

Abstract
Fibroblast growth factor receptor 3 (FGFR-3) is expressed in the lower crypt epithelium, where stem cells of the intestine reside. The role of FGFR-3 signaling in regulating features of intestinal morphogenesis was examined in FGFR-3-null (FGFR-3−/−) mice. FGFR-3−/− mice had only about half the number of intestinal crypts and a marked decrease in the number of functional clonogenic stem cells, as assessed by an in vivo microcolony-forming assay, compared with wild-type littermates. A marked deficit in allocation of progenitor cells to Paneth cell differentiation was noted, although all the principal epithelial lineages were represented in FGFR-3−/− mice. The total cellular content and nuclear localization of β-catenin protein were reduced in FGFR-3−/− mice, as was expression of cyclin D1 and matrix metalloproteinase-7, major downstream targets of β-catenin/T cell factor-4 (Tcf-4) signaling. Activation of FGFR-3 in Caco-2 cells, an intestinal epithelial cell line, abrogated the fall in β-catenin/Tcf-4 signaling activity that is normally observed in these cells as cultures become progressively more confluent. These findings are consistent with the hypothesis that, during intestinal development, FGFR-3 signaling regulates crypt epithelial stem cell expansion and crypt morphogenesis, as well as Paneth cell lineage specification, through β-catenin/Tcf-4-dependent and -independent pathways.
Keywords: fibroblast growth factors, β-catenin, T cell factor-4, intestinal morphogenesis
development of the normal intestinal mucosa requires specific inductive signals for guiding formation of normal architectural relationships and regional patterns of epithelial differentiation. The fetal murine intestinal epithelium undergoes a morphological reorganization, changing from a poorly differentiated pseudostratified cuboidal epithelium to a simple stratified columnar epithelium, beginning at embryonic day 14.5 (13, 29, 46). Nascent villi then form by upward growth of mesenchymal tissue underlying the monolayer of columnar epithelial cells. At this developmental stage, the proliferative unit of the mature intestinal epithelium, the crypt, has not yet formed. Rather, replicating cells are found in the fetal homolog of the crypt, the intervillus epithelium, also populated by descendants of multiple pluripotent stem cells (16, 42). Formation of small intestinal crypts first begins during early postnatal life by the morphological invagination of the intervillus epithelium into the underlying mesenchyme. Newly formed crypts subsequently increase in number through expansion of the stem cell population and subsequent crypt fission, a process that is largely completed by 4–5 wk after birth.
In the adult mouse small intestinal epithelium, cell replication and differentiation are tightly coupled to cell position along the crypt-to-villus axis. All four principal differentiated cell types, enterocytes, enteroendocrine cells, goblet cells, and Paneth cells, are derived from one or more multipotent stem cells located in each intestinal crypt (8, 12, 41, 51). Transit-replicating cells are rapidly proliferating (cell cycle time is ∼12–13 h in the mouse) descendants of this stem cell and undergo four to six divisions within a zone of proliferation located in the midportion of each crypt. Differentiation occurs as progeny of the transit cell population migrate in vertically coherent bands onto the villus epithelium or toward the base of the crypt (8, 37, 38).
Fibroblast growth factors (FGFs), a family of ≥22 proteins, have been shown to provide inductive signals that control diverse events during normal embryonic tissue development, such as cell fate and differentiation, cell migration, and spatial pattern formation (25, 33, 44). We recently found that one of the receptors for this important family of growth factors, FGFR-3, is highly expressed by undifferentiated crypt epithelial cells in the developing intestine. Additionally, potential ligands for this receptor are highly expressed during crypt morphogenesis in the suckling mouse intestine (50). These findings led us to ask whether FGFR-3-mediated signaling plays a critical role in regulating morphogenic events and/or functional differentiation of crypt epithelial cells during intestinal development (50).
Signaling through β-catenin and the T cell factor (Tcf)/lymphocyte enhancer binding factor (Lef) family of transcription factors is important in regulating epithelial stem cell fate and replication during intestinal development and in intestinal neoplasia (2, 5, 7, 26–28). This is illustrated in mice bearing a mutation in Tcf-4 that shows depletion of the stem cells in the intervillus epithelium and failure to form crypts (26). Furthermore, β-catenin/Tcf activation regulates the expression of a number of genes important in cell cycle regulation, cell fate determination, and Paneth cell differentiation (35, 36, 47). Recent studies also demonstrate that β-catenin/Tcf-4 signaling may control cell positioning along the crypt-to-villus axis in the intestine, including Paneth cell positioning, by regulating the expression of ephrins and their receptors on intestinal epithelial cells (4). Although much attention has focused on Wnt(s) as the canonical upstream mediator of β-catenin/Tcf-4 signaling, recent studies suggest that other growth factors, such as FGF2, can activate this transcription complex through cross talk with the β-catenin signaling pathway (20).
We examined the consequence of FGFR-3 gene disruption on morphogenic events during normal intestinal development to define the functional role of FGFR-3 signaling and its relationship to the β-catenin/Tcf signaling pathway. We report that crypt formation begins at the normal developmental time during postnatal intestinal development. However, 1) normal numbers of intestinal crypts fail to develop in FGFR-3-null (FGFR-3−/−) mice; 2) the number of crypt epithelial stem cells is reduced in FGFR-3−/− mice; 3) Paneth cell specification and/or differentiation is reduced in FGFR-3−/− mice; 4) the cellular distribution and/or stability of β-catenin is altered in FGFR-3−/− mice; and 5) signaling through FGFR-3 sustains high levels of β-catenin/Tcf-4-mediated transcriptional activity in intestinal epithelial cells.
MATERIALS AND METHODS
Animals.
Mice with a targeted disruption of the FGFR-3 gene were kindly provided by Dr. David Ornitz (14). Deletion of the 3′ end of the exon encoding the Ig-like domain II, along with the entire Ig-like domain III and the transmembrane domain, generated these FGFR-3−/− mice (14). These mice are available through The Jackson Laboratory (strain designation B6;129S-Fgfr3tm1Dor/J). Since only a few FGFR-3−/− mice survive to breeding age (14), our colony was maintained using heterozygous (FGFR-3+/−) breeding pairs. Only nonsibling mice were used to establish breeder pairs to avoid inbreeding of potentially linked modifier alleles. Paired FGFR-3−/− wild-type littermates were used as controls at all times. Mice were maintained on a 12:12-h light-dark cycle and fed standard laboratory chow ad libitum. In our colony, we observed that most FGFR-3−/− mice died at ∼28 days after birth. A set of physical and behavioral criteria approved by the University of Virginia Animal Care and Use Committee was used to closely monitor the health status of FGFR-3−/− mice, starting just after birth. FGFR-3−/− mice used in these studies appeared healthy and had normal stool consistency at all ages.
Intestinal tissue was dissected as described elsewhere (43), and segments from each region of the gut were fixed in Bouin's fixative and processed for paraffin embedding and immunohistochemical analysis. For isolation of total RNA and protein, some intestinal segments were snap frozen in liquid nitrogen and stored until use. Total cellular RNA was prepared using the RNeasy Midi Kit (Qiagen, Valencia, CA). For protein isolation, frozen tissue segments (∼0.05–0.18 g of tissue) were homogenized in 1 ml of lysis buffer containing 50 mM Tris·HCl (pH 7.4), 0.5% Triton X-100, and 1× complete protease inhibitor cocktail (Roche, Nutley, NJ). Samples were centrifuged at 12,000 g for 8 min at 4°C. The supernatant Triton-soluble fraction was collected, and protein concentration was determined using the Coomassie Plus Protein Assay Kit (Pierce, Rockford, IL).
Quantitation of crypt numbers within the intestine.
Crypt numbers were determined separately in the proximal, middle, and distal thirds of the small intestine to account for potential regional differences in crypt number. For each segment of the small intestine, crypts were counted in well-oriented transverse cross sections obtained at 5-mm intervals and in a longitudinal section obtained from the middle of each region. The total number of intestinal crypts was obtained by the following equations
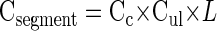 |
and
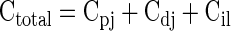 |
where Csegment is the total number of crypts in any one intestinal tissue segment, Cc is the number of crypts per cross-sectional circumference determined for at least six sites per region, Cul is the number of crypts per unit length determined by counting crypts in longitudinal sections, and L is the measured length of the whole fixed segment from which the rest of the crypt counts were determined. The total number of crypts (Ctotal) in the intestine was then determined by summing the total crypts obtained for each segment (Cpj + Cdj + Cil).
Crypt stem cell survival.
At 3.5 days after irradiation, the number of regenerating crypts was measured using a modification of the microcolony assay, as previously described (10, 40, 52). Using a Gammacel 40 cesium irradiator, we assessed crypt survival by subjecting FGFR3−/− and wild-type mice to γ-irradiation, and intestinal segments were harvested 3.5 days after treatment. Cells were labeled in the S phase by administration of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU, 120 mg/kg) and 12 mg/kg 5-fluoro-2′-deoxyuridine intraperitoneally to the irradiated mice 2 h before euthanasia (9, 15, 18). Cells incorporating BrdU were visualized by immunohistochemistry with goat anti-BrdU, as described elsewhere (9). A surviving crypt was defined as one containing five or more BrdU-positive cells, as previously described (10). At least six complete, well-oriented cross sections were scored for each mouse. Fractional crypt survival was defined as the ratio of the mean number of surviving crypts in intestinal cross sections from irradiated mice to the number of crypts per cross section from the same region of the intestine in unirradiated mice of the same strain and age.
Real-time RT-PCR.
Expression of cyclin D1, β-catenin, Muc2, chromogranin, lysozyme, matrix metalloproteinase (MMP)-7, cryptdin 5, Indian hedgehog (Ihh), peroxisome proliferator-activated receptor-β (PPARβ), and Sox9 mRNA in mouse small intestinal segments was quantified by real-time PCR analysis using a sequence detection system (PRISM SDS7000, Applied Biosystems, Foster City, CA). For reverse transcription, random hexamers (1 μg) and 10 ng of total RNA were used in a final reaction volume of 20 μl containing 200 U of Superscript (Invitrogen Life Technologies, Carlsbad, CA). β-Catenin primers were 5′-AGCCGAGATGGCCCAGAAT-3′ and 5′-AAGGGCAAGGTTCGAATCAA-3′. Cyclin D1 sequences were 5′-ATCCGCAAGCATGCACAGA-3′ and 5′-GGGTTGGA AATG AACTTCACATCT-3′. PCR was performed in triplicate for 40 cycles using 10% of the volume of the first-strand synthesis in a total volume of 50 μl, which included 25 μl of SYBR Green Master Mix (Applied Biosystems) and primers at a final concentration of 250 nM. Muc2, chromogranin, lysozyme, MMP-7, cryptdin 5, Ihh, and PPARβ mRNA levels were determined using the Celera Assay on Demand kit (Applied Biosystem). The cycle threshold (ΔΔCT) method was used to quantify relative mRNA levels (User Bulletin 2, Applied Biosystems), with 18S RNA as the reference and internal standard. The TaqMan primer-probe set for 18S RNA with the VIC/TAMRA detection system was used to simultaneously measure 18S RNA in replicates of samples taken for the cyclin D1, β-catenin, Muc2, chromogranin, lysozyme, MMP-7, cryptdin 5, Ihh, PPARβ, and Sox9 quantifications.
Histology and immunohistochemical methods.
Expression of intestinal fatty acid-binding protein (IFABP), serotonin, peptide YY (PYY), and β-catenin was examined in Bouin-fixed or 10% neutral buffered formalin-fixed, deparaffinized sections of proximal jejunum and ileum from FGFR-3−/− and wild-type mice. Primary antibodies were rabbit anti-IFABP (12) and rabbit anti-PYY (1:250 and 1:800 dilution, respectively; Peninsula Laboratories, Belmont, CA), rabbit anti-serotonin (1:2,000 dilution; Diasorin, Stillwater, MN), mouse anti-β-catenin (1:50 dilution; Transduction Laboratories, San Jose, CA), and rabbit anti-lysozyme (1:2,000 dilution; Dako, Carpinteria, CA). Expression of FGFR-3 in Bouin-fixed and deparaffinized intestinal sections was examined using a polyclonal anti-FGFR-3 (1:1,000 dilution; C-15, catalog no. sc-123, Santa Cruz Biotechnology, Santa Cruz, CA). Bound anti-IFABP and anti-FGFR-3 were visualized with a Cy3-conjugated donkey anti-rabbit IgG (1:400 and 1:500 dilution, respectively; Jackson ImmunoResearch Laboratories, West Grove, PA). The anti-β-catenin reaction was performed on sections subjected to antigen retrieval, as described elsewhere (47), and visualized using the Envision+ kit (Dako). Bound anti-PYY and anti-serotonin were visualized with fluorescein-conjugated tyramide signal amplification (DuPont NEN Life Sciences Products, Boston, MA) after incubation with biotin-labeled donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories) followed by reaction with streptavidin-horseradish peroxidase. The anti-lysozyme reaction was visualized by diaminobenzidine precipitation after incubation with biotin SP-labeled donkey anti-rabbit IgG (1:1,000 dilution; Jackson ImmunoResearch Laboratories). Antibody specificities were determined by replacement of the primary antibodies with an irrelevant rabbit or mouse IgG (data not shown).
Electron microscopy.
Electron microscopy was performed through the University of Virginia Advanced Microscopy Core. Briefly, segments of proximal jejunum and ileum were rapidly excised and placed in a small amount of chilled fixative composed of 4% (wt/vol) paraformaldehyde and 2.5% (wt/vol) glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), fixed by immersion at 4°C overnight, postfixed for 1 h in 1% (wt/vol) osmium tetroxide, and embedded in epoxy resin (EPON, Electron Microscopy Sciences). Ultrathin (70- to 80-nm-thick) sections, prepared on a Leica Ultracut ultramicrotome using a Diatome diamond knife, were contrast stained with lead citrate and uranyl acetate. The sections were examined with a transmission electron microscope (JEOL 1230, Japan Electron Optics, Tokyo, Japan) operating at 80 kV and equipped with a digital camera (model SIA 12-C, Scientific Instruments and Applications, Duluth, GA).
Transient transfection protocol.
Caco-2 cells between passages 6 and 16 (American Type Culture Collection) were grown to 80–85% confluence in MEM with Earle's balanced salt solution containing 0.1 mM nonessential amino acids and 1.5 g/l sodium bicarbonate and supplemented with 10% bovine growth serum (Hyclone, Logan, UT) and 15 mM HEPES (pH 7.4). Cells transferred to Opti-MEM I (GIBCO, Carlsbad, CA) were transfected using siLentFect reagent (Bio-Rad, Hercules, CA). Cells were transfected with 3 μg of the Tcf reporter plasmid TOPFlash and 3 μg of one of the following plasmids: 1) Renilla, an irrelevant DNA plasmid, 2) pUC18, an empty vector, 3) a constitutively active mutant of FGFR-3, K650E, that lacks the extracellular ligand binding domain (kindly provided by D. J. Donoghue), or 4) a wild-type FGFR-3 construct. Some cultures that were transfected with TOPFlash or Renilla received daily additions of FGF9 (50 ng/ml final concentration; R & D Systems, Minneapolis, MN) starting at 24 h after transfection and ending at 120 h after transfection (3 days after confluence). Luciferase activity was assayed with the Dual Glo Luciferase Assay System (Promega, Madison, WI) at 48, 72, 96, and 120 h after transfection, at which times cultures were confluent and at 1, 2, and 3 days after confluence, respectively. Samples were read using the Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA).
Western blot analysis of cellular β-catenin.
The Triton X-100-soluble cellular protein fraction (40 μg) was resolved by SDS-PAGE and transferred to polyvinylidene difluoride microporous membranes (Immobilon-P Transfer membrane, Millipore, Billerica, MA) for 1 h at 100 V using a Bio-Rad minigel transfer apparatus. To prevent nonspecific antibody binding to the membranes, we treated the blots for 1 h at room temperature with 1× PBS-0.1% Tween containing 5% dry milk solids. β-Catenin was detected with rabbit polyclonal anti-β-catenin (1:1,000 dilution; Santa Cruz Biotechnology) and visualized by horseradish peroxidase-conjugated donkey anti-mouse (1:5,000 dilution; ECL kit, Amersham, Pittsburgh, PA).
Statistics.
Data were analyzed by pairwise t-tests using the pooled estimate of variance and Bonferroni's correction of the P values for multiple comparisons. Differences were considered significant at P ≤ 0.05.
RESULTS
Phenotypic effects of FGFR-3 signaling.
We examined the role of FGFR-3 in regulating morphogenesis and/or functional differentiation of the intestinal epithelium. In wild-type mice, FGFR-3 is highly expressed on the surface of a subset of epithelial cells in the lower half of crypts present in the suckling mouse intestine (50). In contrast, immunoreactive FGFR-3 was not seen in epithelial cells of mice homozygous for a targeted disruption of the FGFR-3 gene, i.e., FGFR-3−/− mice (Fig. 1, A and B). As previously reported, mice homozygous for a targeted disruption of the FGFR-3 gene (FGFR-3−/−) were found to develop marked bone abnormalities of varying severity that included kinked tails, kyphosis, and curvature and overgrowth of long bones (14). FGFR-3−/− mice were similar in body weight to their wild-type littermates at birth and for the first 4 days of life. However, beginning at ∼7 days after birth, they failed to show normal weight gain during the suckling period compared with their wild-type littermates (Fig. 1C). Thus, by 21 days of age, FGFR-3−/− mice were ∼50% the size of their wild-type littermates. When FGFR-3−/− mice were allowed to age, most died at ∼28 days after birth from as yet unknown causes. Those individual FGFR-3−/− mice that survived past 28 days and into adulthood showed a spurt in growth, so that their body weight gain approached that of control mice. The intestinal length of FGFR-3−/− mice was consistently shorter than that of their wild-type littermates (Fig. 1D, Table 1). By 21 days after birth, the length of the small intestine in FGFR-3−/− mice was reduced to ∼60% of that in their wild-type littermates.
Fig. 1.

Phenotypic characteristics of fibroblast growth factor (FGF) receptor-3 (FGFR-3)-null (FGFR-3−/−) and wild-type (wt/wt) mice. Immunohistochemical detection of FGFR-3 expression in 14-day-old small intestine of wild-type (A) and FGFR-3−/− (B) mice shows that FGFR-3−/− mice do not express FGFR-3. C: body weight of wild-type and FGFR-3−/− (FR3−/−) mice at 0–30 days of age. D: photograph of intestines from 21-day-old FGFR-3−/− and wild-type (FR3+/+) mice showing difference in total gut length.
Table 1.
Body weight, small intestine length, and number of crypts in 21-day-old FGFR-3−/− mice and their wild-type littermates
| Genotype | Body Wt, g | Small Intestine Length, cm | Crypts per Small Intestine, ×10−5 |
|---|---|---|---|
| wt/wt | 11.2±1.4 | 27.6±1.3 | 4.08±0.08 |
| FGFR-3−/− | 5.2±2.0* | 17.3±1.2† | 1.98±0.03† |
Values are means ± SE. FGFR-3−/−, fibroblast growth factor receptor-null mice; wt/wt, wild-type littermates. Significantly different from wt/wt:
P < 0.001;
P < 0.005.
Number of intestinal crypts is regulated by FGFR-3 signaling.
We next examined whether FGFR-3−/− mice form normal numbers of intestinal crypts, since the number of crypts increases markedly in the suckling mouse intestine and FGFR-3 is highly expressed in the developing intestine during this time period (50). To determine whether FGFR-3 signaling regulates crypt formation, we quantified the number of small intestinal crypts in FGFR-3−/− and wild-type mice at various developmental time points. Crypt formation in the small intestine was first observed 4 days after birth in FGFR-3−/− and wild-type mice. The number of crypts per cross section was significantly reduced in the jejunum of FGFR-3−/− mice as early as 7 days of age (51.5 ± 4.5 vs. 78.0 ± 6.1 in wild-type mice). By 10 days after birth, the number of crypts per cross section in the jejunum was reduced 34% in the jejunum and 37% in the ileum of FGFR-3−/− mice (Fig. 2A). The deficit in crypt number persisted through 21 days of age in the jejunum and ileum of FGFR3−/− mice (Fig. 2A). However, these differences are an underestimate of the reduction in the total numbers of small intestinal crypts, since the small intestine is substantially shorter in FGFR-3−/− than wild-type mice (Fig. 1D, Table 1). When the difference in intestinal length is taken into account, the total number of crypts in each region of the small intestine of 21-day-old FGFR-3−/− mice was markedly reduced (Fig. 2B). The total number of crypts in the entire small intestine was only 48% of that in wild-type littermates (Table 1). A reduction in the number crypts per cross section was still observed in the small number of FGFR-3−/− mice that survived well into adulthood (114.7 ± 3.1 vs. 135.5 ± 3.1 in wild-type littermate jejunum and 101.7 ± 5.3 vs. 112.2 ± 3.1 in wild-type littermate ileum), although the magnitude of this reduction was less pronounced than at 10–21 days after birth (Fig. 2). In wild-type and FGFR-3−/− mice, crypt fission was initiated at the base of the crypt and shared a common crypt lumen (Fig. 2, C vs. D).
Fig. 2.

Crypt formation in FGFR-3−/− and wild-type mice. A: number of crypts per cross section in hematoxylin-eosin-stained sections from proximal jejunum and ileum of 10- and 21-day-old mice. Six separate cross sections were scored for each of 5–6 mice per group, and results were averaged. B: total number of crypts in each region of small intestine in 21-day-old FGFR-3−/− and wild-type mice. Values are means ± SE (n = 4 mice per group). *P < 0.05. **P < 0.01. †P < 0.005. Photographs of hematoxylin-eosin-stained sections of proximal jejunum from wild-type (C) and FGFR-3−/− (D) mice show crypts undergoing fission.
Accrual of crypt epithelial stem cells is regulated by FGFR-3 signaling.
Since FGFR-3−/− mice form fewer crypts than wild-type controls, we examined whether the number of clonogenic crypt stem cells was also reduced. Since highly specific markers for crypt epithelial stem cells have yet to be identified, we used an in vivo functional assay to quantify clonogenic stem cell numbers in FGFR-3−/− and wild-type mice. This assay is based on the capacity of stem cells that survive radiation-induced injury to regenerate crypt-like foci or “microcolonies” of replicating epithelial cells that are scored histologically 3–4 days after irradiation. The number of surviving crypts per intestinal cross section is a function of the number of clonogenic stem cells in each crypt before irradiation and the sensitivity of individual stem cells to radiation-induced cell death. Crypt stem cell survival was examined in 21-day-old FGFR-3−/− and wild-type mice (Fig. 3A). Crypt survival was reduced by almost sixfold in the proximal jejunum and ileum of FGFR-3−/− compared with wild-type mice. When these data are normalized to the number of crypts per cross section in unirradiated FGFR-3−/− and control mice, the fractional survival of crypts is still markedly reduced in FGFR-3−/− mice [Fig. 3B; for proximal jejunum, 1.5% vs. 7.3% in wild-type mice (P < 0.05); for ileum, 0.7% vs. 5.6% in wild-type littermates (P < 0.05)]. This decrease in crypt survival could be due to a significantly reduced number of clonogenic stem cells per crypt in FGFR-3−/− mice or a decreased ability of stem cells to repair potential lethal radiation-induced damage. To distinguish between these two possibilities, we used the method described by Roberts et al. (39) to measure the capacity of the clonogenic stem cell population in FGFR-3−/− and wild-type mice to recover from radiation-induced damage. The results demonstrate that the capacity of crypt stem cells to recover from radiation-induced damage is not reduced in FGFR-3−/− mice compared with wild-type controls (Fig. 3C). Rather, FGFR-3−/− mice show a modest increase in stem cell recovery after radiation-induced damage. Therefore, the reduction in crypt survival is not a consequence of defective injury repair in the crypt stem cell population; rather, the total number of stem cells is significantly lower in FGFR-3−/− mice.
Fig. 3.

Clonogenic stem cell survival in FGFR-3−/− mice. A: number of surviving crypts per cross section in proximal jejunum and ileum determined by microcolony assay 3.5 days after 14-Gy γ-irradiation of 19-day-old FGFR-3−/−, FGFR-3+/−, and FGFR-3+/+ mice. Values are means ± SD (n = 6 mice per group). *P < 0.05. †P < 0.01. B: fractional crypt survival determined by normalization to number of crypts in unirradiated FGFR-3−/−, FGFR-3+/−, and FGFR-3+/+ mice. Values are means ± SD (n = 6 mice per group). *P < 0.05. C: ability of stem cells to recover from radiation-induced damage (recovery factor) 84 h after irradiation in 21-day-old mice. Recovery factor was assessed as ratio of crypt stem cell survival in mice that received two 7-Gy γ-irradiation dose fractions separated by a 5-h recovery period to survival in mice that received a single dose of 14 Gy γ-irradiation.
Effects of FGFR-3 signaling on transit amplifying cells of the crypt.
During postnatal intestinal development, FGFR-3 is expressed in a subset of replicating cells in the lower crypt (49). To examine whether signaling through FGFR-3 has a role in regulating proliferation of the transit amplifying cell population in the crypt, we examined the patterns of crypt epithelial replication in various regions of the intestine in FGFR-3−/− and wild-type mice (Fig. 4). BrdU-labeled cells in the S phase are seen predominantly in the lower two-thirds of the crypt epithelium in wild-type and FGFR-3−/− mice (Fig. 4, A and B). Quantitation of the number of BrdU-labeled epithelial cells within crypts of 14-day-old FGFR-3−/− mice showed a small, but significant, reduction in the number of cells in the S phase in the proximal small intestine (duodenum and proximal jejunum) compared with wild-type controls (Fig. 4C). However, by 21 days of age, this difference was no longer apparent (data not shown). A modest reduction in villus height was also observed in the small intestine of 14-day-old FGFR-3−/− mice, possibly reflecting the decreased output of cells from the proliferative compartment (Fig. 4D). In suckling mice, the depth of small intestinal crypts in FGFR-3−/− mice was not significantly different from that in wild-type littermates at any age (data not shown). Apoptosis is believed to be one mechanism whereby the crypt stem cell number and the size of the replicating transit cell population are regulated. However, we observed no differences in apoptosis, as measured by caspase-3 immunohistochemistry, between FGFR-3−/− and wild-type mice (data not shown).
Fig. 4.

Effect of FGFR-3 on epithelial cell replication and villus height in mouse small intestine. Photomicrographs of jejunal crypts of 14-day-old wild-type (A) and FGFR-3−/− (B) mice show incorporation of 5-bromo-2′-deoxyuridine (BrdU) detected by anti-BrdU antibody. C: number of cells in S phase as determined by BrdU incorporation and immunohistochemistry in 20 crypts per segment for each mouse at 14 days of age. Values are means ± SE (n = 7 mice in each group). *Significantly different from wt/wt: P < 0.05 for proximal jejunum; P < 0.01 for duodenum. D: villus height in various segments of small intestine of 14-day-old mice. Values are means ± SE (n = 6 mice per group). *P < 0.05 vs. wt/wt in proximal jejunum. †P < 0.005 vs. wt/wt in distal jejunum.
FGFR-3-mediated signaling regulates epithelial lineage allocation and patterns of cellular differentiation.
Regional patterns of epithelial differentiation were examined using a variety of markers of intestinal differentiation from birth through 28 days of age. All the principal differentiated cell types were represented in the small intestine (Fig. 5) and colon (data not shown) of FGFR-3−/− mice. Paneth and goblet cells were appropriately positioned along the crypt-to-villus axis (Fig. 5, C and F) in FGFR-3−/− mice. Furthermore, an examination of the regional expression of a number of neuroendocrine cell products produced in the gut revealed that the localization and number of neuroendocrine cells containing PYY, serotonin (Fig. 5, D and E), and secretin (not shown) in the small intestine and colon (data not shown) were not different between FGFR-3−/− and wild-type mice. However, absorptive enterocytes expressing IFABP, a differentiation marker normally restricted to villus-associated enterocytes, were occasionally intermixed with undifferentiated proliferating cells in the intervillus epithelium and in nascently forming crypts of 4- to 7-day-old FGFR-3−/− mice (Fig. 5B).
Fig. 5.

Differentiation of intestinal epithelium in FGFR-3−/− and wild-type mice. A and B: expression of intestinal fatty acid-binding protein (IFABP, red fluorescence) in jejunum of a 4-day-old FGFR-3−/− mouse. A: expression of IFAPB in all villus-associated enterocytes. Magnification ×40. B: occasional cells in intervillus epithelium expressing IFABP (arrows). Magnification ×200. C: Paneth cells (arrows) in ileum of 14-day-old FGFR-3−/− mouse. Magnification ×400. D: peptide YY (PYY)-expressing cells (arrowhead) in ileum of 4-day-old FGFR-3−/− mouse. Magnification ×400. E: serotonin-containing neuroendocrine cells in jejunum (arrowheads) of 4-day-old FGFR-3−/− mouse. Magnification ×200. F: goblet cells (arrows) in jejunum of 10-day-old FGFR-3−/− mouse.
Number of Paneth cells that form in the developing intestine is regulated by FGFR-3.
The expression of marker genes characteristic of each of the secretory lineages was determined at various times after birth. The timing of initial appearance of all secretory cell subtypes was similar in FGFR-3−/− and wild-type mice (Fig. 6A). However, FGFR-3−/− mice displayed a marked reduction in the expression of Paneth cell markers that became evident by 21 days of age. Lysozyme mRNA levels were nearly fourfold lower, while cryptdin 5 mRNA levels were almost threefold lower, in FGFR-3−/− than in wild-type mice at 21 days of age (Fig. 6A). By contrast, no significant differences were detected in mRNA levels of Muc2 or chromogranin, goblet and enteroendocrine cell markers, respectively, between FGFR-3−/− and wild-type mice. To determine whether the reduced expression of lysozyme and cryptdin 5 mRNA in FGFR-3−/− mice was due to an actual decrease in Paneth cell numbers, Paneth cell distribution in the small intestine at 21 days of age was examined by immunohistochemistry using an antibody to Paneth cell lysozyme (Fig. 6, B and C). The number of Paneth cells was greatly reduced throughout the small intestine of 14- to 21-day-old FGFR-3−/− mice, with some crypts totally devoid of Paneth cells (Fig. 6C), compared with wild-type littermates (Fig. 6B). The number of Paneth cells in the small intestine was also reduced in the small number of FGFR-3−/− mice that survived past weaning, but to a lesser extent than in suckling mice (data not shown). However, the ultrastructural appearance of Paneth cells that did form in FGFR-3−/− mice was similar to that in the wild-type mice, with well-formed secretory granules in both strains of mice (Fig. 6, D and E).
Fig. 6.

Secretory cell lineage allocation in intestinal epithelium of FGFR-3−/− and wild-type mice. A: mRNA levels of secretory cell markers determined by real-time RT-PCR in RNA isolated from total terminal ileum of FGFR-3−/− and wild-type mice at 7–21 days of age. Although a significant reduction in Paneth cell lysozyme and cryptdin 5 expression was observed in FGFR-3−/− compared with wild-type mice, chromogranin and Muc2 expression was not different. Values are means ± SE (n = 4 mice per group). P ≤ 0.001 for lysozyme and cryptdin 5 for FGFR-3−/− vs. wild-type mice at 21 days of age. B and C: immunohistochemical visualization of Paneth cells in cross sections of terminal ileum from 21-day-old wild-type and FGFR-3−/− mice, respectively, with use of an antibody to lysozyme. D and E: electron micrographs of ultrathin sections of ileum of 21-day-old wild-type and FGFR-3−/− mice, respectively.
Regulation of Paneth cell lineage allocation by FGFR-3-mediated signaling is not dependent on Sox9 or Ihh signaling.
Studies by Varnat et al. (48) showed reduced numbers of Paneth cells in the duodenum of PPARβ-null mice. Their data suggest that Paneth cell maturation is regulated by PPARβ through inhibition of Ihh signaling. More recent data also implicated Sox9 in the regulation of Paneth cell lineage specification in the intestine and colon (3, 32). Thus Sox9-null mice are virtually devoid of Paneth cells. Therefore, we examined whether PPARβ and Ihh, as well as Sox9, expression was perturbed during postnatal intestinal development in our FGFR-3−/− mice (Fig. 7). Similar to the findings of Varnat et al., we observed an increase in PPARβ expression in the ileum of 14- and 21-day-old wild-type and FGFR-3−/− mice (Fig. 7A). In wild-type mice, this increase was greater than fivefold, whereas FGFR-3−/− mice showed a threefold increase in PPARβ mRNA levels. However, Ihh mRNA levels were not different between FGFR-3−/− and wild-type mice at any age, and these levels remained fairly constant between 7 and 21 days of age (Fig. 7B). Similarly, there was no significant difference in the level of Sox9 expression in the ileum between wild-type and FGFR-3−/− mice at any time point (Fig. 7C).
Fig. 7.

Expression of peroxisome proliferator-activated receptor-β (PPARβ), Indian hedgehog (Ihh), and Sox9 in developing ileum of FGFR-3−/− and wild-type mice. PPARβ (A), Ihh (B), and Sox9 (C) mRNA levels were measured by real-time PCR in RNA isolated from ileum of 7, 14, and 21-day-old FGFR-3−/− and wild-type mice. Values are means ± SE (n = 3–5 mice per group).
Signaling through FGFR-3 alters the distribution and level of β-catenin in the crypt epithelium.
Activation of the nuclear β-catenin/Tcf-4 signaling pathway has been shown to be important not only for the regulation and maintenance of intestinal stem cells, but also for Paneth cell lineage allocation (3, 32). Activation of the β-catenin-Tcf-4 transcriptional complex requires nuclear translocation of β-catenin from cytoplasmic pools and subsequent formation of a complex with Tcf-4. We examined the intracellular localization of β-catenin in FGFR-3−/− and wild-type mice by immunohistochemistry. A marked reduction in crypt epithelial cells with nuclear β-catenin staining was noted in FGFR-3−/− mice compared with wild-type controls (Fig. 8, A and B). The overall intensity of anti-β-catenin reactivity was also less in the intestine of FGFR-3−/− mice (Fig. 8, A and B). To determine whether the total intracellular pool of β-catenin was reduced in the intestine of FGFR-3−/− mice, we examined β-catenin protein expression in tissue lysates from small intestine by Western blotting using an antibody to the carboxy terminus of β-catenin. Two closely migrating ∼97-kDa bands were detected in tissue lysates from control C57BL6/j and wild-type FGFR-3 mice (Fig. 8C). The intensity of both of these bands was significantly less in protein extracts from the small intestine of FGFR-3−/− mice, indicating reduced cellular β-catenin protein in these mice. However, the noted differences in protein expression between FGFR-3−/− and wild-type mice were not due to transcriptional regulation, since β-catenin mRNA levels, as assessed by real-time PCR, were not different (Fig. 8D).
Fig. 8.

Expression of β-catenin and genes regulated by β-catenin/T cell factor (Tcf) in small intestine. A and B: immunohistochemical localization of β-catenin in sections of small intestine of 14-day-old wild-type and FGFR-3−/− mice, respectively. Nuclear localization of β-catenin (arrow) was observed in wild-type mice but was rare in FGFR-3−/− mice. C: Western blot analysis of β-catenin in lysates of small intestinal segments of C57BL6/j (B6), FGFR-3−/−, and FGFR-3+/+ mice. D: real-time PCR of total RNA isolated from small intestine of 14-day-old FGFR-3−/− and FGFR-3+/+ mice. Levels of β-catenin mRNA were determined by quantitative real-time PCR. Values are means ± SE (n = 5 mice per group). P = not significant. E: cyclin D1 and matrix metalloproteinase-7 (MMP-7) mRNA levels measured by real-time PCR in RNA isolated from 21-day-old FGFR-3−/− and wild-type mice. Values (means ± SE) are expressed relative to 18S rRNA × 10−3 (n = 3–5 mice per group).
Since cyclin D1 and MMP-7 are known to be major downstream gene targets upregulated by the active nuclear β-catenin-Tcf-4 complex, we also measured the mRNA levels of these genes by real-time PCR (Fig. 8E) in total RNA preparations of small intestine. Consistent with the decreased β-catenin nuclear translocation and implied lower activity of the β-catenin/Tcf-4 pathway, FGFR-3−/− mice displayed significantly decreased transcript levels of cyclin D1 and MMP-7 relative to their wild-type controls.
Activation of the Tcf-4 signaling pathway is maintained by signaling through FGFR-3.
Subconfluent Caco-2 cultures have high Tcf-4 activity that is dramatically reduced in postconfluent cultures, at which time cells display many features of differentiated absorptive enterocytes (30). Using the Tcf reporter construct TOPFlash, we examined the effect of FGFR-3 signaling on Tcf-4 pathway activation in Caco-2 cells (Fig. 9). The addition of FGF9, a ligand for FGFR-3, to Caco-2 cultures resulted in a sustained and significantly higher expression of the Tcf-4 reporter by 3 days after confluence than in cultures treated only with vehicle, which displayed a 10-fold decrease in TOPFlash luminescence signal (Fig. 9A). The effect of FGF9 was specific for the TOPFlash reporter plasmid, since the control plasmid, Renilla, did not respond (Fig. 9A). Since FGF9 can bind to multiple FGF receptors, we then examined whether signaling solely through FGFR-3 could maintain Tcf-4 activity in postconfluent Caco-2 cells. Tcf-4 activity at 4 days after confluence was nearly fourfold greater in cells cotransfected with TOPFlash and K650E, a ligand-independent, constitutively active FGFR-3 construct, than in untransfected control cells or cells cotransfected with wild-type FGFR-3 or with the empty vector (pUC18), all of which lost Tcf-4 activity during this time (Fig. 9B).
Fig. 9.

Signaling via FGFR-3 maintains β-catenin/Tcf activity in Caco-2 cells. A: luciferase activity in cell lysates from Caco-2 cells transiently transfected with pTOPFlash or Renilla and treated daily with FGF9 (50 ng/ml) starting at 24 h after transfection and harvested 48, 72, 96, and 120 h later (0, 1, 2, and 3 days after confluence, respectively). Cells were cotransfected with an FGFR-3 construct bearing the K650E mutation conferring constitutive activation of the receptor, pUC18 empty vector, or a wild-type FGFR-3 construct. B: β-catenin/Tcf activity expressed relative to luciferase activity from pTOPFlash or Renilla in unstimulated Caco-2 cells. Values are means ± SE (n = 3 culture wells per group for each of 3 independent experiments).
DISCUSSION
In this study, we used mice with a disruption of the FGFR-3 gene to investigate the role of this receptor in regulating morphogenic events involved in postnatal intestinal development. Our previous findings that FGFR-3 and its cognate ligands FGF1, FGF2, and FGF9 are coordinately expressed at high levels during postnatal intestinal development suggested that this signaling pathway mediates the morphogenic events at this time (50). Although our prior study did not define the specific role(s) of FGFR-3 in regulating developmental events in the intestine, the observation that expression of FGFR-3 was restricted to undifferentiated epithelial cells within the lower portion of the crypt, a region known to house the epithelial stem cell population, further suggested that signaling through FGFR-3 is important in mediating some aspect of epithelial morphogenesis. We now show that signaling through FGFR-3 plays a critical role in regulating accrual of a sufficient number of epithelial stem cells and crypts during mouse postnatal intestinal development, as well as in modulating the specification of the Paneth cell lineage. Our data further suggest that FGFR-3 signaling may modulate some features of intestinal epithelial morphogenesis in the mouse through interaction with the β-catenin/Tcf-4 pathway.
The deficit in crypt number observed in the intestine of suckling FGFR-3−/− mice could result from a failure of any of several morphogenic events that occur during intestinal development. The initial failure of the intervillus epithelium to invaginate into the underlying mesenchyme would result in failure of crypt formation. However, the timing of the onset of crypt formation is identical in FGFR-3−/− and wild-type mice, and the intervillus epithelium is not persistent in the intestine of FGFR-3−/− mice, suggesting that this morphogenic process is not impaired in the absence of signaling through FGFR-3. The number of intestinal crypts subsequently increases through a process of crypt bifurcation or fission that, in the mouse, is largely completed by 4–5 wk after birth (31). The regulation of crypt fission is not fully understood; however, it is clear that the number of crypt stem cells must expand to support segregation of at least one stem cell to each daughter crypt during this process. Some investigators have suggested that crypt fission is triggered when the number of stem cells in a crypt exceeds a critical threshold (31, 38). If the decrease in crypt number observed in FGFR-3−/− mice was solely the result of impaired or reduced branching morphogenesis without a concomitant reduction in stem cell proliferation, then the number of stem cells per crypt would be expected to be increased. Alternatively, a decreased number of crypts could result if FGFR-3−/− mice were unable to expand the crypt stem cell population at a rate sufficient to support the level of crypt fission observed in wild-type mice. To distinguish between these alternatives, we used a well-characterized in vivo functional assay for estimating the number of potential clonogenic stem cells per crypt (10, 19, 21, 40). Our data showing that the number of clonogenic stem cells per crypt is decreased in the small intestine of FGFR-3−/− mice imply that the reduction in the number of crypts in these mice is not merely due to a failure of branching morphogenesis during intestinal development.
The decreased number of clonogenic stem cells per crypt in FGFR-3−/− mice could result from several distinct mechanisms. One possibility is that FGFR-3 regulates the proliferation of stem cells. Signaling through FGFR-3 has been shown to regulate proliferation of intestinal epithelial cell lines (23, 30). Replication of a clonogenic stem cell can give rise, symmetrically, to two daughters that retain stem cell characteristics or, alternatively, to one stem cell and a daughter progenitor cell whose progeny are committed to a differentiation pathway. Therefore, the probability of symmetric stem cell division must be sufficiently high to support the increase in the number of stem cells needed to permit crypt fission without depleting the number of clonogens per crypt. Hence, it is possible that lack of signaling through FGFR-3 could inhibit the rate of stem cell proliferation or alter the balance between symmetric and asymmetric stem cell division and commitment of stem cell progeny to differentiate. FGFR-3 signaling can regulate the balance between proliferation and commitment to differentiation in other tissues, such as bone, during normal development (34). Determining whether either of these mechanisms explains the deficit in crypt number in FGFR-3−/− mice requires methods for direct quantification of the rate of proliferation of intestinal stem cells and the commitment of their progeny to enter the intestinal epithelial differentiation pathway. Finally, it is also possible that the lack of FGFR-3 during embryogenesis may have resulted in the formation of fewer intervillus epithelial stem cells. Because of the lack of reliable stem cell markers in the developing intestine, it is difficult to entirely rule out this possibility.
The small and transient reduction in the number of replicating cells per crypt in the proximal intestine of FGFR-3−/− mice suggests that signaling through FGFR-3 does not regulate the replication of the transit amplifying cells during crypt morphogenesis. In a recent study, Arnaud-Dabernat et al. (1) found a modest increase in the number of replicating transit amplifying cells and the size of the crypt in adult FGFR-3−/− × NOD/shi F2 mice homozygous for the FGFR-3 gene disruption. This conclusion initially seems at odds with our demonstration that FGFR-3 is necessary for the elaboration of an adequate number of crypt stem cells, as reflected in the decreased number of intestinal crypts in the intestine of our FGFR-3−/− mice. However, there is a major difference in the biological context between the two studies with respect to the intestinal crypt cell populations under examination. Our studies focused on the consequence of FGFR-3 deletion during stem cell proliferation and crypt morphogenesis, dynamic processes that take place during postnatal intestinal development. Arnaud-Dabernat and colleagues examined the consequences of FGFR-3 deletion on the replication of transit amplifying cells in the unperturbed adult intestine, a developmental stage at which expression of FGFR-3 in crypt epithelial cells is low, the number of crypts is static, and crypt stem cells are relatively quiescent compared with the suckling mouse intestine (50). It is possible that the response of the transit amplifying cell population of the adult intestine to FGFR-3 signaling is different from that of stem cells of the developing intestine.
The onset of crypt formation occurred at ∼4 days after birth in FGFR-3−/− and wild-type mice, and their intestines were morphologically similar. All the principal differentiated cell types found in the small bowel and colon were represented in FGFR-3−/− mice. However, FGFR-3−/− mice accrued a significantly reduced number of Paneth cells throughout the small intestine, whereas other secretory cell lineages were unaffected. This reduction in the number of Paneth cells was reflected in diminished mRNA levels of the Paneth cell markers MMP-7, lysozyme, and cryptdin 5 and confirmed by immunohistochemistry using antibodies to Paneth cell lysozyme and cryptdin 5. Recent studies suggest that Paneth cell lineage allocation and differentiation/maturation are regulated by distinct, but interacting, pathways. Sox9, a high-mobility group-box transcription factor expressed in intestinal crypt cells, including Paneth cells, has been identified as a regulator of Paneth and goblet cell lineage specification in the intestine and colon (3, 32). Sox9-null mice lack Paneth cells in the small intestine, as determined by immunohistochemistry using antibodies to a variety of markers, including lysozyme, which is considered to be an early marker of Paneth cell specification (3, 32). By contrast, van Es et al. (47) demonstrated that the Wnt/Frizzled-5 signaling pathway regulates intestinal Paneth cell maturation (differentiation) and positioning, but not the number of Paneth cells. Although there were fewer Paneth cells in the intestine of FGFR-3−/− mice, the expression of Sox9 was not different between these mice and their littermate controls at any time during the developmental period examined. Additionally, whereas Sox9 null mice were reported to have enlarged crypts and increased numbers of cells incorporating BrdU, crypts of FGFR-3−/− mice did not appear larger than those of wild-type mice and FGFR-3−/− mice had only a small and transient increase in S-phase cells in the proximal gut.
Finally, PPARβ has also been shown to regulate Paneth cell homeostasis through modulation of the Ihh signaling pathway (48). Adult PPARβ-null mice have reduced numbers of lysozyme-staining Paneth cells. Although Wnt and Notch pathway functionality appeared normal, there was a significant increase in the expression of Ihh in the intestine of the PPARβ-null mice. Inhibition of Ihh signaling directly or through activation of PPARβ resulted in increased Paneth cell numbers and expression of Paneth cell products (48). Of particular relevance for our studies, Varnat et al. (48) observed about a fivefold increase in PPARβ expression in the developing duodenum between 15 and 21 days after birth. In view of these latter findings, we examined PPARβ expression in the intestine of FGFR-3−/− and wild-type mice. In agreement with the data of Varnat et al., PPARβ expression was increased in the ileum of wild-type and FGFR-3−/− mice between 14 and 21 days after birth. However, lack of FGFR-3 signaling did not alter the expression of Ihh during intestinal development. Taken together, these findings suggest that FGFR-3 signaling represents an additional pathway that can mediate Paneth cell lineage allocation and/or differentiation directly by modulating signaling via the β-catenin-Tcf complex or indirectly through other, as yet unidentified, effector molecules.
In this study, we observed that cellular levels and nuclear localization of β-catenin are reduced in the crypt epithelium of suckling mice. Signaling through β-catenin and the Tcf/Lef family of transcription factors plays a central role in regulating epithelial stem cell fate and replication during intestinal development and in intestinal neoplasia (2, 5, 7, 26, 28). Its key role in intestinal development is underlined by the observation that mice homozygous for a disrupted Tcf-4 gene lack replicating cells within the intervillus region of the small intestine and do not form crypts (26). The β-catenin/Tcf-4 pathway is also implicated in the regulation of colonic epithelial cell proliferation and differentiation (30), consistent with its well-documented role in regulating epithelial cell proliferation through the modulation of cyclin D1 and c-Myc (45). Our findings that FGFR-3−/− mice have a reduced number of crypt epithelial stem cells, coupled with the observation that some differentiated cells fail to appropriately segregate from the undifferentiated cell population, raise the possibility that FGFR-3 mediates its effects on intestinal epithelial development through regulation of Tcf/β-catenin activity. The uncoupling of differentiation with position along the crypt-to-villus axis for enterocytes and Paneth cells in the small intestine has been observed in EphB2/EphB3-knockout mice (4). Levels of EphB2 and EphB3, along with their ephrin ligands, are regulated in the crypt epithelium through the β-catenin/Tcf-4 signaling pathway. Thus the mispositioning of differentiated absorptive enterocytes within the intervillus epithelium and in nascently forming crypts in FGFR-3−/− mice also raises the possibility that FGFR-3 signaling might regulate expression of EphB2/EphB3 directly or by modulation of β-catenin/Tcf-4 activity. However, the appropriate localization of Paneth cells to the crypt base in FGFR-3−/− mice suggests that the regulation of cell positioning in the intestine is complex and may involve other, as yet unidentified, factors.
The reduction in the total cellular level and nuclear localization of β-catenin in FGFR-3−/− mice at 14 days of age, a critical time for expansion of the number of small intestinal crypts, further supports the hypothesis that FGFR-3 signaling may modulate β-catenin/Tcf-4 activity during epithelial morphogenesis. The regulation of intracellular pools of β-catenin is complex and occurs at multiple levels (6). The reduced level of β-catenin in the intestinal epithelium of FGFR-3−/− mice was not due to downregulation of transcription, since mRNA levels were similar between FGFR-3−/− and wild-type mice, suggesting that FGFR-3 signaling modulates β-catenin protein stability and/or distribution/localization. The absence of detectable nuclear β-catenin in crypt epithelial cells of FGFR-3−/− mice, coupled with the finding of lower levels of cyclin D1 and MMP-7, major downstream gene targets of β-catenin/Tcf-4 transcriptional regulation, strongly supports the hypothesis that FGFR-3 signaling modulates the transcriptional activity of the β-catenin-Tcf-4 complex during intestinal development. Furthermore, our in vitro studies provide a direct demonstration that signaling through FGFR-3 can modulate β-catenin/Tcf-4 activity. The introduction of a ligand-independent, constitutively active FGFR-3 construct into Caco-2 cells, as well as ligand-mediated activation of FGFR-3, resulted in continued high Tcf-4 activity at a time when Caco-2 cell cultures would normally downregulate this activity. Although the molecular details of the signaling cascade linking FGFR-3 signal transduction to modulation of β-catenin/Tcf-4 activity remain to be elucidated, the demonstration that FGFR-3 can interact with this developmentally important transcription complex opens a new and intriguing avenue of investigation for characterizing the mechanisms by which FGFs and their receptors participate in the orchestration of intestinal development.
Finally, it also is possible that FGFR-3 regulates intestinal crypt formation and lineage allocation through the modulation of stem cell number via pathways that are independent of Tcf-4, since ligand binding of FGFR-3 can result in the activation of multiple signaling cascades (17, 22). The effects of FGFR-3 signaling are cell and context specific (24). Thus additional studies are required to identify which signaling pathways are activated by FGFR-3 in the crypt epithelium and how these signaling cascades regulate morphogenic events during intestinal development.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants R01 DK-064751 and P01 DK-57880 to S. M. Cohn and through the Molecular Biology and Morphology Cores of the University of Virginia NIH/NIDDK Digestive Diseases Research Core Center (Grant DK-50306).
REFERENCES
- 1.Arnaud-Dabernat S, Yadav D, Sarvetnick N. FGFR3 contributes to intestinal crypt cell growth arrest. J Cell Physiol 216: 261–268, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Barker N, Huls G, Korinek V, Clevers H. Restricted high level expression of Tcf-4 protein in intestinal and mammary gland epithelium. Am J Pathol 154: 29–35, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, Blache P, Jay P. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 178: 635–648, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, Clevers H. β-Catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111: 251–263, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 103: 311–320, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Brantjes H, Barker N, van Es J, Clevers H. TCF: Lady Justice casting the final verdict on the outcome of Wnt signaling. Biol Chem 383: 255–261, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 11: 3286–3305, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian theory of the origin of the four epithelial cell types. Am J Anat 141: 537–562, 1974. [DOI] [PubMed] [Google Scholar]
- 9.Cohn SM, Lieberman MW. The use of antibodies to 5-bromo-2′-deoxyuridine for the isolation of DNA sequences containing excision-repair sites. J Biol Chem 259: 12456–12462, 1984. [PubMed] [Google Scholar]
- 10.Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest 99: 1367–1379, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohn SM, Simon TC, Roth KA, Birkenmeier EH, Gordon JI. Use of transgenic mice to map cis-acting elements in the intestinal fatty acid binding protein gene (Fabpi) that control its cell lineage-specific and regional patterns of expression along the duodenal-to-colonic and crypt-to-villus axes of the gut epithelium. J Cell Biol 119: 27–44, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colony PC, Neutra MR. Macromolecular transport in the fetal rat intestine. Gastroenterology 89: 294–306, 1985. [DOI] [PubMed] [Google Scholar]
- 14.Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12: 390–397, 1996. [DOI] [PubMed] [Google Scholar]
- 15.Green RP, Cohn JM, Sacchettini JC, Jackson KE, Gordon JI. The mouse intestinal fatty acid binding protein genes: nucleotide sequence, pattern of developmental and regional expression, and proposed structure of its protein product. DNA Cell Biol 11: 31–41, 1992. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths DFR, Davies SJ, Williams D, Williams GT, Williams ED. Demonstration of somatic mutation and crypt clonality by X-linked enzyme histochemistry. Nature: 461–463, 1988. [DOI] [PubMed]
- 17.Hart KC, Robertson SC, Donoghue DJ. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol Biol Cell 12: 931–942, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauft SM, Kim SH, Schmidt GH, Pease S, Rees S, Harris S, Roth KA, Hansbrough JR, Cohn SM, Ahnen DJ, Wright NA, Goodlad RA, Gordon JI. Expression of SV40 T antigen in the small intestinal epithelium of transgenic mice results in proliferative changes in the crypt and re-entry of villus-associated enterocytes into the cell cycle but has no apparent effect on cellular differentiation programs and does not cause neoplastic transformation. J Cell Biol 117: 825–839, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendry JH, Roberts SA, Potten CS. The clonogen content of murine intestinal crypts: dependence on radiation dose used in its determination. Radiat Res 132: 115–119, 1992. [PubMed] [Google Scholar]
- 20.Holnthoner W, Pillinger M, Groger M, Wolff K, Ashton AW, Albanese C, Neumeister P, Pestell RG, Petzelbauer P. Fibroblast growth factor-2 induces Lef/Tcf-dependent transcription in human endothelial cells. J Biol Chem 277: 45847–45853, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Houchen CW, George RJ, Sturmoski MA, Cohn SM. FGF-2 enhances intestinal stem cell survival and its expression is induced after radiation injury. Am J Physiol Gastrointest Liver Physiol 276: G249–G258, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Kanai M, Goke M, Tsunekawa S, Podolsky DK. Signal transduction pathway of human fibroblast growth factor receptor 3. Identification of a novel 66-kDa phosphoprotein. J Biol Chem 272: 6621–6628, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Kanai M, Rosenberg I, Podolsky DK. Cytokine regulation of fibroblast growth factor receptor 3 IIIb in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 272: G885–G893, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Kannan K, Givol D. FGF receptor mutations: dimerization syndromes, cell growth suppression, and animal models. IUBMB Life 49: 197–205, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Klambt C, Glazer L, Shilo BZ. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev 6: 1668–1678, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 19: 379–383, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Korinek V, Barker N, Willert K, Molenaar M, Roose J, Wagenaar G, Markman M, Lamers W, Destree O, Clevers H. Two members of the Tcf family implicated in Wnt/β-catenin signaling during embryogenesis in the mouse. Mol Cell Biol 18: 1248–1256, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lickert H, Domon C, Huls G, Wehrle C, Duluc I, Clevers H, Meyer BI, Freund JN, Kemler R. Wnt/β-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Dev Suppl 127: 3805–3813, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Madara JL, Neutra MR, Trier JS. Junctional complexes in the fetal rat small intestine during morphogenesis. Dev Biol 86: 170–178, 1981. [DOI] [PubMed] [Google Scholar]
- 30.Mariadason JM, Bordonaro M, Aslam F, Shi L, Kuraguchi M, Velcich A, Augenlicht LH. Down-regulation of β-catenin TCF signaling is linked to colonic epithelial cell differentiation. Cancer Res 61: 3465–3471, 2001. [PubMed] [Google Scholar]
- 31.Maskens AP, Dujardin-Loitis R. Kinetics of tissue proliferation in colorectal mucosa during post-natal growth. Cell Tissue Kinet 14: 467–477, 1981. [DOI] [PubMed] [Google Scholar]
- 32.Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, Clevers H, de Crombrugghe B. SOX9 is required for the differentiation of Paneth cells in the intestinal epithelium. Gastroenterology 133: 539–546, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol 2: REVIEWS3005, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 16: 1446–1465, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res 306: 357–363, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 17: 1709–1713, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potten CS, Loeffler M. A comprehensive model of the crypts of the small intestine of the mouse provide insight into the mechanisms of cell migration and the proliferation hierarchy. J Theor Biol 127: 381–391, 1987. [DOI] [PubMed] [Google Scholar]
- 38.Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110: 1001–1020, 1990. [DOI] [PubMed] [Google Scholar]
- 39.Roberts SA, Hendry JH, Potten CS. Deduction of the clonogen content of intestinal crypts: a direct comparison of two-dose and multiple-dose methodologies. Radiat Res 141: 303–308, 1995. [PubMed] [Google Scholar]
- 40.Roberts SA, Potten CS. Clonogen content of intestinal crypts: its deduction using a microcolony assay on whole mount preparations and its dependence on radiation dose. Int J Radiat Biol 65: 477–481, 1994. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt GH, Wilkinson MM, Ponder BAJ. Cell migration pathway in the intestinal epithelium: an in situ marker system using mouse aggregation chimeras. Cell 40: 425–429, 1985. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt GH, Winton DJ, Ponder BAJ. Development of the pattern of cell renewal in the crypt villus unit of chimeric mouse intestine. Development 103: 785–790, 1988. [DOI] [PubMed] [Google Scholar]
- 43.Sweetser DA, Birkenmeier EH, Hoppe PC, McKeel DW, Gordon JI. Mechanisms underlying generation of gradients in gene expression within the intestine: an analysis using transgenic mice containing fatty acid binding protein/human growth hormone fusion genes. Genes Dev 2: 1318–1332, 1988. [DOI] [PubMed] [Google Scholar]
- 44.Templeton T, Hauschka S. FGF mediated aspects of skeletal muscle growth and differentiation are controlled by a high affinity receptor, FGFR1. Dev Biol 154: 169–181, 1992. [DOI] [PubMed] [Google Scholar]
- 45.Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Trier JS, Moxey PC. Morphogenesis of the small intestine during fetal development. CIBA Found Symp 70: 3–29, 1979. [DOI] [PubMed] [Google Scholar]
- 47.van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, Clevers H. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7: 381–386, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Varnat F, Heggeler BB, Grisel P, Boucard N, Corthesy-Theulaz I, Wahli W, Desvergne B. PPARβ/δ regulates Paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology 131: 538–553, 2006. [DOI] [PubMed] [Google Scholar]
- 49.Vidrich A, Buzan J, Ilo C, Bradley L, Skaar K, Cohn SM. Signaling through FGFR3 is necessary for expansion of the epithelial stem cell compartment during normal intestinal development (Abstract). Gastroenterology 124: A22, 2003. [Google Scholar]
- 50.Vidrich A, Buzan JM, Ilo C, Bradley L, Skaar K, Cohn SM. Fibroblast growth factor receptor-3 is expressed in undifferentiated intestinal epithelial cells during murine crypt morphogenesis. Dev Dyn 230: 114–123, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Winton DJ, Ponder BAJ. Stem cell organization in mouse small intestine. Proc R Soc Lond B 241: 13–18, 1990. [DOI] [PubMed] [Google Scholar]
- 52.Withers HR, Elkind MM. Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol 117: 261–267, 1970. [DOI] [PubMed] [Google Scholar]