we were able to identify that salvage predominantly occurs due to epicardial limitation of the infarct as the lateral boundaries of necrosis generally approximated to the lateral extent of the ischemic AAR. The mean lateral infarct border zone was 5 mm, which did not vary between partial thickness and transmural infarcts. For all subjects, a mean of 82% of the width of the AAR was infarcted, and in transmural infarctions, the lateral border zones represented only 12% of the total ischemic bed area. The width of the lateral infarct border zone in an ex vivo human study (21) has been reported as 1.7 ± 3.0 mm, which is slightly narrower than in our series. The differences may be partly attributable to the lower spatial resolution that is achievable with noninvasive imaging techniques but also the fact that the histological studies were of completed fatal infarcts where no therapeutic reperfusion was attempted. Small differences in pain to balloon time may result in a significant increase in infarct transmurality and the extent of MVO (38, 39); therefore, there has been extensive interest in the development of cardioprotective strategies to limit the progress of necrosis around the time of reflow (42). Our findings have shown that it is the transmural extent of necrosis that is the predominant determinant of infarct size after PPCI, and so there is limited potential for improving salvage at the narrow lateral infarct border zones.
The physiological correlates of the peri-infarct border zone using CMRI have been described in animal models and human studies (31). There is histopathological evidence in animal models that the area of edema on T2-weighted CMRI corresponds to ischemic tissue that is potentially recoverable (3, 17) and shows close correlation to AAR measurements using the fluorescein method (14). T2-weighted imaging performed up to 1 wk after reperfusion can accurately determine the myocardium at risk as it was before reperfusion compared with 99mTc tetrofosmin SPECT (11). T2 imaging also has a potential advantage in demonstrating myocardial ischemia in cases of combined vascular beds (21), where the ischemic zone may not conform to the entire distribution of a single coronary artery. The changes in T2 relaxation are a consequence of increased mobile water content due to intracellular Na+ accumulation (36) and cell swelling (18). Reperfusion hemorrhage causes shortening of T2 relaxation, and foci of signal hypointensity correspond to histological evidence of hemorrhage (6, 40). Late gadolinium enhancement is confined to the region of necrosis in acute infarctions, but excludes areas of severe transient ischemia in the border zone, and is a consequence of altered contrast kinetics resulting from a loss of sarcomere membrane integrity (20). Regions of nonenhancement within the infarct core indicate the presence of MVO (7, 25, 37). Combining the two techniques of T2-weighted imaging and late enhancement provides a method for distinguishing irreversibly injured myocardium from ischemic but potentially viable myocardium.
The observation that after coronary artery occlusion there is confluent necrosis that evolves as a transmural wavefront from the subendocardium to subepicardium has been described in canine models (33, 34). The mechanism for this gradient is suspected to be due to the distribution of epicardial collateral blood flow (41), which produces a sharp boundary between ischemic and necrotic tissue (33). The size of the ischemic vascular bed depends on the anatomic boundaries of the occluded coronary artery, but the extent of necrosis within that zone is variable (21). Although the lateral boundaries of the infarct are established shortly after occlusion of the culprit coronary artery (21), the transmural advance of necrosis may be arrested by timely coronary reperfusion (34). In our series, the ischemic subendocardium was always infarcted, and this was independent of the duration of coronary occlusion. In canines, the rate at which subendocardial to subepicardial myocyte injury occurs is primarily related to the gradient of ischemia maintained by the transmural distribution of coronary collateral flow. There is significant variability in the development of vascular collateral networks in different animal models (22) and in patients with coronary artery disease (32). Compared with canine models, the subjects in our study had relatively poor development of epicardial collateral vessels, and two subjects developed transmural infarctions despite the presence of partial collateral filling. A study (22) on sheep, which also develop only limited collateral networks, has shown that it is the midmyocardium that is the most susceptible to ischemia. Although our findings showed that severe myocardial injury, demonstrated by microvascular obstruction and interstitial hemorrhage, was observed in the midmyocardium, there was no evidence of endocardial resistance to infarction.
We have demonstrated that wall motion abnormalities are present not only in the infarcted territory but also in the zone of myocardium that lies adjacent to the infarct. It is known that there is abnormal contraction in the peri-infarct region (5) and that this may be due to abnormal cardiac myocardial energetics (16). However, we have shown that the lateral ischemic border zone is narrow, and so abnormal contraction in the peri-infarct region may not simply be a direct result of ischemic injury. Our findings are consistent with a previous report (5) of disturbed mechanical interactions between normal and ischemic myocardium that extend well beyond the infarct zone. MVO and hemorrhage are associated with poorer systolic function within the infarct, and this may reflect the severity of ischemic injury and necrosis that has occurred.
Study limitations.
Although both the size of the infarct and the extent of edema gradually diminish after coronary occlusion, the areas of signal abnormality on T2-weighted and late enhancement imaging are stable within the first week after infarction (20, 28). While retrospective determination of the ischemic AAR is a unique advantage of CMRI, the delay between PPCI and CMRI may introduce a potential inaccuracy to the assessment of myocardial salvage. It is also not possible to speculate, based on the numbers used in this study, to what extent the degree of myocardial salvage is due to coronary intervention or to the natural infarct limitation that is sometimes observed in untreated infarctions (21). The relationship between pain to balloon time and normalized infarct size after PPCI may be nonlinear (9); however, our study was underpowered to confirm the association between myocardial salvage and coronary occlusion time. Finally, there may be a variation between the actual onset of coronary occlusion and the history of chest pain offered by the patient, and so this measure has limited accuracy in determining the precise duration of myocardial ischemia, which is common to all human studies of STEMI.
Conclusions.
Myocardial salvage is largely determined by epicardial limitation of the infarct within the ischemic AAR after PPCI. The lateral boundaries of necrosis approximate to the lateral extent of the ischemic bed, and systolic wall motion abnormalities extend well beyond the infarct border zone. The narrow lateral border zones in partial and full thickness infarcts indicate that cardioprotective strategies may have limited potential for improving myocardial salvage at these infarct margins.
GRANTS
This work was primarily funded by the Medical Research Council (UK) (to D. P. O'Regan, R. Ahmed, Y. Tan, C. Neuwirth, G. Durighel, J. V. Hajnal, and S. A. Cook). In addition, S. A. Cook is funded by the British Heart Foundation (UK) and the Fondation Leducq (France). We are also grateful for support from the National Institute for Health Research Biomedical Research Centre funding scheme.
REFERENCES
- 1.Abdel-Aty H, Simonetti O, Friedrich MG. T2-weighted cardiovascular magnetic resonance imaging. J Magn Reson Imaging 26: 452–459, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Aletras AH, Tilak GS, Natanzon A, Hsu LY, Gonzalez FM, Hoyt RF Jr, Arai AE. Retrospective determination of the area at risk for reperfused acute myocardial infarction with T2-weighted cardiac magnetic resonance imaging: histopathological and displacement encoding with stimulated echoes (DENSE) functional validations. Circulation 113: 1865–1870, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Arganda-Carreras I, Sánchez Sorzano CO, Marabini R, Carazo JM, Ortiz-de Solorzano C, Kybic J. Consistent and elastic registration of histological sections using vector-spline regularization. In: Computer Vision Approaches to Medical Image Analysis. Berlin: Springer, 2006, p. 85–95.
- 5.Ashikaga H, Mickelsen SR, Ennis DB, Rodriguez I, Kellman P, Wen H, McVeigh ER. Electromechanical analysis of infarct border zone in chronic myocardial infarction. Am J Physiol Heart Circ Physiol 289: H1099–H1105, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basso C, Corbetti F, Silva C, Abudureheman A, Lacognata C, Cacciavillani L, Tarantini G, Marra MP, Ramondo A, Thiene G, Iliceto S. Morphologic validation of reperfused hemorrhagic myocardial infarction by cardiovascular magnetic resonance. Am J Cardiol 100: 1322–1327, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bogaert J, Kalantzi M, Rademakers FE, Dymarkowski S, Janssens S. Determinants and impact of microvascular obstruction in successfully reperfused ST-segment elevation myocardial infarction. Assessment by magnetic resonance imaging. Eur Radiol 17: 2572–2580, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Boxt LM, Hsu D, Katz J, Detweiler P, Mclaughlin S, Kolb TJ, Spotnitz HM. Estimation of myocardial water content using transverse relaxation time from dual spin-echo magnetic resonance imaging. Magn Reson Imaging 11: 375–383, 1993. [DOI] [PubMed] [Google Scholar]
- 9.Busk M, Kaltoft A, Nielsen SS, Bottcher M, Rehling M, Thuesen L, Botker HE, Lassen JF, Christiansen EH, Krusell LR, Andersen HR, Nielsen TT, Kristensen SD. Infarct size and myocardial salvage after primary angioplasty in patients presenting with symptoms for <12 h vs. 12–72 h. Eur Heart J. In press. [DOI] [PubMed]
- 10.Bydder M, Larkman DJ, Hajnal JV. Combination of signals from array coils using image-based estimation of coil sensitivity profiles. Magn Reson Med 47: 539–548, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Carlsson M, Ubachs J, Heiberg E, Hedstrom E, Jovinge S, Arheden H. Myocardium at risk and myocardial salvage after acute infarction in humans; quantification by magnetic resonance imaging. J Cardiovasc Magn Reson 11: O29, 2009. [Google Scholar]
- 12.Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D'Agostino RB, O'Donnell CJ. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116: 39–48, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich MG, Abdel-Aty H, Taylor A, Schulz-Menger J, Messroghli D, Dietz R. The salvaged area at risk in reperfused acute myocardial infarction as visualized by cardiovascular magnetic resonance. J Am Coll Cardiol 51: 1581–1587, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Dorado D, Oliveras J, Gili J, Sanz E, Perez-Villa F, Barrabes J, Carreras MJ, Solares J, Soler-Soler J. Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc Res 27: 1462–1469, 1993. [DOI] [PubMed] [Google Scholar]
- 15.Heiberg E, Ugander M, Engblom H, Gotberg M, Olivecrona GK, Erlinge D, Arheden H. Automated quantification of myocardial infarction from MR images by accounting for partial volume effects: animal, phantom, and human study. Radiology 246: 581–588, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Hu Q, Wang X, Lee J, Mansoor A, Liu J, Zeng L, Swingen C, Zhang G, Feygin J, Ochiai K, Bransford TL, From AHL, Bache RJ, Zhang J. Profound bioenergetic abnormalities in peri-infarct myocardial regions. Am J Physiol Heart Circ Physiol 291: H648–H657, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Ibanez B, Prat-Gonzalez S, Speidl WS, Vilahur G, Pinero A, Cimmino G, Garcia MJ, Fuster V, Sanz J, Badimon JJ. Early metoprolol administration before coronary reperfusion results in increased myocardial salvage: analysis of ischemic myocardium at risk using cardiac magnetic resonance. Circulation 115: 2909–2916, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Jennings RB, Murry CE, Steenbergen C, Reimer KA. Development of cell injury in sustained acute ischemia. Circulation 82: 2–12, 1990. [PubMed] [Google Scholar]
- 19.Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 361: 13–20, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Kim RJ, Fieno DS, Parrish TB, Harris K, Chen EL, Simonetti O, Bundy J, Finn JP, Klocke FJ, Judd RM. Relationship of MRI delayed contrast enhancement to irreversible injury, infarct age, and contractile function. Circulation 100: 1992–2002, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Lee JT, Ideker RE, Reimer KA. Myocardial infarct size and location in relation to the coronary vascular bed at risk in man. Circulation 64: 526–534, 1981. [DOI] [PubMed] [Google Scholar]
- 22.Leshnower BG, Sakamoto H, Hamamoto H, Zeeshan A, Gorman JH, Gorman RC. Progression of myocardial injury during coronary occlusion in the collateral-deficient heart: a non-wavefront phenomenon. Am J Physiol Heart Circ Physiol 293: H1799–H1804, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Look DC, Locker DR. Time saving in measurement of NMR and EPR relaxation times. Rev Sci Instrum 41: 250–251, 1970. [Google Scholar]
- 24.Lotan CS, Miller SK, Cranney GB, Pohost GM, Elgavish GA. The effect of postinfarction intramyocardial hemorrhage on transverse relaxation time. Magn Reson Med 23: 346–355, 1992. [DOI] [PubMed] [Google Scholar]
- 25.Lund GK, Stork A, Saeed M, Bansmann MP, Gerken JH, Muller V, Mester J, Higgins CB, Adam G, Meinertz T. Acute myocardial infarction: evaluation with first-pass enhancement and delayed enhancement MR imaging compared with 201Tl SPECT imaging. Radiology 232: 49–57, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Malladi R, Sethian JA. Level set methods for curvature flow, image enhancement, and shape recovery in medical images. In: Proceedings of Conferences on Visualization and Mathematics. Berlin, Germany: Springer-Verlag, 1995, p. 329–345.
- 27.Ndrepepa G, Mehilli J, Schwaiger M, Schuhlen H, Nekolla S, Martinoff S, Schmitt C, Dirschinger J, Schomig A, Kastrati A. Prognostic value of myocardial salvage achieved by reperfusion therapy in patients with acute myocardial infarction. J Nucl Med 45: 725–729, 2004. [PubMed] [Google Scholar]
- 28.Nilsson JC, Nielsen G, Groenning BA, Fritz-Hansen T, Sondergaard L, Jensen GB, Larsson HB. Sustained postinfarction myocardial oedema in humans visualised by magnetic resonance imaging. Heart 85: 639–642, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nordmann AJ, Hengstler P, Harr T, Young J, Bucher HC. Clinical outcomes of primary stenting versus balloon angioplasty in patients with myocardial infarction: a meta-analysis of randomized controlled trials. Am J Med 116: 253–262, 2004. [DOI] [PubMed] [Google Scholar]
- 30.O'Regan DP, Ahmed R, Karunanithy N, Neuwirth RN, Tan Y, Durighel G, Hajnal JV, Nadra I, Corbett SJ, Cook SA. Reperfusion hemorrhage following acute myocardial infarction: assessment with T2* mapping and effect on measuring the area at risk. Radiology 251: 916–922, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Pennell D Myocardial salvage: retrospection, resolution, and radio waves. Circulation 113: 1821–1823, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Pohl T, Seiler C, Billinger M, Herren E, Wustmann K, Mehta H, Windecker S, Eberli FR, Meier B. Frequency distribution of collateral flow and factors influencing collateral channel development: functional collateral channel measurement in 450 patients with coronary artery disease. J Am Coll Cardiol 38: 1872–1878, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest 40: 633–644, 1979. [PubMed] [Google Scholar]
- 34.Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 56: 786–794, 1977. [DOI] [PubMed] [Google Scholar]
- 35.Rentrop KP, Cohen M, Blanke H, Phillips RA. Changes in collateral channel filling immediately after controlled coronary artery occlusion by an angioplasty balloon in human subjects. J Am Coll Cardiol 5: 587–592, 1985. [DOI] [PubMed] [Google Scholar]
- 36.Rochitte CE, Kim RJ, Hillenbrand HB, Chen El, Lima JAC. Microvascular integrity and the time course of myocardial sodium accumulation after acute infarction. Circ Res 87: 648–655, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Rochitte CE, Lima JA, Bluemke DA, Reeder SB, McVeigh ER, Furuta T, Becker LC, Melin JA. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 98: 1006–1014, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Tarantini G, Cacciavillani L, Corbetti F, Ramondo A, Marra MP, Bacchiega E, Napodano M, Bilato C, Razzolini R, Iliceto S. Duration of ischemia is a major determinant of transmurality and severe microvascular obstruction after primary angioplasty: a study performed with contrast-enhanced magnetic resonance. J Am Coll Cardiol 46: 1229–1235, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Thiele H, Kappl MJ, Linke A, Erbs S, Boudriot E, Lembcke A, Kivelitz D, Schuler G. Influence of time-to-treatment, TIMI-flow grades, and ST-segment resolution on infarct size and infarct transmurality as assessed by delayed enhancement magnetic resonance imaging. Eur Heart J 28: 1433–1439, 2007. [DOI] [PubMed] [Google Scholar]
- 39a.TIMI Study Group. The Thrombolysis in Myocardial Infarction (TIMI) trial. Phase I findings. TIMI Study Group. N Engl J Med 312: 932–936, 1985. [DOI] [PubMed] [Google Scholar]
- 40.van den Bos EJ, Baks T, Moelker AD, Kerver W, van Geuns RJ, van der Giessen WJ, Duncker DJ, Wielopolski PA. Magnetic resonance imaging of haemorrhage within reperfused myocardial infarcts: possible interference with iron oxide-labelled cell tracking? Eur Heart J 27: 1620–1626, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Weisse AB, Kearney K, Narang RM, Regan TJ. Comparison of the coronary collateral circulation in dogs and baboons after coronary occlusion. Am Heart J 92: 193–200, 1976. [DOI] [PubMed] [Google Scholar]
- 42.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 357: 1121–1135, 2007. [DOI] [PubMed] [Google Scholar]