

SUMMARY
The requirement for cholesterol is greater in developing tissues (fetus, placenta, and yolk sac) as compared to adult tissues. Here, we compared cholesterol-induced suppression of sterol synthesis rates in the adult liver to the fetal liver, fetal body, placenta, and yolk sac of the Golden Syrian hamster. Sterol synthesis rates were suppressed maximally in non-pregnant adult livers when cholesterol concentrations were increased. In contrast, sterol synthesis rates were suppressed only marginally in fetal livers, fetal bodies, placentas, and yolk sacs when cholesterol concentrations were increased. To begin to elucidate the mechanism responsible for the blunted response of sterol synthesis rates in fetal tissues to exogenous cholesterol, the ratio of sterol regulatory element-binding protein (SREBP) cleavage-activating protein (SCAP) to Insig-1 was measured in these same tissues since the ratio of SCAP to the Insigs can impact SREBP processing. The fetal tissues had anywhere from a 2- to 6-fold greater ratio of SCAP to Insig-1 than did the adult liver, suggesting constitutive processing of the SREBPs. As expected, the level of mature, nuclear SREBP-2 was not different in the fetal tissues with different levels of cholesterol whereas it was different in adult livers. These findings indicate that the suppression of sterol synthesis to exogenous sterol is blunted in developing tissues and the lack of response appears to be mediated at least partly through relative levels of Insigs and SCAP.
Keywords: cholesterol, Smith-Lemli-Opitz, HMG-CoA reductase, fetus, SREBP-2, IUGR
INTRODUCTION
During embryonic and fetal development, there is substantial requirement for cholesterol due to the rapid cell division that occurs to maintain significant growth. The necessity of synthesized cholesterol by the fetus and/or embryo is depicted best by fetuses and/or embryos unable to synthesize cholesterol. There are 7 known defects in the sterol biosynthetic pathway, all of which will lead to congenital malformations [1–3]. The most common of the defects is the Smith-Lemli-Opitz syndrome (SLOS). Individuals with this syndrome have mutations in 3β-hydroxysterol Δ7-reductase (DHCR7) and convert minimal to no 7-dehydrocholesterol to cholesterol, depending on the type of mutation [4, 5]. The deficiency of cholesterol can lead to a wide range of congenital defects, ranging from the mild (minor physical abnormality with behavioral and learning disabilities) to the severe (lethal with multiple major congenital anomalies) [6, 7]. Not surprising, the severity of the syndrome is correlated with circulating sterol concentrations [8].
The fetus and/or embryo has two sources of cholesterol: endogenous and exogenous [9–11]. The endogenous sterol is synthesized de novo. As might be expected of a compound essential for growth, fetal sterol synthesis rates are relatively elevated [12–14]. Since the fetus does not come in direct contact with the maternal circulation, its exogenous source of cholesterol originates in the placenta and yolk sac and is secreted and/or effluxed to the fetal circulation [9]. The cholesterol secreted and/or effluxed is that which was synthesized in the placenta and yolk sac and/or that which was taken up from the maternal circulation by the placenta and yolk sac, transported across cells and secreted and/or effluxed. Though both endogenous and exogenous sources make significant contributions to the fetal cholesterol, it is likely that a greater percentage of fetal sterol originates from de novo synthesis [15, 16].
The synthesis of cholesterol is a multi-step pathway with the rate-limiting step occurring with the conversion of HMG-CoA to mevalonate, a reaction catalyzed by HMG-CoA reductase (HMGR). HMGR is regulated at several levels and by multiple effectors, including nonsterol intermediates as well as cholesterol [17, 18]. The major transcription factor that elicits the sterol-mediated response is the sterol regulatory element-binding protein-2 (SREBP-2) [19–21]. There are three SREBPs, SREBP-1a, SREBP-1c, and SREBP-2. The SREBPs are synthesized as inactive, precursor proteins which are inserted into the ER membrane where they bind to the SREBP cleavage-activating protein (SCAP). Under conditions of depleted cholesterol, SCAP escorts the SREBPs to the Golgi. Once in the Golgi, two different proteases cleave the SREBPs to their mature forms. The SREBPs are translocated to the nucleus where they bind to sterol regulatory elements within promoter regions of HMGR and other proteins involved in the maintenance of sterol balance such as HMG-CoA synthase and the LDL receptor [19]. When cellular cholesterol is in excess, SCAP undergoes a conformational change that allows it to bind to Insig-1 and -2 [22–24]. Since the Insigs are resident membrane proteins of the ER, the SCAP:SREBP complex is retained within the ER and the SREBPs are not processed to their mature forms [22–24]. This processing of the SREBPs can be affected markedly by changing the ratio of SCAP to the Insigs [25, 26]. Though SREBP-2 drives sterol synthesis in adult tissues, SREBP-1a drives sterol synthesis in cell culture and when overexpressed [27].
Though the regulation of sterol biosynthesis has been studied extensively in adult tissues, very little research has examined its regulation during development. To gain a better understanding of the regulation of sterol biosynthesis during this rapid time of growth, the relationship between cholesterol concentrations and sterol synthetic rates in the fetal liver and body, the placenta, and the yolk sac were compared to the same relationship in the non-pregnant adult liver of the Golden Syrian hamster. Hamsters were used in these studies because 1) hamsters are readily responsive to dietary cholesterol [14, 28], 2) embryonic and extraembryonic fetal tissue cholesterol concentrations can be readily changed in hamsters [29] and 3) the gestational age of hamsters is very precise [29]. We found that sterol synthesis rates were maximally suppressed in the adult liver but only marginally suppressed in each of the extraembryonic and embryonic fetal tissues in the presence of excess tissue cholesterol. The ratios of SCAP mRNA to Insig-1 mRNAs were also markedly different between the adult and fetal tissues, and were 2–6-fold greater in the fetal tissues. Thus, we hypothesize that the differences in responsiveness of sterol synthesis rates to cholesterol concentrations between adult and fetal tissues are at least partly due to constitutive processing of SREBP-2.
MATERIAL AND METHODS
Animals and diets
Male and female hamsters (Charles River Laboratories, Inc.) weighing between 110 and 120 g were maintained in a temperature and humidity controlled room and fed a pelleted chow (diet #7012, Harlan Teklad). After at least one week of acclimation, females were split into two different groups. In one group, females were fed diets consisting of 0, 0.05, 0.10, 0.15, 0.20, or 0.25 % added cholesterol (wt/wt) in ground chow (#7012, Harlan Teklad); diets had an inherent cholesterol concentration of 0.00013% (wt/wt). After 3 weeks, studies were performed in the non-pregnant females; livers from non-pregnant females were used instead of livers from pregnant females due to the marked and potentially confounding effects of pregnancy on the maternal liver [30]. In the second group, females were fed diets consisting of 0, 0.12, 0.5, or 2.0% added cholesterol (wt/wt), levels that were known to affect whole fetus sterol synthesis, in ground chow for 3 weeks and mated [29]. At 11.5 days post-conception (dpc), studies were performed; hamsters have a gestational period of 15.5 days.
Sterol synthesis rates
Animals were injected with 50 (pregnant) or 15 (non-pregnant) mCi of [3H]H2O (GE Healthcare). After 1 h, adult livers, placentas, yolk sacs, fetal livers, and fetal bodies were collected, saponified, and digitonin-precipitable sterols (DPS) were isolated [31, 32]. The rates of sterol synthesis are expressed as the nmol of [3H]H2O incorporated into DPS per h per g tissue. Synthesis rates in fetal livers and bodies were corrected for the time it took for [3H]H2O to equilibrate between the fetus and dam [31, 33].
Cholesterol concentrations
Tissues were saponified in alcoholic base. Cholesterol was extracted with petroleum ether, and concentrations were determined by gas liquid chromatography using stigmastanol (Sigma) as an internal standard [34].
Immunoblots
Fetal and adult livers, fetal bodies, placentas, and yolk sacs were homogenized. Nuclei and microsomes were isolated as described [35, 36], except samples were stored in 10% glycerol. Protein concentrations were measured by the Lowry assay, and equal amounts of protein of like samples pooled. Proteins were separated on Bio-Rad 4–15% Tris-HCL Ready gels under denaturing conditions, and transferred to nitrocellulose membranes. Blots containing membrane-bound proteins were pre-incubated in a blocking buffer containing 5% dry milk and 0.1% Tween. Blots of nuclei were incubated with anti-SREBP-2 IgG (a gift from Dr. J. Horton, University of Texas Southwestern Medical Center) followed by an incubation with donkey anti-rabbit secondary antibody (GE Healthcare). Chemiluminescence from ECL Plus was detected by a Storm 450 Phosphoimager, and band densities were quantitated by Image J software. Blots were stripped with Restore Western Blot Stripping Buffer [37] and reprobed with CREB (Cell Signaling Technology, Inc.), to standardize protein loading. Blots of microsomes were incubated with anti-Insig-1 (Santa Cruz Biotechnology) or anti-SCAP (Santa Cruz Biotechnology) followed by incubations with appropriate secondary antibodies. Bands were detected, blots stripped, and reprobed with β-actin (Bioscience Research Reagents). Pooled samples were analyzed at least twice (n=3–4 per set).
Real time PCR
Samples of adult, neonatal, and fetal livers, fetal bodies, placentas, and yolk sacs were collected. Tissue RNA was isolated using TRIazol [38] and stored in FORMAzol®. RNA was treated with RNase-free DNase I and reverse transcribed to cDNA by SuperScript II reverse transcriptase using random hexamers. PCR assays were performed on a Bio-Rad iCycler iQ real-time PCR Detection System with SYBR green as our fluorophore. A serial dilution of a randomly-picked sample was used to generate a standard curve for each gene examined. This standard curve was used to calculate the relative levels of mRNA for the gene of interest and the reference/housekeeping gene (cyclophilin). Primers used were modified from those previously reported (Table I; SCAP, Insig-1, and cyclophilin) [39–41] or were similar to those previously reported (SREBP-1a) [42].
TABLE I.
Nucleotide sequences of hamster-specific primers used for quantitative real time PCR
| mRNA | Sequences of forward and reverse primers (5′-3′) |
|---|---|
| Insig-1 | CCCAGATTTCTTCTATATTCG |
| CCCATAGCTAACTGTCGTCC | |
| SCAP | CTGCTGCTACCCTCTGCTGAAG |
| CTTGTTTGTGGTCAGAGTC | |
| Cyclophilin | ATTCATGTGCCAGGGTGGTGACT |
| TCAGTCTTGGCGGTGCAGAT |
Statistical analyses
Data are presented as means ± SEM. Linear regression models were run treating dietary cholesterol as the independent variable and cholesterol concentration as the dependent variable. Only the first three data points were used to determine if the slope of the line was significantly different from zero since the first three data points correspond to the dietary concentration we are most interested in. Plus, inclusion of all points will skew results and end in a positive line due to the marked changes that occur with higher cholesterol concentrations. Student’s t-tests were conducted when comparing mean levels between two groups. When increasing amounts of cholesterol were fed, cholesterol concentrations or synthesis rates were compared between livers of dams fed 0 versus 0.05% cholesterol, 0.05 versus 0.10% cholesterol, 0.10 versus 0.15% cholesterol, etc. In fetal tissues, comparisons were between 0 and 0.12% cholesterol, 0.12 and 0.50% cholesterol, and 0.5 and 2.0% cholesterol. Significance was considered P<0.05.
RESULTS
The goal of these studies was to delineate differences in regulation of sterol biosynthesis between adult and fetal tissues and to begin to elucidate the mechanism(s) responsible for any differences in regulation. We studied the adult liver, the fetal liver and body, the placenta, and the yolk sac. In the adult non-pregnant female, diets were fed that contained from 0 to 0.25% added cholesterol. Hepatic cholesterol concentrations were 1.93 ± 0.10 mg/g in dams fed ground chow (Fig. 1A, left panel). Cholesterol concentrations did not increase in livers of dams fed 0.05% or 0.10% added cholesterol. In support of this, the slope of the line using cholesterol concentrations in dams fed 0, 0.05, and 0.10% cholesterol as the dependent variable and dietary cholesterol as the independent variable was not different from zero (Table II). Hepatic cholesterol concentrations increased 2.2-fold, 4.3-fold and 6.2-fold in livers of females fed 0.15, 0.20, or 0.25% cholesterol (P<0.05), respectively, as compared to those fed no added cholesterol. Even though hepatic cholesterol concentrations were not increased when hamsters were fed 0.05% cholesterol, sterol synthesis rates decreased 51% (Fig. 1A, right panel; P<0.05). Rates were not significantly different with 0.10% dietary cholesterol but decreased further with 0.15% dietary cholesterol (P<0.05) and reached a nadir of maximal suppression when 0.20 or 0.25% cholesterol was fed (P<0.05).
Figure 1.

Hepatic cholesterol concentrations and sterol synthesis rates in tissues of pregnant and non-pregnant females fed increasing amounts of cholesterol. Non-pregnant females were fed 0, 0.05, 0.10, 0.15, 0.20, and 0.25% (wt/wt) added cholesterol. Hepatic cholesterol concentrations (A, left panel) were measured by GLC and sterol synthesis rates (A, right panel) were measured in vivo using [3H]H2O. Data are presented as means ± SEM (n=3–6). Pregnant females were fed 0, 0.12, 0.50, and 2.00% (wt/wt) added cholesterol prior to and during pregnancy. Cholesterol concentrations (left panel) and sterol synthesis rates (right panel) were measured in fetal livers (B), fetal bodies (C), placentas (D), and yolk sacs (E) of dams at 11.5 dpc. Data are presented as means ± SEM (n=3–6). * depicts significant differences (P<0.01) or ** depicts trends to be significantly different (0.05<P<0.07) between values from tissues of dams fed consecutive amounts of cholesterol, i.e. dams fed 0.10 versus 0.15% cholesterol.
TABLE II.
Cholesterol concentrations as dependent variables in the adult liver and fetal tissues
| Tissue | Slope | SE | P value vs. 0 |
|---|---|---|---|
| Adult liver | 1.11 | 1.84 | 0.555 |
| Fetal liver | 1.29 | 0.20 | <0.001 |
| Fetal body | 0.54 | 0.27 | 0.063 |
| Placenta | 1.28 | 0.39 | 0.007 |
| Yolk sac | 4.38 | 0.73 | <0.001 |
Slopes were determined using values obtained in the livers of non-pregnant females fed 0, 0.05, and 0.10% cholesterol or in tissues of pregnant dams fed 0, 0.12, and 0.5% cholesterol.
To study the relationship between cholesterol concentrations and sterol synthesis rates in fetal tissues, females were fed diets which would result in increased cholesterol concentrations in the embryonic and extraembryonic fetal tissues [29]; maternal cholesterol concentrations increased 15–300% with dietary cholesterol [29]. Cholesterol concentrations in tissues of dams fed 0.12% cholesterol were not significantly different from concentrations in tissues of dams fed no added cholesterol. However, concentrations were increased in the fetal livers (Fig. 1B, left panel), fetal bodies (Fig. 1C, left panel), placentas (Fig. 1D, left panel), and yolk sacs (Fig. 1E, left panel) of dams fed 0.5% cholesterol. Importantly, when cholesterol concentrations were plotted as the dependent variable and dietary cholesterol as the independent variable, the slopes of all the lines were significantly different from zero, or at least tended to be different from zero (Table II). Sterol synthesis rates were also measured in these same fetal tissues. Synthesis rates decreased in each of the fetal tissues of dams fed 0.12 or 0.5% cholesterol (P<0.05). Synthesis rates were suppressed to a nadir in the fetal livers (Fig. 1B, right panel) and bodies (Fig. 1C, right panel) of dams fed 0.5% cholesterol. The nadir in these fetal tissues (≈500 nmol/h per g) was much greater than the nadir in the adult liver (18 nmol/h per g). Synthesis rates were also suppressed to a nadir in the placentas (Fig. 1C, right panel) of dams fed 0.5% cholesterol, though the nadir of this tissue was ≈200 nmol/h per g. Rates did not reach a nadir in the yolk sac but decreased continuously in yolk sacs (Fig. 1E, right panel) of dams fed increasing amounts of cholesterol.
To better understand the differences in regulation between the adult liver and the fetal tissues (fetal liver and body, placenta, and yolk sac), relative sterol synthesis rates were plotted against relative cholesterol concentrations in each tissue; sterol synthesis rates and cholesterol concentrations in hamsters fed no added cholesterol were set at 100% and all other values presented as a percentage of the original rates or concentrations. As seen in Figure 2 and Table II, sterol synthesis rates decreased prior to an increase in cholesterol concentrations in the adult liver. In contrast, rates and concentrations changed simultaneously in the fetal liver and body, the placenta, and the yolk sac. To determine if the differences in response were merely due to a relatively smaller increase in cholesterol concentrations in the fetal tissues, the relative sterol synthesis rates of tissues with ≈2-fold increase in cholesterol concentration were compared (Fig. 3). Regardless of the fact that all tissues presented in this figure had ≈2-fold increase in cholesterol concentrations, sterol synthesis rates were suppressed to 8% of original rates in the adult liver whereas rates were suppressed to only 29 to 59% in fetal tissues.
Figure 2.

Relationship between sterol synthesis rates and cholesterol concentrations in the adult liver and fetal tissues. Sterol synthesis rates and cholesterol concentrations in tissues of dams fed no added cholesterol were set to 100%; data originated in Figure 1. Relative changes in synthesis rates and cholesterol concentrations in tissues of dams fed cholesterol were calculated and plotted. The box depicts tissues with ≈2-fold increases in cholesterol concentrations.
Figure 3.

Sterol synthesis rates in tissues with increased cholesterol concentrations. Relative sterol synthesis rates from tissues with ≈2-fold increase in cholesterol concentration were plotted; data corresponds to the box in Figure 2. The fold increase of cholesterol concentrations in tissues of non-pregnant and pregnant dams fed cholesterol compared to values in non-pregnant and pregnant dams fed no cholesterol are in parentheses. Note that there are two values for yolk sacs corresponding to yolk sacs of dams fed 0.5 and 2.0% cholesterol.
The question now becomes - why are the fetal tissues less responsive to exogenous sterol as compared to the adult tissue? We hypothesized that the blunted response to exogenous sterol in the fetal tissues was the result of constitutive processing of SREBP-2 due to different ratios of SCAP to the Insigs [25, 26] and/or higher SREBP-1a levels [27]. To test this hypothesis, SCAP and Insig-1 mRNA levels were measured in the adult livers and the fetal tissues; only Insig-1 was measured in these studies since Insig-2 is not expressed in the rodent liver until after parturition [43]. The relative amounts of Insig-1 mRNA were 11.6% (P<0.001) in the fetal liver, 23.1% (P<0.001) in the placenta, and 50.2% (P=0.01) in the yolk sac, as compared to amounts in the adult liver (Fig. 4A). The amounts of SCAP mRNA were 98.2% (P=ns) in the fetal liver, 125.4% (P=ns) in the placenta, and 40.4% (P=0.057) in the yolk sac, as compared to amounts in the adult liver (Fig. 4A). The ratio of SCAP to Insig-1 (Fig. 4B) was 5.8-fold greater in the fetal liver (P=0.006), 2.8-fold greater in the yolk sac (P=0.037), and 1.8-fold greater in the placenta (P=ns) as compared to the adult liver. Additionally, the relative mRNA levels in whole fetuses of dams fed 0 and 2% cholesterol were similar: Insig-1 (0.24±0.01 vs 0.21±0.01 for 0 and 2% cholesterol, respectively) and SCAP (0.35±0.04 and 0.32±0.01 for 0 and 2% cholesterol, respectively). Protein levels of Insig-1 and SCAP in relation to β-actin were measured as well (Fig. 5). For Insig-1, very little protein was detected for the fetal body and fetal liver. The amount of Insig-1 with respect to β-actin expression in the extraembryonic tissues were ≈62% of that in the adult liver. For SCAP, there were relatively similar amounts to slightly more SCAP in relation to β-actin in the fetal liver (128%), fetal body (116%), placenta (102%), and yolk sac (98%) as compared to adult liver. The relative levels of SREBP-1a mRNA levels were measured in the adult and fetal livers as an alternative mechanism for a lack of change in sterol synthesis rates. Interestingly, the adult liver (1.95±0.08) had more SREBP-1a mRNA as compared to the fetal liver (1.17±0.17).
Figure 4.
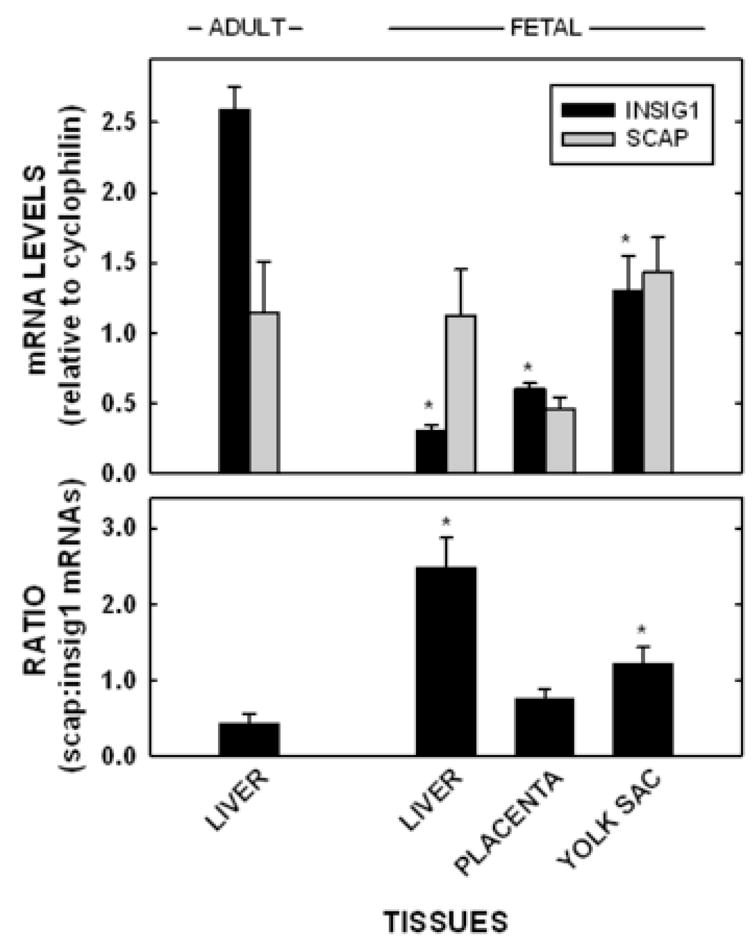
Insig-1 and SCAP mRNA levels in the adult liver and fetal tissues. Tissues were collected from non-pregnant and pregnant (11.5 dpc) females fed no added cholesterol. The Insig-1 and SCAP mRNA levels were measured by real time PCR and presented as means ± SEM (n=3–4) using cyclophilin as our reference/housekeeping gene (A). The ratio of SCAP:Insig-1 mRNA levels were calculated and presented (B). In both panels, * depicts significant differences from the adult liver (P<0.05).
Figure 5.
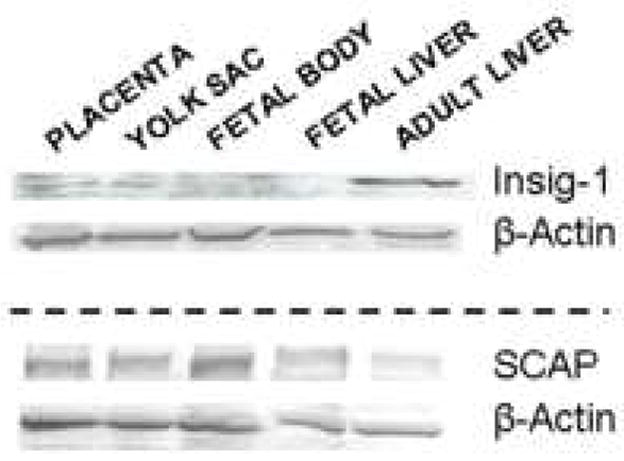
Insig-1 and SCAP expression levels in embryonic and extraembryonic fetal tissues and adult livers. Fetal tissues were collected at 11.5 dpc and adult livers were collected 3 weeks after being fed ground chow. β-Actin was used to equalize protein loading.
Finally, we wanted to measure the relative levels of mature, nuclear SREBP-2 protein levels in the fetal tissues with different levels of cholesterol. As expected in tissues with constitutive processing, protein expression levels were similar between fetal tissues of dams fed 0 or 2% cholesterol (Fig. 6A). Also expected, was a lack of effect of diet on HMGR or HMG synthase in the fetal tissues (data not shown). Not surprisingly, adult livers with a 2-fold increase in cholesterol concentration (0.15% dietary cholesterol) demonstrated ≈45% decrease in mature, nuclear SREBP-2 protein levels (Fig. 6B).
Figure 6.
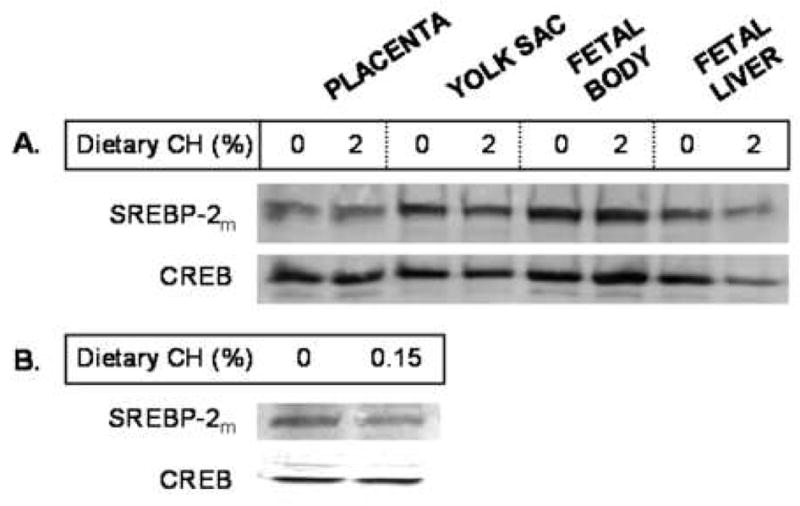
Mature SREBP-2 (SREBP-2m) expression levels in nuclei of embryonic and extraembyronic fetal tissues and adult livers with different cholesterol concentrations. Embryonic and extraembryonic fetal tissues were collected from females treated as described in Figure 1 except only 0 and 2% cholesterol was fed (A). At 11.5 dpc, tissues were collected and assayed for mature, nuclear SREBP-2 in the nuclei using CREB to equilibrate protein loading. Livers from non-pregnant females fed 0.15% cholesterol for 3 weeks were also assayed for mature, nuclear SREBP-2 in the nuclei (B).
DISCUSSION
The regulation of sterol synthesis is complex and multivalent [17, 18]. This complex regulation allows for a greater than 200-fold variation in sterol synthesis rates to occur over relatively short periods of time [18]. This ability to finely regulate sterol production is beneficial to the adult tissues where the need for cholesterol is low, and excess sterol can lead to cell death and/or acceleration of disease processes. In fetal tissues, the ability to rapidly suppress and activate sterol synthesis rates may not be advantageous since the need for cholesterol is considerable and constant. In the current studies, cholesterol-induced suppression of sterol synthesis rates was blunted in the fetal tissues as compared to adult tissues. First, sterol synthesis rates were suppressed maximally in adult livers (18 nmol/h per g) whereas sterol synthesis rates were suppressed only marginally in each of the embryonic and extraembryonic fetal tissues (187–644 nmol/h per g). Second, cholesterol concentrations increased in fetal tissues simultaneously with a decrease in synthesis rates whereas cholesterol concentrations increased in the adult liver only when synthesis rates were suppressed maximally. While the relationship between synthesis rates and cholesterol concentrations in the adult liver have been described previously [44], these are the first to demonstrate a simultaneous change when synthesis is only marginally suppressed. Simply put, in the adult liver, synthesis rates are suppressed to compensate for exogenous sterol and concentrations increase only when synthesis rates can not be reduced further. In contrast, sterol synthesis rates in fetal tissues can not be suppressed significantly and so concentrations increase.
Several mechanisms exist which could be responsible for the apparent dysregulation of sterol synthesis in the fetal tissues and possibly other rapidly growing tissues, including constitutive processing of SREBP-2, decreased degradation of SREBP-2, and decreased degradation of HMGR.
Constitutive processing of SREBP-2
The hallmark of constitutive processing of the SREBPs is elevated sterol synthesis rates, even in the presence of increased cholesterol concentration. Others have demonstrated that the processing of SREBP-2 can be manipulated by changing the amount of SCAP relative to the amount of Insigs in cells. When SCAP was overexpressed, SREBP-2 was constitutively processed, even in the presence of sterol [26]. Likewise, overexpression of the Insigs resulted in maintaining SREBP-2 in the Golgi [25]. Based on our data, specifically the fact that the SCAP to Insig-1 ratio is ≈2–6-fold greater in the fetal tissues as compared to the adult liver and the fact that nuclear, mature SREBP-2 levels do not decrease in fetal tissues with increased cholesterol concentrations whereas levels do change in adult tissues with similar fold increases in cholesterol concentration, at least part of the reason for the lack of change in sterol synthesis rates in fetal tissues could be due to constitutive processing of SREBP-2. The ratio of SCAP to Insig-1 was greater in all fetal tissues, though the greatest difference (≈6-fold) occurred between the fetal and adult livers. It appeared that differences in protein may have been even greater than differences in mRNA levels. Why might the Insig-1 levels, especially protein, be so low in the fetal tissues in comparison to the adult liver? Either the relatively different expression levels of SCAP or Insig-1 between fetal tissues and adult liver is hard-wired to ensure high sterol synthesis rates or other effectors are involved in their regulation. Since insulin can affect Insig levels [45], it is possible that the low levels of insulin in the rodent fetus [43] can impact upon Insig expression levels. This would not explain why Insig-1 levels are low in the placenta and yolk sac since these tissues come in contact with the maternal circulation, though it has been hypothesized that all tissues are insulin resistant during gestation [46, 47]. Also, Insig-1 is stabilized when sterols are present [22, 23]. If there is no abundance of sterols due to their requirement for membrane formation during rapid growth, Insig-1 may be ubiquintated and degraded rapidly during the fetal period as seen by low protein levels [48], supporting our hypothesis that the fetus is in a negative sterol balance [49]. Regardless, other factors besides the ratio of SCAP to Insig-1 must be involved in responsiveness of sterol synthesis rates to exogenous sterol since a correlation did not exist between the ratio of mRNA of these two proteins and the degree of suppressed synthesis rates in the fetal liver, placenta, and yolk sac.
SREBP-2 degradation
The lack of marked change in sterol biosynthesis in the fetal tissues could also be due to an inability to degrade SREBP-2. SREBP-2 is degraded via the ubiquitin-proteosome pathway after its phosphorylation and ubiquination [50–52]. Though the ubiquitin-proteosome system for degradation is active during development [53, 54], it seems that the SREBPs would not be readily modified and/or degraded during this time of rapid growth since sterol synthesis rates remain relatively active in the presence of cholesterol. Lack of SREBP-2 degradation may be the result of a change in proteosome activity [55] or rate of de-ubiquitination [41].
HMGR degradation
Finally, the blunted effect of exogenous sterol in the fetal tissues could be the result of an inability to degrade HMGR. As with the SREBPs and other proteins, HMGR is targeted for degradation once it is ubiquitinated [41, 56]. While HMGR can be ubiquinated in the presence of sterol [41], it is further ubiquinated in the presence of both sterol plus geranylgeraniol, implying involvement of a geranylgeranylated protein [57], or plus lanosterol [58]. These more recent data support the observations that maximal suppression of sterol synthesis rates occurs only in the presence of cholesterol plus a mevalonate-derived product [18, 59], which may be absent in a tissue in a negative sterol balance and/or proliferating rapidly as during development [49]. Additionally, a lack of HMGR degradation in the fetal tissues could be due to the fact that HMGR degradation is accelerated by binding to Insig-1 [41]. The relatively low levels of Insig-1 mRNAs we detected in the early/mid fetal tissues would favor a lack of HMGR ubiquitination and therefore a lack of degradation of HMGR.
Summary
To summarize, it appears that there is a dysregulation of sterol synthesis rates in the embryonic and extraembryonic fetal tissues. The dysregulation might occur to ensure a steady supply of cholesterol for the essential growth- and development-related processes that take place in these tissues during gestation. The lack of regulation may occur through multiple processes, including constitutive processing of SREBP-2.
What are some of the cholesterol-dependent growth- and development-related processes that take place in the fetus and/or placenta? The most noted growth-related role of cholesterol is that it is a major component of cellular membranes. As might then be expected in persons unable to synthesize significant amounts of cholesterol, SLOS infants and children have diminished growth rates [6], which can be improved with dietary cholesterol [60–62]. It would be expected that fetal growth rates are also reduced. Fetal growth rates could be further impacted upon if placental growth rates were altered; a small placenta often times lead to placental insufficiency, the leading cause of intrauterine growth restriction (IUGR) [63–65]. The ability to maintain placental growth rates through a constant supply of membrane components could be a novel way to ensure adequate placental, and thereby fetal, growth rates. In addition to its role as a membrane substrate, cholesterol and closely related sterols can affect various signaling events. First, the sterols activate Sonic hedgehog (Shh) [66, 67]. Shh is a key patterning protein involved in forebrain as well as limb and organ development [68]. A lack of Shh activity, possibly due to low levels of sterols [69], leads to a number of congenital defects in mice and humans alike [66–72]. Second, changes in cholesterol concentrations could affect composition of membrane microdomains where abundant signaling originates. While it has been demonstrated that signaling will change in membranes of SLOS individuals [73, 74], due at least partly to a redistribution of proteins into various microdomains [75], the full extent of a change in signaling during development is unknown. Thus, a steady supply of cholesterol during development through relatively unresponsive, elevated sterol synthesis rates aids in the maintenance of a number of sterol-dependent developmental processes and growth.
Acknowledgments
The authors wish to thank Catherine Wilke for her excellent technical assistance. These studies were supported by grant HD 34089 from the National Institute of Health.
Abbreviations
- SLOS
Smith-Lemli-Opitz syndrome
- DHCR7
3β-hydroxysterol Δ7-reductase
- HMGR
HMG-CoA reductase
- SREBP
sterol regulatory element binding protein
- SCAP
SREBP cleavage-activating protein
- DPS
digitonin-precipitable sterols
- Shh
Sonic hedgehog
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nwokoro NA, Wassif CA, Porter FD. Genetic disorders of cholesterol biosynthesis in mice and humans. Mol Genet Metab. 2001;74:105–119. doi: 10.1006/mgme.2001.3226. [DOI] [PubMed] [Google Scholar]
- 2.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 3.Porter FD. Malformation syndromes due to inborn errors of cholesterol synthesis. J Clin Invest. 2002;110:715–724. doi: 10.1172/JCI16386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irons M, Elias ER, Salen G, Tint GS, Batta AK. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341:1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- 5.Shefer S, Salen G, Batta AK, Honda A, Tint GS, Irons M, Elias ER, Chen TC, Holick MF. Markedly inhibited 7-dehydrocholesterol-delta 7-reductase activity in liver microsomes from Smith-Lemli-Opitz homozygotes. J Clin Invest. 1995;96:1779–1785. doi: 10.1172/JCI118223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porter FD. RSH/Smith-Lemli-Opitz Syndrome: A multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol Genet Metab. 2000;71:163–174. doi: 10.1006/mgme.2000.3069. [DOI] [PubMed] [Google Scholar]
- 7.Kelley RI, Hennikam RCM. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tint GS, Salen G, Batta AK, Shefer S, Irons M, Elias ER, Abuelo DN, Johnson VP, Lambert M, Lutz R. Correlation of severity and outcome with plasma sterol levels in variants of the Smith-Lemli-Opitz syndrome. J Pediatr. 1995;127:82–87. doi: 10.1016/s0022-3476(95)70261-x. [DOI] [PubMed] [Google Scholar]
- 9.Woollett LA. Maternal cholesterol in fetal development: transport of cholesterol from the maternal to the fetal circulation. Am J Clin Nutr. 2005;82:1155–1161. doi: 10.1093/ajcn/82.6.1155. [DOI] [PubMed] [Google Scholar]
- 10.Lichtenberg MH, Wilke CS, McConihay JA, Granholm NA, Woollett LA. Yolk sac cholesteryl ester secretion rates can be manipulated in the Golden Syrian hamster: effect of yolk sac cholesterol concentrations. Biochim Biophys Acta. 2005;1735:214–221. doi: 10.1016/j.bbalip.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Schmid KE, Davidson WS, Myatt L, Woollett LA. The transport of cholesterol across a placental cell monolayer: Implications for net transport of sterol from the maternal to fetal circulation. J Lipid Res. 2003;44:1909–1918. doi: 10.1194/jlr.M300126-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Carr BR, Simpson ER. Cholesterol synthesis in human fetal tissues. J Clin Endocrinol Metab. 1982;55:447–452. doi: 10.1210/jcem-55-3-447. [DOI] [PubMed] [Google Scholar]
- 13.Levin MX, Pitt AJA, Schwartz AL, Edwards PA, Gordon JI. Developmental changes in the expression of genes involved in cholesterol biosynthesis and lipid transport in human and rat fetal and neonatal livers. Biochim Biophys Acta. 1989;1003:293–300. doi: 10.1016/0005-2760(89)90235-x. [DOI] [PubMed] [Google Scholar]
- 14.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34:1637–1659. [PubMed] [Google Scholar]
- 15.Yoshida S, Wada Y. Transfer of maternal cholesterol to embryo and fetus in pregnant mice. J Lipid Res. 2005;46:2168–2174. doi: 10.1194/jlr.M500096-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res. 2006;47:1535–1541. doi: 10.1194/jlr.M600141-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanish M, Goldstein JL, Brown MS. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzymes. J Biol Chem. 1988;263:8929–8937. [PubMed] [Google Scholar]
- 18.Brown MS, Goldstein J. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 19.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton JD, Shimomura I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol. 1999;10:143–150. doi: 10.1097/00041433-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Osborne TF. Sterol regulatory element binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 22.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 24.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the pruified embrane region of SCAP: Mechanism for a sterol-sensing domain. Mol Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, Liang G. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J Clin Invest. 2004;113:1168–1175. doi: 10.1172/JCI20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang T, Goldstein JL, Brown MS. Overexpression of membrane domain of SCAP prevents sterols from inhibiting SCAP/SREBP exit from endoplasmic reticulum. J Biol Chem. 2000;275:29881–29886. doi: 10.1074/jbc.M005439200. [DOI] [PubMed] [Google Scholar]
- 27.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spady DK, Dietschy JM. Interaction of dietary cholesterol and triglycerides in the regulation of hepatic low density lipoprotein transport in the hamster. J Clin Invest. 1988;81:300–309. doi: 10.1172/JCI113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McConihay JA, Horn PS, Woollett LA. The effect of maternal hypercholesterolemia on fetal sterol metabolism in the Golden Syrian hamster. J Lipid Res. 2001;42:1111–1119. [PubMed] [Google Scholar]
- 30.Yao L, Dawson PA, Woollett LA. Increases in biliary cholesterol-to-bile acid ratio in pregnant hamsters fed low and high levels of cholesterol. Am J Physiol. 2003;284:G263–G268. doi: 10.1152/ajpgi.00332.2002. [DOI] [PubMed] [Google Scholar]
- 31.Woollett LA. Origin of cholesterol in the fetal Golden Syrian hamster: contribution of de novo sterol synthesis and maternal-derived lipoprotein cholesterol. J Lipid Res. 1996;37:1246–1257. [PubMed] [Google Scholar]
- 32.Dietschy JM, Spady DK. Measurement of rates of cholesterol synthesis using tritiated water. J Lipid Res. 1984;25:1469–1476. [PubMed] [Google Scholar]
- 33.Belknap WM, Dietschy JM. Sterol synthesis and low density lipoprotein clearance in vivo in the pregnant rat, placenta, and fetus. Sources for tissue cholesterol during fetal development. J Clin Invest. 1988;82:2077–2085. doi: 10.1172/JCI113829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turley SD, Herndon MW, Dietschy JM. Reevaluation and application of the dual-isotope plasma ratio method for the measurement of intestinal cholesterol absorption in the hamster. J Lipid Res. 1994;35:328–339. [PubMed] [Google Scholar]
- 35.Botolin D, Jump DB. Selective proteolytic processing of rat hepatic sterol regulatory element binding protein-1 (SREBP-1) and SREBP-2 during postnatal development. J Biol Chem. 2003;278:6959–6962. doi: 10.1074/jbc.M212846200. [DOI] [PubMed] [Google Scholar]
- 36.Jelinek DF, Andersson S, Slaughter CA, Russell DW. Cloning and regulation of cholesterol 7β-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J Biol Chem. 1990;265:8190–8197. [PMC free article] [PubMed] [Google Scholar]
- 37.Jegathesan J, Liebenthal JA, Arnett MG, Clancy RL, Pierce JD. Apoptosis: Understanding the new molecular pathway. Med Nurs. 2004;13:371–376. [PubMed] [Google Scholar]
- 38.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 39.Shimomura I, Bashmakov Y, Shimano H, Horton JD, Goldstein JL, Brown MS. Cholesterol feeding reduces nuclear forms of sterol regulatory element binding proteins in hamster liver. Proc Natl Acad Sci USA. 1997;94:12354–12359. doi: 10.1073/pnas.94.23.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yabe D, Xia ZP, Adams CM, Rawson RB. Three mutations in sterol-sensing domain of SCAP block interaction with Insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci USA. 2002;99:16672–16677. doi: 10.1073/pnas.262669399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 42.Field FJ, Born E, Mathur SN. Fatty acid flux suppresses fatty acid synthesis in hamster intestine independently of sterol regulatory element-binding protein-1 expression. J Lipid Res. 2003;44:1199–1208. doi: 10.1194/jlr.M300013-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Bobard A, Hainault I, Ferré P, Foufelle F, Bossard P. Differential regulation of sterol regulatory element-binding protein 1c transcriptional activity by insulin and liver X receptor during liver development. J Biol Chem. 2005;280:199–206. doi: 10.1074/jbc.M406522200. [DOI] [PubMed] [Google Scholar]
- 44.Turley SD, Spady DK, Dietschy JM. Identification of a metabolic difference accounting for the hyper- and hyporesponder phenotypes of cynomologus monkey. J Lipid Res. 1997;38:1598–1611. [PubMed] [Google Scholar]
- 45.Bortoff KD, Zhu CC, Hrywna Y, Messina JL. Insulin induction of pip92, CL-6 and novel mRNAs in rat hepatoma cells. Endocrine. 1997;7:199–207. doi: 10.1007/BF02778142. [DOI] [PubMed] [Google Scholar]
- 46.Sugden MC, Holness MJ. Potential role of peroxisome proliferator-activated receptor-alpha in the modulation of glucose-stimulated insulin secretion. Diabetes. 2004;53:S71–S81. doi: 10.2337/diabetes.53.2007.s71. [DOI] [PubMed] [Google Scholar]
- 47.Herrera E, Munoz C, Lopez-Luna P, Ramos P. Carbohydrate-lipid interactions during gestation and their control by insulin. Braz J Med Biol Res. 1994;27:2499–2519. [PubMed] [Google Scholar]
- 48.Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol Chem. 2006;281:39308–39315. doi: 10.1074/jbc.M608999200. [DOI] [PubMed] [Google Scholar]
- 49.Schmid KE, Woollett LA. Differential effects of polyunsaturated fatty acids on sterol synthesis rates in adult and fetal tissues of the hamster: Consequence of altered sterol balance. Am J Physiol. 2003;285:G796–G803. doi: 10.1152/ajpgi.00226.2003. [DOI] [PubMed] [Google Scholar]
- 50.Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem. 2001;276:36431–36437. doi: 10.1074/jbc.M105200200. [DOI] [PubMed] [Google Scholar]
- 51.Sundquist A, Ericsson J. Transcription-dependent degradation controls the stability of the SREBP family of transcription factors. Proc Natl Acad Sci. 2003;100:13833–13838. doi: 10.1073/pnas.2335135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Punga T, Bengoechea-Alonso MT, Ericsson J. Phosphorylation and ubiquitination of the transcription factor sterol regulatory element-binding protein-1 in response to DNA binding. J Biol Chem. 2006;381:25278–25286. doi: 10.1074/jbc.M604983200. [DOI] [PubMed] [Google Scholar]
- 53.An JY, Seo JW, Tasaki T, Lee MJ, Varshavsky A, Kwon YT. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc Natl Acad Sci. 2006;103:6212–6217. doi: 10.1073/pnas.0601700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowerman B, Kurtz T. Degrade to create: developmental requirements for ubiquitin-mediated proteolysis during early C. elegans embryogenesis. Development. 2006;133:773–784. doi: 10.1242/dev.02276. [DOI] [PubMed] [Google Scholar]
- 55.Riddle TM, Kuhel DG, Woollett LA, Ficktenbaum CJ, Hui DY. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J Biol Chem. 2001;276:37514–37519. doi: 10.1074/jbc.M104557200. [DOI] [PubMed] [Google Scholar]
- 56.Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated uiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 57.Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of Insig-1 to its sterol-sensing domain. Mol Cell. 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 58.Song BL, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005;1:179–189. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- 60.Elias ER, Irons MB, Hurley AD, Tint GX, Salen G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS) Am J Med Genet. 1997;68:305–310. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 61.Irons M, Elias ER, Abuelo D, Bull MJ, Greene CL, Johnson VP, Keppen L, Schanen C, Tint GS, Salen G. Treatment of Smith-Lemli-Opitz syndrome: results of a multi-center trial. Am J Med Genet. 1997;68:311–314. [PubMed] [Google Scholar]
- 62.Nwokoro NA, Mulvihill JJ. Cholesterol and bile acid replacement therapy in children and adults with Smith-Lemli-Opitz (SLO-RSH) syndrome. Am J Med Genet. 1997;68:315–321. doi: 10.1002/(sici)1096-8628(19970131)68:3<315::aid-ajmg13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 63.Molteni RA. Placental growth and fetal/placental weight (F/P) ratios throughout gestation - Their relationship to patterns of fetal growth. Semin Perinat. 1984;8:94–100. [PubMed] [Google Scholar]
- 64.Heinonen S, Taipale P, Saarikoski S. Weights of placentae from small-for-gestational age infants revisited. Placenta. 2001;22:399–404. doi: 10.1053/plac.2001.0630. [DOI] [PubMed] [Google Scholar]
- 65.Pardi G, Marconi AM, Cetin E. Placental-fetal interrelationship in IUGR fetuses-A review. Placenta. 2002;23:S136–S141. doi: 10.1053/plac.2002.0802. [DOI] [PubMed] [Google Scholar]
- 66.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 67.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 68.Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Wesphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 69.Cooper MK, Wassif CA, Krabowiak PA, Taipale J, Gong R, Kelley RI, Porter FD, Beachy PA. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nature Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 70.Marti E, Bovolenta P. Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neuro. 2002;25:89–96. doi: 10.1016/s0166-2236(02)02062-3. [DOI] [PubMed] [Google Scholar]
- 71.Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nature Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 72.Kelley RL, Roessler E, Hennekam RC, Feldman GL, Kosaki K, Jones MC, Palumbos JC, Muenke M. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: Does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet. 1996;66:478–484. doi: 10.1002/(SICI)1096-8628(19961230)66:4<478::AID-AJMG22>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 73.Tulenko TM, Boeze-Battaglia K, Mason RP, Tint GS, Steiner RD, Connor WE, Labelle EF. A membrane defect in the pathogenesis of the Smith-Lemli-Opitz syndrome. J Lipid Res. 2006;47:134–143. doi: 10.1194/jlr.M500306-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Kovarova M, Wassif CA, Odom S, Liao K, Porter FD, Rivera J. Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J Exp Med. 2006;203:1161–1171. doi: 10.1084/jem.20051701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keller RK, Arnold TP, Fliesler SJ. Formation of 7-dehydrocholesterol-containing membrane rafts in vitro and in vivo, with relevance to the Smith-Lemli-Opitz syndrome. J Lipid Res. 2004;45:347–355. doi: 10.1194/jlr.M300232-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]