

Abstract
Thiamin is essential for the normal function of the endocrine pancreas, but very little is known about uptake mechanism(s) and regulation by beta cells. We addressed these issues using mouse-derived pancreatic beta-TC-6 cells, and freshly isolated primary mouse and human pancreatic islets. Results showed that thiamin uptake by beta-TC-6 cells involves a pH (but not Na+)-dependent carrier-mediated process that is saturable at both the nanomolar (apparent Km = 37.17 ± 9.9 nM) and micromolar (apparent Km = 3.26 ± 0.86 μM) ranges, cis-inhibited by thiamin structural analogs, and trans-stimulated by unlabeled thiamin. Involvement of carrier-mediated process was also confirmed in primary mouse and human pancreatic islets. Both THTR-1 and THTR-2 were found to be expressed in these mouse and human pancreatic preparations. Maintaining beta-TC-6 cells in the presence of a high level of thiamin led to a significant (P < 0.01) decrease in thiamin uptake, which was associated with a significant downregulation in level of expression of THTR-1 and THTR-2 at the protein and mRNA levels and a decrease in transcriptional (promoter) activity. Modulators of intracellular Ca2+/calmodulin- and protein-tyrosine kinase-mediated pathways also altered thiamin uptake. Finally, confocal imaging of live beta-TC-6 cells showed that clinical mutants of THTR-1 have mixed expression phenotypes and all led to impairment in thiamin uptake. These studies demonstrate for the first time that thiamin uptake by the endocrine pancreas is carrier mediated and is adaptively regulated by the prevailing vitamin level via transcriptional mechanisms. Furthermore, clinical mutants of THTR-1 impair thiamin uptake via different mechanisms.
Keywords: THTR-1, THTR-2, thiamin uptake, transport regulation, TRMA
thiamin (vitamin b1) plays an essential role in normal cellular functions, growth, and development. In its coenzyme form, i.e., thiamin pyrophosphate, the vitamin plays a vital role in metabolism and energy production reactions that include the decarboxylation of pyruvic acid and α-ketoglutamic acid and the utilization of pentose in the hexose monophosphate shunt (3). Furthermore, because thiamin bridges the glycolytic and the pentose phosphate metabolic pathways that are critical for creating chemical reducing power in cells, the vitamin is also considered as having a role in reducing cellular oxidative stress (4, 10). Thus low intracellular levels of thiamin will lead to impairment in energy metabolism and to a propensity for oxidative stress. In addition, a reduction in intracellular thiamin leads to apoptosis (19, 21, 36, 22). Clinically, thiamin deficiency in humans leads to a variety of abnormalities including neurological and cardiovascular disorders (3, 42, 45). Systemic thiamin deficiency and suboptimal levels represent significant nutritional problems in both developed and developing countries and occur in a variety of conditions as in patients with diabetes mellitus (34, 43, 44) and chronic alcoholism (8, 17, 40, 41). Thiamin deficiency also occurs at localized levels, leading to pathologies in the affected tissues as in the case of patients with thiamin-responsive megaloblastic anemia (TRMA; see below) (1, 21). In contrast to the negative effects of thiamin deficiency and suboptimal levels, optimization of thiamin level appears to have the potential of preventing diabetic retinopathy and blocking tissue damage caused by hyperglycemia of diabetes (12). It is also effective in the treatment of many of the clinical symptoms associated with TRMA (1, 18, 21, 29). Thus studies that are aimed at understanding the mechanisms involved in the maintenance and regulation of normal thiamin cellular homeostasis are of significant physiological and nutritional importance.
The pancreas contains high levels of thiamin (23), and, because cells of this vital organ cannot synthesize thiamin, they must obtain the vitamin from their surrounding environment via transport across cell membrane. Thiamin is important for both the endocrine and the exocrine functions of this organ (24, 25, 35). With regard to the endocrine function, thiamin deficiency leads to a marked impairment in insulin synthesis and secretion (24, 25). Of relevance to the latter is the development of diabetes mellitus in patients with TRMA (1, 18, 21, 29), an autosomal recessive disorder that is caused by mutations in thiamin transporter-1 (THTR-1; product of the SLC19A2 gene) (6, 9, 16). These mutations lead to impairment in cellular thiamin accumulation and the development of localized thiamin deficiency in pancreatic beta cells (and other affected tissues). This in turn leads to derangements in cellular metabolism, cell stress, and apoptosis (1, 21, 36). Supplementation of TRMA patients with high doses of thiamin brings about a marked improvement in the clinical symptoms of the disease including a reduction or cessation in the need for exogenous insulin (1, 18, 21, 29).
Despite the importance of thiamin for the normal functions of the pancreas, there is little known about the cellular and molecular mechanisms involved in the transport of this vitamin into pancreatic cells and their regulation. Also not known is the cell biology of THTR-1 mutants found in TRMA patients in pancreatic beta cells, which are a major target of TRMA pathology. Addressing these issues is of significant physiological and nutritional importance because of the need of the vitamin for the normal function of this vital organ and because such knowledge may assist in the designing of effective strategies to optimize thiamin pancreatic homeostasis, especially in conditions associated with thiamin deficiency and suboptimal levels. Here we report the results of our investigations into the mechanism(s) and regulation of thiamin uptake by pancreatic beta cells and islets. We used mouse-derived culture pancreatic beta-TC-6 cells and freshly isolated primary mouse and human pancreatic islets as models in our investigations.
MATERIALS AND METHODS
[3H]Thiamin (specific activity 555 GBq/mmol; radiochemical purity >98%) was obtained from American Radio-labeled Chemicals (St. Louis, MO). All other chemicals, routine reagents, and kits were of the analytical or molecular biology grade and were purchased from commercial vendors.
Culturing of Pancreatic Beta-TC-6 Cells and Uptake Studies
Mouse-derived pancreatic beta-TC-6 insulinoma cells (passages 10-22) were obtained from American Type Tissue Collection (ATCC; Rockville, MD) and maintained in DMEM growth medium in the presence of 15 nM thiamin. Uptake was examined (unless otherwise stated) at 37°C in Krebs-Ringer buffer (in mM: 133 NaCl, 4.93 KCl, 1.23 MgSO4, 0.85 CaCl2, 5 glucose, 5 glutamine, 10 HEPES, and 10 MES; pH 8.0) by a rapid filtration technique (14), as described by us previously (10, 30). Labeled and unlabeled thiamin was added to the incubation medium at the onset of incubation and uptake was examined during the initial linear period (i.e., 10 min). The reaction was terminated by the addition of 1 ml of ice-cold buffer followed by filtration through 0.45-μm nitrocellulose filters (Millipore; Billerica, MA) and washing with 5 ml of ice-cold buffer. Filters were then placed in scintillation vials containing Econo-Safe scintillation fluid, and radioactivity was counted in a scintillation counter. Protein content of cell digests was measured in parallel wells by using a Bio-Rad Dc protein assay kit (Hercules, CA).
In examining the effect of thiamin level in the culture medium, beta-TC-6 cells were maintained (8–10 days) in culture medium supplemented with the usual 15 nM or with 12 μM thiamin then used for uptake investigation. In examining the effect of modulators of intracellular regulatory pathways, the modulators were added to the incubation buffer 1 h prior to uptake investigations. In determining the metabolic form of 3H radioactivity taken up by pancreatic beta-TC-6 cells following incubation (10 min) with [3H]thiamin, cells were homogenized in 100% ethanol and centrifuged, and the supernatant was applied onto a cellulose precoated thin-layer chromatography plates and run with a solvent system of isopropanol:acetate buffer (0.5 M, pH 4.5):water (65:15:20 vol/vol/vol) (2).
Isolation of Mouse Primary Pancreatic Islets, Processing of Human Primary Pancreatic Islets, and Uptake Studies
Mouse islets [which contain a high proportion (>90%) of beta cells; Ref. 13] were isolated on the day of experiment by stationary digestion and the Ficoll gradient method as described previously (11). Five to six mice were euthanized and the pancreas was dissected out (without fat contamination). Pancreas was minced and digested with HEPES (25 mM) containing collagenase V (1 mg/ml) and DNase I (0.1 mg/ml) for 40 min at 37°C. The digested tissue was filtered through a nylon mesh with a pore size of 600 μm, and islet cells were purified on a discontinuous Ficoll gradient in the order of 25, 23, 20, and 11% in HBSS. The gradient was centrifuged for 15 min at 2,500 RPM and 4°C, and islets were taken from the interface of 20 and 11% Ficoll gradient. The islet cells were then washed with HBSS and identified by dithizone staining. After purification, islets were maintained in DMEM with 10% FBS for 5 h prior to uptake studies. The viability of the mouse pancreatic islets (tested by Trypan blue exclusion) was ∼80%. Uptake of [3H]thiamin was examined at 37°C by a rapid filtration technique. With regard to freshly isolated primary human pancreatic islets, the islets were obtained from two adult organ donors (National Disease Research Interchange, NDRI; Philadelphia, PA). The viability of the human pancreatic islets tested in our laboratory by Trypan blue exclusion immediately before use for transport investigations was ∼73%. The islets were centrifuged at 1,500 rpm for 8 min, washed with ice-cold Krebs-Ringer buffer, centrifuged again, and then resuspended in Krebs-Ringer buffer and used for uptake investigations as described earlier. The Institutional Animal Care and Use Committee (IACUC) of Veterans Affairs Long Beach and University of California Irvine approved the experimental procedures used for mice in this study.
Semiquantitative and Real-Time PCR Analysis
Oligo(dT) primers and 3 μg of total RNA isolated from cultured pancreatic beta-TC-6 cells and primary mouse and human pancreatic islets were used for the first-strand cDNA synthesis using an Invitrogen Superscript synthesis system. Primers for semiquantitative PCR utilized in this study were specific for the open reading frame (ORF) of mouse THTR-1 (forward: 5′- GTTCCTCACGCCCTACCTTC-3′, reverse: 5′-GCATGAACCACGTCACAATC-3′), THTR-2 (forward: 5′-GTCAGCCCAGAACACTATCA-3′, reverse: 5′-CCAGTACATTGTTCCAGTGG CT-3′), and β-actin (forward: 5′-AGCCAGACCGTCTCCTTGTA-3′, reverse: 5′-TAGAGAGGGCCCACCACAC-3′). Regarding semiquantitative and real-time PCR primers for human THTR-1, the forward primer used was 5′-AGCCAGACCGTCTCCTTGTA-3′, and the reverse was 5′-TAGAGAGGGCCCACCACAC-3′. For human THTR-2, the forward primer was 5′-TTCCTGGATTTACCCCACTG-3′ and the reverse primer was 5′-GTATGTCCAAACGGGGAAGA; for human β-actin, the forward was 5′-CATCCTGCGTCTGGACCT-3′ and the reverse was 5′-TAATGTCACGCACGATTTCG-3′. The conditions for semiquantitative PCR were 95°C for 30 s, annealing at 58°C for 15 s, and extension at 72°C for 30 s (29 and 34 cycles for mTHTR-1 and mTHTR-2, respectively). The products were analyzed on 2% agarose gels, the images were captured via an Eagle Eye II system, and the amplified RT-PCR products were normalized to amplified β-actin controls. Real-time quantitative PCR was performed by using a SYBR Green PCR kit (Qiagen, Valencia, CA). Data was normalized relative to β-actin and calculated by use of a relative relationship method supplied by iCycler (Bio-Rad; Hercules, CA).
Assessment of Promoter Activity
The SLC19A2 (−2250 to −36) and SLC19A3 (−1957 to +59) promoter-luciferase full-length reporter constructs used in this study were generated and described by us previously (20, 26, 28). Pancreatic beta-TC-6 cells were cotransfected in 12-well plates at ∼50–80% confluency with 2 μg of each promoter construct and 100 ng of the Renilla transfection control plasmid Renilla luciferase-thymidine kinase (pRL-TK) (Promega, Madison, WI). Transfection was performed with Lipofectamine (Invitrogen) according to the manufacturer's instructions. Cells were then maintained in control (15 nM thiamin) or thiamin-oversupplemented (12 μM) growth medium (6–8 days) and transfected, and then (48 h later) Renilla-normalized firefly luciferase activity was determined by the Dual Luciferase Assay system (Promega).
Live Cell Confocal Imaging of hTHTR1-GFP and Mutants in Pancreatic Beta-TC-6 Cells
The hTHTR1-GFP fusion construct (37) was used as a template to introduce insertions or deletions of missense clinical mutations in the ORF of hTHTR1 by Quick Change Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) as described previously (37, 38). For the newly identified clinical hTHTR-1 mutant, i.e., G172R (1), the forward primer used was 5′-GTGGGCTTTACAGTGCGCTCTGTCCTAGGGCAA-3′ and the reverse primer was 5′-TTGCCCTAGGACAGAGCGCACTGTAAAGCCCAC-3′. All other mutants used in this study were generated and sequenced previously (38, 39). Mutants were transiently transfected into pancreatic beta-TC-6 cells grown on coverslips, and live cell imaging was performed (24 to 48 h later) by using a Nikon C1 confocal scanner head attached to a Nikon inverted phase-contrast microscope. Fluorophores were excited by using the 488-nm line from an Ar ion laser (green) and 543-nm line from a HeNe ion laser (red), and emitted fluorescence was monitored with 515 ± 30-nm short-pass [green fluorescent protein (GFP)] or 570 ± 50+-nm long-pass (FM4–64) filters.
Data Presentation and Statistical Analysis
Uptake values are means ± SE of multiple (at least three) individual uptake determinations and are expressed in femtomoles or picomoles per milligram protein per unit time. Student's t-test and ANOVA were used in statistical analysis. P < 0.05 was considered statistically significant. Quantitative variations in the absolute amounts of thiamin uptake between different cell patches were observed, and, thus, appropriate controls were run simultaneously with each set of experiments and data were expressed as percentage relative to such controls. Kinetic parameters of the saturable uptake processes of thiamin [i.e., maximal velocity (Vmax) and apparent Km] were calculated as described by us previously (2, 39) by the Wilkinson method (46). Western blotting, semiquantitative RT-PCR, and live confocal imaging studies were all performed on at least three separate occasions with comparable results and representative experiments are presented in this report. Promoter activity is presented as mean ± SE of at least three independent experiments and given as fold over pGL3-Basic expression set arbitrarily at 1.
RESULTS
Thiamin Uptake by Mouse Cultured Pancreatic Beta-TC-6 Cells and by Freshly Isolated Mouse Pancreatic Islets: General Characterization
Uptake of thiamin (15 nM) by mouse cultured pancreatic beta-TC-6 cells as a function of time at pH 8.0 was linear (r2 = 0.98) for up to 12 min of incubation (rate = 7.9 fmol·mg protein−1·min−1; not shown). By thin-layer chromatography, the metabolic form of the transported 3H-labeled substrate following 10-min incubation with 0.16 μM [3H]thiamin was determined and found to be mostly (94%) in the form of intact (unmetabolized) thiamin. On the basis of these two findings, a 10-min incubation time was chosen in all subsequent uptake experiments.
Thiamin (15 nM) uptake by pancreatic beta-TC-6 cells was pH dependent. Uptake at buffer pH 8.0 was more than 2.3-fold higher (P < 0.01) than uptake at pH 5.0 (Fig. 1). Thus we performed all subsequent studies at buffer pH 8.0. The role of extracellular Na+ in thiamin uptake was also tested by examining the effect of isoosmotic replacement of Na+ with K+ or with mannitol. The results showed similar thiamin (15 nM) uptake in the presence and absence of Na+ (202.5 ± 11.8, 195.4 ± 5.6, and 200.3 ± 17 fmol·mg protein−1·10 min−1 in the presence of Na+ and in its absence but presence of K+ and mannitol, respectively). The role of Na+ in thiamin uptake was further tested by examining the effect of pretreating (for 30 min) pancreatic beta-TC-6 cells with the Na+-K+-ATPase inhibitor ouabain (1 mM) on thiamin (15 nM) uptake. Again no significant inhibition in thiamin uptake was observed (198.3 ± 6.4 and 219.8 ± 12.6 fmol·mg protein−1·10 min−1 for control and ouabain pretreated cells, respectively). This indicates that thiamin uptake by pancreatic beta-TC-6 cells is Na+ independent. Energy dependence of the thiamin (15 nM) uptake process of pancreatic beta-TC-6 cells was tested by examining the effect of pretreating (1 mM; 30 min) the cells with the metabolic inhibitors 2,4-dinitrophenol (DNP) and iodoacetate. A significant inhibition (P < 0.01 for both) in thiamin uptake was observed (210.1 ± 13.6, 86.8 ± 6.2, and 100.2 ± 9.7 fmol·mg protein−1·10 min−1 for control and in cells pretreated with DNP and iodoacetate, respectively).
Fig. 1.

Effect of incubation buffer pH on the thiamin uptake by pancreatic beta-TC-6 cells. Confluent cell monolayers were incubated at 37°C in Krebs-Ringer buffer of varying pH (5 to 8.5). [3H]thiamin (15 nM) was added to the incubation medium at the onset of a 10-min incubation, i.e., initial rate. Values are means ± SE of at least 3 separate uptake determinations. When not shown, error bars are smaller than the symbol.
We also examined the effect of incubation temperature and buffer pH on thiamin (15 nM) uptake by freshly isolated primary mouse pancreatic islets. In line with our observations with cultured pancreatic beta-TC-6 cells, a significantly (P < 0.01) higher thiamin uptake was observed at 37°C compared with 4°C (22.38 ± 1.96 and 8.02 ± 0.54 fmol·mg protein−1·10 min−1, respectively). Thiamin uptake by freshly isolated primary mouse pancreatic islets was also found to be significantly (P < 0.05) higher at buffer pH 8.0 compared with pH 5.0 (29.76 ± 0.49 and 21.23 ± 2.63 fmol·mg protein−1·10 min−1, respectively).
Functional Evidence for Existence of a Carrier-Mediated Process for Thiamin Uptake by Mouse Pancreatic Beta-TC-6 Cell and by Freshly Isolated Primary Mouse and Human Pancreatic Islets
Figure 2 shows the results on the initial rate of thiamin uptake (10 min) as a function of substrate concentration in the incubation medium over a wide concentration range that spans the nanomolar (15–80 nM) and micromolar (0.1–20 μM) ranges. Saturation was observed at both ranges with apparent Km and Vmax (calculated as described in methods) of 37.17 ± 9.90 nM and 216.40 ± 25.50 fmol·mg protein−1·10 min−1, respectively, for the nanomolar component and 3.26 ± 0.86 μM and 16.87 ± 1.48 pmol·mg protein−1·10 min−1, respectively, for the micromolar component. These findings suggest the involvement of two functional transport systems for thiamin uptake in pancreatic beta-TC-6 cells.
Fig. 2.

Uptake of thiamin by pancreatic beta-TC-6 cells as a function of substrate concentration. Confluent cell monolayers were incubated at 37°C in Krebs-Ringer buffer, pH 8.0, in the presence of nanomolar (A; 15–80 nM) and micromolar (B; 0.1–20 μM) concentrations of thiamin. Uptake by the carrier-mediated system was calculated as described in methods. Values are means ± SE of at least 3 separate uptake determinations. When not shown, error bars are smaller than the symbol.
We also examined the effect of unlabeled thiamin (100 μM) on the initial rate of [3H]thiamin (15 nM) uptake by mouse cultured pancreatic beta-TC-6 cells and by freshly isolated primary mouse pancreatic islets. The results showed a significant (P < 0.01 for both) inhibition in [3H]thiamin uptake by unlabeled thiamin in both these mouse pancreatic preparations (for beta-TC-6 cells, 125.18 ± 2.65 and 63.44 ± 1.6 fmol·mg protein−1·10 min−1; for mouse primary pancreatic islets, 28.5 ± 4.3 and 10.1 ± 2.2 fmol·mg protein−1·10 min−1 in the absence and presence of 100 μM unlabeled thiamin, respectively). Similarly, uptake of [3H]thiamin (0.6 μM) by freshly isolated primary human pancreatic islets was significantly (P < 0.01) inhibited by unlabeled thiamin (354.3 ± 59.60 and 90.7 ± 3.60 fmol·mg protein−1·10 min−1 for control and in the presence of 100 μM unlabeled thiamin, respectively).
We also tested the effect of the thiamin structural analogs triphenyltetrazolium chloride (TTC), oxythiamin, and pyrithiamin (all at 100 μM) on the initial rate of [3H]thiamin (15 nM) uptake by pancreatic beta-TC-6 cells. The results showed all compounds to cause a significant (P < 0.01 for all) reduction in thiamin uptake (211.2 ± 1.8, 134.3 ± 5.0, 149.7 ± 4.0, and 175.8 ± 7.0 fmol/mg of protein·10 min−1 for control and in the presence of TTC, oxythiamin, and pyrithiamin, respectively). Similarly, uptake of [3H]thiamin (15 nM) by freshly isolated mouse primary pancreatic islets was significantly (P < 0.05) inhibited by the thiamin structural analog oxythiamin (100 μM) (16.63 ± 1.70 and 8.78 ± 1.47 fmol·mg protein−1·10 min−1 for control and in the presence of oxythiamin, respectively).
Potential trans-stimulation of [3H]thiamin efflux by unlabeled thiamin was also examined in pancreatic beta-TC-6 cells. This was performed by preloading the pancreatic beta-TC-6 cells with [3H]thiamin (i.e., incubation with 45 nM [3H]thiamin for 10 min at 37°C) followed by incubation of the preloaded cells (for 10 min) in Krebs-Ringer buffer in the absence and presence of 1 mM unlabeled thiamin. The results showed a significantly (P < 0.01) lower cellular content of 3H radioactivity in cells incubated in the presence of unlabeled thiamin compared with those incubated in its absence (396.4 ± 63.5 and 916.1 ± 95.6 fmol·mg protein−1·10 min−1, respectively).
Effect of Membrane Transport Inhibitors on Thiamin Uptake by Mouse Pancreatic Beta-TC-6 Cells
We examined the effect of the sulfhydryl-group inhibitor p-chloromercuribenzene sulfonate (p-CMBS; 0.5 mM; pretreatment for 30 min) on the initial rate of thiamin (15 nM) uptake by beta-TC-6 cells; we also tested whether the reducing agent DTT (10 mM; 30 min) could reverse any potential effect of p-CMBS on thiamin uptake. The results showed that pretreatment with p-CMBS leads to a significant (P < 0.01) inhibition in thiamin uptake and that DTT causes a significant (P < 0.01) reversal in that inhibition (186.73 ± 5.3, 64.8 ± 2.8 and 104.19 ± 4.4 fmol·mg protein−1·10 min−1 in control cells, those pretreated with p-CMBS alone, and those pretreated with p-CMBS then with DTT, respectively).
We have also examined the effect of amiloride (1 mM), a compound that has been shown to interfere with thiamin uptake in a variety of cellular systems (2, 32, 39), on the initial rate of thiamin (15 nM) uptake and observed a significant (P < 0.01) inhibition in uptake by pancreatic beta-TC-6 cells (184.9 ± 3.93 and 153.2 ± 3.1 fmol·mg protein−1·10 min−1 for control and in the presence of amiloride, respectively).
Thiamin Transporters Expression in Mouse Pancreatic Beta-TC-6 Cells and in Freshly Isolated Mouse and Human Pancreatic Islets
To further understand the mechanism of thiamin uptake by beta cells, we examined (by means of semiquantitative PCR or real-time quantitative PCR) the expression of the thiamin transporters THTR-1 and THTR-2 in mouse pancreatic beta-TC-6 cells and in freshly isolated mouse and human pancreatic islets (see methods). THTR-1 and THTR-2 are the main thiamin transporters in both mouse and human tissues. The results showed that both transporters are expressed in pancreatic beta-TC-6 cells (Fig. 3A), as well as in mouse (Fig. 3B) and human (Fig. 3C) primary pancreatic islets. Interestingly, the level of expression of THTR-1 was found to be significantly (P < 0.01 for all) higher than that of THTR-2 in all of these pancreatic preparations.
Fig. 3.
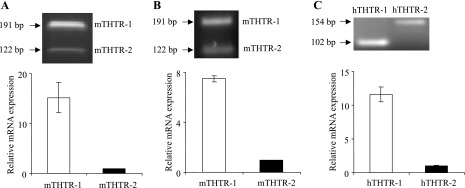
Expression of thiamin transporter-1 and -2 (THTR-1 and THTR-2) mRNA in cultured pancreatic beta-TC-6 cells and freshly isolated primary mouse and human pancreatic islets. Real-time quantitative PCR was performed by using gene-specific primers for THTR-1 and THTR-2 and reverse transcribed total RNA isolated from pancreatic beta-TC-6 cells (A) and primary mouse (B) and human (C) pancreatic islets. Data were normalized relative to β-actin. Insets: representative RT-PCR gels.
Regulation of the Thiamin Uptake Process of Pancreatic Beta-TC-6 Cells
Adaptive regulation by extracellular substrate level.
We then asked the question whether the thiamin uptake process of the pancreatic beta-TC-6 cells is under adaptive regulation by extracellular substrate level. We measured the initial rate of [3H]thiamin uptake by confluent cell monolayers maintained (for 8–10 days) in culture media oversupplemented with thiamin (12 μM) and in control culture medium (15 nM thiamin). The results showed a significant (P < 0.01) downregulation in [3H]thiamin uptake by cells maintained in the thiamin-oversupplemented culture medium compared with those maintained in the control medium (82.8 ± 8.7 and 229.1 ± 16.0 fmol/mg of protein·10 min−1, respectively). These findings suggest that the thiamin uptake process of mouse pancreatic beta-TC-6 cells is under adaptive regulation by the prevailing vitamin level.
To gain insight into the molecular mechanism(s) involved in this adaptive regulation, we examined (by means of Western blotting) the effect of thiamin oversupplementation on level of expression of THTR-1 and THTR-2 at the protein level. The results (Fig. 4A) showed a significant (P < 0.01 for both) reduction in the level of THTR-1 and THTR-2 proteins in the thiamin-oversupplemented cells compared with control cells [for THTR-1 (in arbitrary units) 37.6 ± 4.1 and 12.6 ± 4.2 respectively; for THTR-2 (in arbitrary units) 25.85 ± 4.17 and 15.18 ± 4.4, respectively]. As can be seen the reduction in THTR-1 protein level was markedly more pronounced than that of THTR-2. We also examined (by mean of semiquantitative PCR) the effect of thiamin oversupplementation on level of mRNA expression of THTR-1 and THTR-2. The results (Fig. 4B) showed a significant (P < 0.01 for both) reduction in THTR-1 and THTR-2 mRNA levels in cells maintained in thiamin-oversupplemented culture medium compared with those maintained in control medium [for THTR-1 (in arbitrary units) 31.5 ± 1.6 and 19 ± 2 respectively; for THTR-2 (in arbitrary units) 27.5 ± 1.3 and 23 ± 0.8, respectively]; again the reduction in THTR-1 mRNA level was markedly more pronounced than that of THTR-2. We further examined the effect of thiamin level in the culture medium on activity of the SLC19A2 and SLC19A3 promoters (which drive the expression of hTHTR-1 and hTHTR-2, respectively). The results (Fig. 5) showed significant (P < 0.01 for both) reduction in the activity of the SLC19A2 and SLC19A3 promoters in cells maintained in the presence of thiamin-oversupplemented condition compared with those maintained under control condition. Again the change in the activity of the SLC19A2 promoter was more pronounced than the change in the activity of SLC19A3 promoter (Fig. 5). It is also worth noticing here that activity of the SLC19A2 promoter is significantly (P < 0.01) higher than activity of the SLC19A3 promoter in pancreatic beta-TC-6 cells maintained in control growth medium (Fig. 5).
Fig. 4.

Effect of maintaining pancreatic beta-TC-6 cells in a culture medium with different concentrations of thiamin on level of expression of THTR-1 and THTR-2 at the protein (A) and mRNA (B) levels. A: Western blots were run by using membranous fractions of pancreatic beta-TC-6 cells and specific polyclonal antibodies against THTR-1 and THTR-2. The same amounts of proteins (30 μg) from beta-TC-6 cells grown (for 8–10 days) in control (15 nM) and thiamin-oversupplemented (12 μM) culture medium were used. B: RT-PCR products obtained by using RNA from pancreatic beta-TC-6 cells maintained in the presence of different (15 nM and 12 μM) thiamin and gene-specific primers for the open reading frame of THTR-1, THTR-2, and β-actin were analyzed on a 2% agarose gel. Data of a representative experiment are shown.
Fig. 5.

Effect of maintaining pancreatic beta-TC-6 cells in a culture medium with different levels of thiamin on activity of the SLC19A2 and SLC19A3 promoters. Full-length SLC19A2 and SLC19A3 promoters in pGL3-Basic were transiently expressed in cultured pancreatic beta-TC-6 cells maintained (for 8–10 days) under control (15 nM) thiamin-oversupplemented (12 μM) culture medium. Luciferase assays were performed as described by us previously (20, 26, 27). Data are reported as relative luciferase activity normalized to Renilla luciferase activity and represent means ± SE of at least 3 independent experiments. *P < 0.01.
Regulation by intracellular regulatory pathways.
We then investigated whether the thiamin uptake process of mouse pancreatic beta-TC-6 cells is regulated by specific intracellular regulatory pathways [protein kinase A (PKA)-, Ca2+/calmodulin-, and protein tyrosine kinase (PTK)-mediated pathways] using specific modulators of these pathways. The results showed that modulators of the PKA-mediated pathway (dibutyryl cAMP and forskolin) to have no effect on thiamin uptake by pancreatic beta-TC-6 cells (data not shown). On the other hand, modulators of the Ca2+/calmodulin- and PTK-mediated pathways altered thiamin uptake. Specifically, inhibitors of the Ca2+/calmodulin-mediated pathway (trifluoperazine, calmidazolium, and W13) led to a concentration-dependent inhibition in thiamin (15 nM) uptake (Table 1). Similarly, the PTK inhibitor tyrphostin A25 (but not its negative control tyrophostin A1) caused a concentration-dependent inhibition in thiamin (15 nM) (Table 2). Another PTK inhibitor, genistein, also caused a concentration-dependent inhibition in thiamin uptake whereas its negative control showed minimal effect (Table 2).
Table 1.
Effect of modulators of Ca2+/calmodulin mediated pathway on thiamin uptake by beta-TC-6 cells
| Compound | Uptake, fmol·mg protein−1·10 min−1 | P Value |
|---|---|---|
| Control | 238.9±10.6 | |
| Trifluoperazine | ||
| 50 μM | 119.7±6.7 | < 0.01 |
| 100 μM | 37.6±5.6 | < 0.01 |
| Calmidazolium | ||
| 15 μM | 178.3±3.4 | < 0.01 |
| 50 μM | 51.5±4.5 | < 0.01 |
| 100 μM | 29.9±5.6 | < 0.01 |
| W13 | ||
| 100 μM | 117.9±4.8 | < 0.01 |
Cell monolayers were pretreated for 1 h with varying concentrations of the individual modulator. Uptake was examined after10 min incubation (i.e., initial rate) at 37°C in Krebs-Ringer buffer. Values are means ± SE of at least 3 separate uptake determinations. P value comparison was made relative to simultaneously performed controls.
Table 2.
Effect of modulators of PTK mediated pathway on thiamin uptake by beta-TC-6 cells
| Compound | Uptake, fmol·mg protein−1·10 min−1 | P Value* |
|---|---|---|
| Control | 226.8±7.8 | |
| Tyrphostin A25 | ||
| 20 μM | 187.3±2.9 | P < 0.01 |
| 100 μM | 134.6±2.7 | P < 0.01 |
| Tyrphostin A1 | ||
| 100 μM | 221.9±11.4 | NS |
| Genistein | ||
| 20 μM | 152.8±1.4 | P < 0.01 |
| 50 μM | 117.8±7.5 | P < 0.01 |
| Genistin | ||
| 100 μM | 181.1±8.4 | NS |
Cell monolayers were pretreated for 1 h with varying concentrations of the individual modulator. Uptake was examined after10 min incubation (i.e., initial rate) at 37°C in Krebs-Ringer buffer. Values are means ± SE of at least 3 separate uptake determinations. Notice that the negative controls tyrphostin A1 and genistin were either without or with minimal effect. P value comparison was made relative to simultaneously performed controls.
Cell biology of clinical hTHTR-1 mutants in pancreatic beta-TC-6 cells
As mentioned earlier, the cause of the autosomal recessive disorder TRMA is mutations in hTHTR-1 gene (6, 9, 16), and the pancreatic beta cells are a primary target of TRMA pathology of this disorder (1, 18, 21, 29). Because of this fact and our new finding that THTR-1 appears to be the predominant thiamin transporter in pancreatic beta cells, together with the fact that a given mutant may display different phenotype(s) in different cell types (38), i.e., a mutant could be expressed at the cell surface in one cell type but could be retained intracellularly in another, we analyzed the pattern of cellular expression of different clinical THTR-1 mutants found in patients with TRMA in pancreatic beta-TC-6 cells using live cell confocal imaging. The clinical missense mutants examined were P51L, S143F, T158R, and G172R. The results (Fig. 6A) showed that wild-type hTHTR1-GFP was predominantly expressed at the cell membrane (as shown by the overlap with the membrane dye FM4-64) with some being expressed within intracellular vesicular compartments. In contrast, GFP alone was expressed in the cytoplasm (Fig. 6A). With regard to the clinical mutants, they showed a spectrum of expression phenotypes. Mutants P51L and G172R were expressed at the cell membrane (with some expression occurring within intracellular vesicular compartments); mutant S143F was expressed at the cell surface (as well as some expression within intracellular vesicular compartments) but with a lower level of expression efficiency compared with wild-type hTHTR-1 (Fig. 6A); and mutant T158R was predominantly retained within intracellular membranes although some cells (< 5%) expressed the mutant at the cell surface (Fig. 6A). We also examined the consequence of existence of these clinical mutants on the ability of pancreatic beta-TC-6 cells to take up thiamin compared with wild-type hTHTR-1. Cells were transfected with the different mutants or with wild-type hTHTR-1 and [3H]thiamin uptake was measured. Results showed that cells expressing wild-type hTHTR-1 took up significantly (P < 0.01) more thiamin compared with all clinical mutants (Fig. 6). These data indicate that existence of these mutations impairs the ability of pancreatic cells that express them from taking thiamin via the hTHTR-1 system.
Fig. 6.

Distribution of wild-type hTHTR1-GFP and clinical mutants found in thiamin-responsive megaloblastic anemia (TRMA) patients in live pancreatic beta-TC-6 cells. A: lateral (xy) confocal images showing spectrum of expression phenotypes of wild-type and mutant hTHTR-1 constructs together with plasma membrane dye (FM4-64) in live beta-TC-6 cells, 24–48 h after transient transfection. Data of representative experiments are shown. B: uptake of [3H]thiamin (15 nM) in control cells and those transiently expressing green fluorescent protein (GFP), hTHTR1-GFP, and clinical hTHTR1 mutants in pancreatic beta-TC-6 cells. Values are means ± SE of 3 separate uptake determinations and are expressed as percentage relative to simultaneously performed controls. aP < 0.01; bnot significant.
DISCUSSION
As mentioned earlier, thiamin is essential for the normal function of pancreatic beta cells and deficiency of this vitamin negatively impacts insulin synthesis and secretion (24, 25), as well as disturbing intracellular metabolism and oxidative stress (3, 4, 10). In line with these observations is the development of diabetes mellitus in patients with TRMA (1, 18, 21, 29). This disorder is caused by mutations in THTR-1 that lead to the development of localized thiamin deficiency in the affected tissues, which include the endocrine pancreas. Despite the importance of thiamin for the normal function of the endocrine pancreas, there is very little known about the mechanism(s) involved in its uptake by pancreatic beta cells and islets and its regulation. Little is also known about cell biology of the hTHTR-1 mutants found in patients with TRMA in these cells, which are a major pathological target of this disease. Our objective in these investigations was to address these issues using cultured mouse-derived pancreatic beta-TC-6 cells and complementing the findings with studies using freshly isolated mouse and human primary pancreatic islets. Our results showed uptake of thiamin by pancreatic beta-TC-6 cells to be both temperature and pH (but not Na+) dependent and occurred without metabolic alteration in the transported substrate. Similar findings were observed with primary mouse pancreatic islets, thus providing confirmation with native pancreatic preparations.
The uptake process of thiamin by pancreatic beta-TC-6 cells was carrier mediated in nature. Evidence for this include the saturation in the substrate uptake as a function of concentration, the significant cis-inhibition in [3H]thiamin uptake by unlabeled thiamin and by thiamin structural analogs, and the significant trans-stimulation in the efflux of [3H]thiamin from preloaded cells by unlabeled thiamin. Similar to what has been seen in other tissues (e.g., renal epithelial cells), the saturation in thiamin uptake was observed at both the nanomolar and micromolar concentration ranges (2, 39). This, as suggested before, may indicate the involvement of both THTR-1 and THTR-2 in thiamin uptake by pancreatic cells. These transporters operate at the micromolar and nanomolar concentration ranges, respectively (2, 39). Indeed both THTR-1 and THTR-2 are expressed in pancreatic beta cells at the protein and mRNA levels (see below). The existence of a carrier-mediated process for thiamin uptake was also observed in freshly isolated primary mouse and human pancreatic islets, as indicated by significant inhibition in [3H]thiamin in these preparations. Again, this provides confirmation with native mouse pancreatic tissue as well as an extension to the human situation. At the molecular level, both THTR-1 and THTR-2 were found to be expressed in pancreatic beta-TC-6 cells as well as in primary mouse and human pancreatic islets with expression of the former being markedly higher than that of the latter.
Uptake of thiamin by pancreatic beta-TC-6 cells was also found to be energy dependent, as indicated by the significant inhibition in thiamin uptake by metabolic inhibitors. Furthermore, the uptake was sensitive to the inhibitory effect of the sulfhydryl group inhibitor p-CMBS. Inhibition by p-CMBS was significantly reversed by the reducing reagent DTT, suggesting possible involvement of sulfhydryl groups in the function of pancreatic thiamin transport system(s). Of particular interest is the inhibition by amiloride of thiamin uptake by pancreatic cells, an inhibition that has also observed in a number of other cellular systems that include the intestine, kidney, and retinal pigment epithelia (2, 32, 39). This raises the possibility that long-term use of this diuretic may negatively interfere with cellular thiamin homeostasis. Further studies are needed to test this possibility.
Following delineation of the mechanism of thiamin uptake by pancreatic beta cells and islets, we examined potential regulation of the vitamin uptake process by extracellular and intracellular factors. Our results showed thiamin uptake by pancreatic beta-TC-6 cells to be adaptively regulated by the prevailing thiamin level with higher uptake occurring by cells maintained in the presence of low compared with high thiamin levels. This was associated with a markedly higher level of expression of THTR-1 and THTR-2 at the protein and mRNA, as well as with higher activity of the SLC19A2 and SLC19A3 promoters in the former compared with the latter condition. These findings suggest possible involvement of a transcriptional mechanism(s) in the observed adaptive regulation of pancreatic thiamin uptake process. The response of THTR-1 to the changes in thiamin level, however, was more pronounced than the response of THTR-2 at all levels (i.e., at the protein, mRNA and promoter levels). This demonstrates differential regulation of these two thiamin transporters in pancreatic beta cells. The observation that thiamin level differentially regulates THTR-1 and THTR-2 in pancreatic beta cells could provide an understanding of why these cells end up to be a pathological target for TRMA. This is most likely due to the fact that THTR-1 is the predominant thiamin transporter in these cells and that mutations in this transporter, as occur in TRMA, lead to impairment in the ability of these cells to acquire sufficient amount of thiamin (see below). With the limited capability of the cells to upregulate THTR-2 (compared to their ability to upregulate THTR-1) these combined effects will lead to the development of a state of thiamin deficiency. This will result in disturbance in intracellular metabolism, oxidative stress, and apoptosis. The observation that thiamin availability regulates THTR-1 and THTR-2 in pancreatic beta-TC-6 cells is in contrast to what occurs in intestinal epithelial cells. In the latter cell type, changes in thiamin levels regulate only the level of expression of THTR-2 (27). This indicates that different cells use different systems to regulate their thiamin uptake in response to changes in substrate availability.
Possible regulation of thiamin uptake by pancreatic beta cells by intracellular regulatory pathways was also examined in this investigation with special focus on pathways that have been shown to regulate uptake of other nutrients and substrates in other cellular systems (i.e., PKA-, Ca2+/calmodulin-, and PTK-mediated pathways) (5, 15, 31). The results showed that although modulators of the PKA-mediated pathway failed to affect thiamin uptake, modulators (inhibitors) of the Ca2+/calmodulin-mediated pathway caused significant inhibition in the vitamin uptake process. This suggests possible involvement of Ca2+/calmodulin-mediated pathway in regulating thiamin uptake by pancreatic beta cells. It is interesting to mention here that the thiamin uptake process in other cellular systems (e.g., intestinal, renal, and retinal pigment epithelial cells) was also found to be regulated by this intracellular regulatory pathway (2, 32, 39), suggesting possible genes acting in the use of this pathway to regulate thiamin uptake. Our studies also showed possible involvement of a PTK-mediated pathway in the regulation of pancreatic thiamin uptake process. This suggestion is based on the observations that inhibitors of this pathway (but not their negative controls) caused a significant inhibition in thiamin uptake by these cells. Further studies are needed to delineate the molecular mechanisms involved in the regulation of pancreatic thiamin uptake by the above-described intracellular regulatory pathways.
As mentioned earlier, a given mutant of transporter could display different cell biology phenotype in different cells (38). Since pancreatic beta cells are a major pathological target of TRMA, we investigated in the present study the pattern of expression of selective missense clinical (TRMA) hTHTR-1 mutants, including the newly identified but not characterized mutant G172R in pancreatic beta-TC-6 cells (1). We also tested the functionality of these mutants in these cells. Our results showed a spectrum of expression phenotypes with certain mutants being expressed at cell membranes (P51L, G172R), another being retained intracellularly (T158R), and yet another (S143F) being expressed at the cell surface but with low efficiency compared with wild-type transporter. Compared with cells expressing the wild-type hTHTR-1, which showed significant induction in thiamin transport activity, cells expressing the mutants failed to do so.
In summary, this study represents the first characterization of the mechanism and regulation of thiamin uptake by mouse and human pancreatic beta cells and islets. Our studies show the involvement of pH-dependent, carrier-mediated process that appears to involve THTR-1 and THTR-2 and is adaptively regulated by the prevailing substrate level via transcriptionally mediated mechanism(s) that affect the former to a greater extend than the latter transporter. In addition, the process appears to be under the regulation of an intracellular Ca2+/calmodulin- and PTK-mediated pathways. Furthermore, this study shows that clinical THTR-1 mutants display different cell biology phenotypes in pancreatic beta cells, all leading to impairment in thiamin uptake.
GRANTS
This study was supported by grants from the Department of Veterans Affairs and the National Institutes of Health (DK56061 and AA018071).
REFERENCES
- 1.Alzahrani AS, Baitei E, Zou M, Shi Y. Thiamin transporter mutation: an example of monogenic diabetes mellitus. Eur J Endcrinol 155: 787–792, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Ashokkumar B, Vaziri N, Said HM. Thiamin uptake by the human-derived renal epithelial (HEK-293) cells: cellular and molecular mechanisms. Am J Physiol Renal Physiol 291: F796–F805, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Berdanier CD Advanced Nutrition: Micronutrients. Boca Raton, FL: CRC, 1998.
- 4.Calingasan NY, Gandy SE, Baker H, Sheu KF, Smith JD, Lamb BT, Gearhart JD, Buxbaum JD, Harper C, Selkoe DJ, Price DL, Sisodia SS, Gibson GE. Noval neuritic clusters with accumulations of amyloid precursor protein and amyloid precursor-like protein 2 immunoreactivity in brain regions damaged by thiamine deficiency. Am J Pathol 149: 1063–1071, 1996. [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen ME, Reinlib L, Watson AJ, Gorelick F, Rys-Sikora K, Tse M, Rood RP, Czernik AJ, Sharp GW, Donowitz M. Rabbit ileal villus cell brush border Na+/H+ exchange is regulated by Ca2+/calmodulin-dependent protein kinase II, a brush border membrane protein. Proc Natl Acad Sci USA 87: 8990–8994, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz GA, Banikazemi M, Oishi K, Desnick RJ, Gelb BD. Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat Genet 22: 309–312, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Dudeja PK, Tyagi S, Kavilaveettil RJ, Gill R, Said HM. Mechanism of thiamine uptake by human jejunal brush-border membrane vesicles. Am J Physiol Cell Physiol 281: C786–C792, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Fennelly J, Frank O, Baker H, Leevy CM. Peripheral neuropathy of the alcoholic: I. Aetiological role of aneurin and other B-complex vitamins. Br Med J 2: 1290–1292, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet 22: 305–308, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Frederikse PH, Farnsworth P, Zigler JS Jr. Thiamine deficiency in vivo produces fiber cell degeneration in mouse lenses. Biochem Biophys Res Commun 258: 703–707, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Gotah M, Maki T, Kiyoizumi T, Satomi S, Monaco AP. An improved method for isolation of mouse pancreatic islets. Transplantation 40: 437–438, 1985. [DOI] [PubMed] [Google Scholar]
- 12.Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M. Benfotiamine blocks three major pathways of hyperglycemia damage and prevents experimental diabetic retinopathy. Nat Med 9: 294–299, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Helman B Studies on obese-hyperglycemic mice. Ann NY Acad Sci 131: 541–558, 1965. [DOI] [PubMed] [Google Scholar]
- 14.Hopfer U, Nelson K, Perotto J, Isselbacher KJ. Glucose transport in isolated brush border membrane from rat small intestine. J Biol Chem 248: 25–32, 1973. [PubMed] [Google Scholar]
- 15.Kumar CK, Moyer MP, Dudeja PK, Said HM. A protein-tyrosine kinase-regulated, pH-dependent, carrier-mediated uptake system for folate in human normal colonic epithelial cell line NCM460. J Biol Chem 272: 6226–6231, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, Szargel R, McDonald L, Shalata A, Nosaka K, Gregory S, Cohen N. Mutations in SLC19A2 cause thiamine-response megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet 22: 300–304, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Leevy CM, Baker H. Vitamins and alcoholism. Am J Clin Nutr 21: 1325–1328, 1968. [DOI] [PubMed] [Google Scholar]
- 18.Mandel H, Berant M, Hazani A, Naveh Y. Thiamine-dependent beriberi in the thiamine-responsive anemia syndrome. N Engl J Med 311:836–838, 1984. [DOI] [PubMed] [Google Scholar]
- 19.Matsushima K, MacManus JP, Hakim AM. Apoptosis is restricted to the thalamus in thiamine-deficient rats. Neuroreport 8: 867–870, 1997. [PubMed] [Google Scholar]
- 20.Nabokina SM, Said HM. Characterization of the 5′-regulatory region of the human thiamin transporter SLC19A3: in vitro and in vivo studies. Am J Physiol Gastrointest Liver Physiol 287: G822–G829, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Neufeld EJ, Fleming JC, Tartaglini E, Steinkamp MP. Thiamin-responsive megaloblastic anemia syndrome: a disorder of high-affinity thiamin transport. Blood Cells Mol Dis 27: 135–138, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Oishi K, Kamakura S, Isazawa Y, Yoshimatsu T, Kuida K, Nakafuku M, Masuyama N, Gotoh Y. Male infertility due to germ cell apoptosis in mice lacking the thiamin carrier, Tht1. A new insight into the critical role of thiamin in spermatogenesis. Dev Biol 266: 299–309, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Prasannan KG, Sundaresan R, Venkatesan K. Thiamine deficiency and protein secretion by pancreatic slices in vitro. Experientia 33: 169–170, 1977. [DOI] [PubMed] [Google Scholar]
- 24.Rathanaswami P, Pourany A, Sundaresan R. Effects of thiamine deficiency on the secretion of insulin and the metabolism of glucose in isolated rat pancreatic islets. Biochem Int 25: 577–583, 1991. [PubMed] [Google Scholar]
- 25.Rathanaswami P, Sundaresan R. Effects of thiamine deficiency on the biosynthesis of insulin in rats. Biochem Int 24: 1057–1062, 1991. [PubMed] [Google Scholar]
- 26.Reidling JC, Said HM. In vitro and in vivo characterization of the minimal promoter region of the human thiamin transporter SLC19A2. Am J Physiol Cell Physiol 285: C633–C641, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Reidling JC, Said HM. Adaptive regulation of intestinal thiamin uptake process: molecular mechanisms in wild-type and transgenic mice expressing the promoters of the human thiamin transporters hTHTR-1 and hTHTR-2. Am J Physiol Gastrointest Liver Physiol 288: G1127–G1134, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Reidling JC, Subramanian VS, Dudeja PK, Said HM. Expression and promoter analysis of SLC19A2 in the human intestine. Biochim Biophys Acta 1561: 180–187, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Rogers LE, Porter FS, Sidbury JB Jr. Thiamin-responsive megaloblastic anemia. J Pediatr 74: 494–504, 1969. [DOI] [PubMed] [Google Scholar]
- 30.Said HM, Ghishan FK, Redha R. Folate transport by human intestinal brush-border membrane vesicles. Am J Physiol Gastrointest Liver Physiol 252: G229–G236, 1987. [DOI] [PubMed] [Google Scholar]
- 31.Said HM, Ma TY, Grant K. Regulation of riboflavin intestinal uptake by protein kinase A: studies with Caco-2 cells. Am J Physiol Gastrointest Liver Physiol 267: G955–G959, 1994. [DOI] [PubMed] [Google Scholar]
- 32.Said HM, Ortiz A, Kumar CK, Chatterjee N, Dudeja PK, Rubin S. Transport of thiamine in human intestine: mechanism and regulation in intestinal epithelial cell model Caco-2. Am J Physiol Cell Physiol 277: C645–C651, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Said HM Recent advances in carrier-mediated absorption of water-soluble vitamins. Annu Review Physiol 66: 419–446, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Saito N, Kimura M, Kuchiba A, Itokawa Y. Blood thiamine levels in outpatients with diabetes mellitus. J Nutr Sci Vitaminol (Tokyo) 33: 421–430, 1987. [DOI] [PubMed] [Google Scholar]
- 35.Singh M Effect of thiamin deficiency on pancreatic acinar cell function. Am J Clin Nutr 36: 500–504, 1982. [DOI] [PubMed] [Google Scholar]
- 36.Stagg AR, Fleming JC, Baker MA, Sakamoto M, Cohen N, Neufeld EJ. Defective high-affinity thiamine transporters leads to cell death in thiamine-responsive megaloblastic anemia syndrome fibroblasts. J Clin Invest 103: 723–729, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian VS, Marchant JS, Parker I, Said HM. Cell biology of the human thiamine transporter-1 (hTHTR1): intracellular trafficking and membrane targeting. J Biol Chem 278: 3976–3984, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Subramanian VS, Marchant JS, Said HM. Targeting and intracellular trafficking of clinically relevant hTHTR1 mutations in human cell lines. Clin Sci (Lond) 113: 93–102, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian VS, Mohammed ZM, Molina A, Marchant JS, Vaziri ND, Said HM. Vitamin B1 (thiamine) uptake by human retinal pigment epithelial (ARPE-19) cells: mechanism and regulation. J Physiol 582: 73–85, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tallaksen CM, Bell H, Bohmer T. Thiamin and thiamin phosphate ester deficiency assessed by high performance liquid chromatography in four clinical cases of Wernicke encephalopathy. Alcohol Clin Exp Res 17: 712–716, 1993. [DOI] [PubMed] [Google Scholar]
- 41.Tallaksen CME, Bohmer T, Bell H. Blood and serum thiamin and thiamin phosphate esters concentration in patients with alcohol dependence syndrome before and after thiamin treatment. Alcohol Clin Exp Res 16: 320–325, 1992. [DOI] [PubMed] [Google Scholar]
- 42.Tanphaichitr V Thiamin. In: Modern Nutrition in Health and Disease, edited by Shils ME, Olsen JA, Shike M. New York: Lea and Febiger, 1994, 359–375.
- 43.Thornalley PJ, Babaei-Jadidi R, Al Ali H, Rabbani N, Antonysunil A, Larkin J, Ahmed A, Rayman G, Bodmer CW. High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia 125: 771–774, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valerio G, Franzese A, Poggi V, Patrini C, Laforenza U, Tenore A. Lipophilic thiamine treatment in long-standing insulin-dependent diabetes mellitus. Acta Diabetol 36: 73–76. 1999. [DOI] [PubMed] [Google Scholar]
- 45.Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome and Related Neurological Disorders Due to Alcoholism and Malnutrition. Philadelphia, PA: Davis, 1989.
- 46.Wilkinson GN Statistical estimation in enzyme kinetics. Biochem J 80: 324–332, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]