

Introduction
Cystic fibrosis (CF) is a life‐limiting, hereditary, multisystem disease predominantly affecting the lungs and pancreas. Over the last few years, survival of CF patients has increased markedly. This has been attributed to earlier diagnosis, improved patient care delivered by multidisciplinary teams and more effective therapeutic options. It has been suggested that the pathophysiologic basis of CF lies in different gene mutations that encode abnormalities in the structure of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, leading to physiologic defects in ion transport.1 The clinical course in CF is quite heterogeneous, and much of the variability in morbidity and mortality is unexplained by current conceptualization of the disease. Although currently unidentified modifier genes might explain some of this heterogeneity, other factors are probably contributory.
We describe a CF patient with chronic cough whose lung function continued to deteriorate, in spite of escalation of medical treatment and frequent hospitalizations. A change in home circumstances led to a sustained upturn in lung function as well as a reduction in the frequency of pulmonary exacerbations.
Case report
An 8-year‐old pancreatic insufficient CF girl, heterozygous for ΔF508 and a second unidentified mutation was born at 34 weeks of gestation to parents who smoked. She presented with meconium ileus and malrotation requiring ileostomy on day 2. Her sweat chloride was 66 mmol L−1. Pseudomonas aeruginosa was first isolated at 2 years of age and subsequently she had intermittent growths of P. aeruginosa, Staphylococcus aureus and Stenotrophomonas maltophilia. From the age of 4 years, her respiratory status began to deteriorate. She had a chronic productive cough and frequently had pulmonary exacerbations that were associated with reduced exercise tolerance and hypoxaemia. By the age of 5 years, she was being admitted to hospital every three months for intravenous antibiotics, intensive physiotherapy and nutritional optimization. A total implantable venous access device (Portacath) was inserted around this time. Within weeks of an admission, the benefits of her inpatient treatment would wear off, and as a result of her persistent symptoms, further investigations were undertaken; including flexible bronchoscopy which showed normal airway anatomy, and a bronchoalveolar lavage that was negative for viruses, bacteriology and fungi. CT scan of chest (Figure 1) showed mild bronchiectasis with evidence of air trapping due to distal airways disease. Total IgE was 10 IU L−1, RAST for Aspergillus was <0.35 IU mL−1 and pH study showed no evidence of gastro‐oesophageal reflux. Following this therapeutic trials of a number of agents including bronchodilator therapy, steroids (both oral and inhaled), antifungal therapy, anti‐reflux medications, anti‐inflammatory therapy (azithromycin) and mucolytic (DNAase) therapy failed to improve her symptoms.
Figure 1.
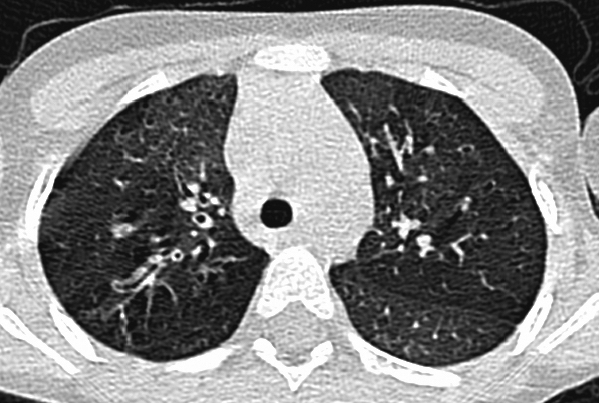
CT scan of the chest showing mild bronchiectasis with evidence of air trapping
At the age of 6.5 years, her mother and brother were sent to prison. As her father was an alcoholic, her care was taken over by her elder sister (aged 21 years). This change in carer and home environment led to marked improvement in treatment adherence; furthermore, there was no longer exposure to environmental tobacco smoke. A dramatic improvement in her symptoms, lung function ( Figure 2) and nutritional status ensued. Hospital admissions (Figure 3) have dramatically reduced from 50 days per year to 14 days in the last two years.
Figure 2.

Lung function over three years
Figure 3.

Hospital days
Discussion
A sustained improvement in lung function as well as a decrease in pulmonary exacerbations accompanied a change in carer and home circumstances. This case reinforces the importance of ‘getting the basics right’, illustrating that before considering escalation of medical therapy, issues of treatment adherence, psychosocial and emotional wellbeing, as well as environmental risk factors should be addressed.
Refinements in treatment of CF over the last few decades have led to great improvements in caregivers' ability to preserve lung function and prolong life, but there is wide variability in the progression of disease. Factor influences on disease severity and outcome can be grouped into three major categories: genetic, which includes all biological factors intrinsic to the individual patient; environmental, which includes exposures that are secondary to socioeconomic and demographic factors; and healthcare‐related, which includes prescribed medical interventions and patient adherence to therapy.
A better understanding of psychosocial and environmental risk factors and how they mediate adverse outcomes could provide insight into the pathophysiology of CF lung disease. Poor phenotype–genotype correlation is reported for CF lung disease.2,3 Although certain genotypes are clearly associated with normal pancreatic function, genotype is a poor predictor of pulmonary disease and eventual patient outcome in the general population of CF patients.4 Effect of CF modifiers genes may explain some of the disease heterogeneity, however in this case the effect of environmental factors appears to be highly significant.
Socioeconomic status (SES) is an important determinant of outcome in many chronic diseases5,6 and may also play a significant role in CF. Decreased survival has been found among CF patients of lower social class.7–9 A cohort analysis of paediatric CF patients in the US using Medicaid status as a proxy for low SES concluded that low SES is associated with significantly poorer outcomes in children with CF. Medicaid patients had more than three‐fold greater risk of death, and survivors had significantly worse pulmonary function and growth.10 These findings are generally congruent with those of other studies that have found worse health outcomes in indigent populations.5
Overall, the adverse effect of poverty on health in CF is probably mediated by many of the same factors that have been shown to be relevant in other chronic diseases.5 Nutritional status is dependent upon patient and parent self‐management skills, which in turn are linked to maternal education and understanding of dietary recommendations, both of which may be suboptimal in disadvantaged families. Nutritional inadequacies associated with poverty might worsen pulmonary function as well as growth, by compromising immunologic responses to lung infection. Exposure to indoor or outdoor air pollution is more prevalent in the poor and may hasten the progression of pulmonary disease. Cigarette smoking is more prevalent in low SES groups,11 and exposure to environmental tobacco smoke has been linked to decreased pulmonary function in children with CF.12 Respiratory virus infections, particularly with respiratory syncytial virus,13 may occur earlier and more frequently in economically disadvantaged children, initiating airway inflammation at a younger age.
Increased stress is another hypothesized cause of greater disease severity in the poor.14 Psychological stress affects immune function and increases susceptibility to infection.15 While stress might impair immune function or resilience, for children with CF it is probably most important to recognize that parental experience of stress may lead to impaired personal and family function, leading to decreased adherence to treatment.16 Children usually rely on parents or caregivers for daily management of their disease. Adherence might also be worse in dysfunctional families because of less parental involvement and education about the disease.17,18 Treatment adherence for children with CF can be measured by multimethod assessment19 (self‐report, prescription refill history, daily phone diary, electronic monitoring and home visits). Accurately measuring treatment adherence in CF children is an important step in developing effective interventions.
The Children Act 2004 places a legal duty on services to ensure that every child, whatever their background or circumstances, should have the support they need to be healthy and stay safe. It is our paramount responsibility to ensure welfare of all children. It may be appropriate for the multidisciplinary CF team (including psychologist and social worker) to work with the disadvantaged/dysfunctional families and provide appropriate psychosocial intervention and non‐medical support. Supporting the child and family to achieve these goals is the favoured approach, although in extreme cases of neglect, earlier consideration of alternative care arrangements may be of benefit. The fact that, in general, a child is best with the birth family, that the significant trauma for the child and family of such separation, and the difficulty of finding foster care make decisions to remove the child to another family fraught with difficulty. This is reflected in our case by the fact that despite massive improvement in health, our case still wants her mother and can't wait for her to return.
Our case emphasizes that factors other than genotype have a significant effect on CF phenotype. This may have implications regarding therapy for, and further research into the mechanism of CF lung disease. As a public health measure, it might be appropriate to offer this group more intense standard treatment (additional supervised treatment, e.g. antibiotics, physiotherapy at school, mentoring treatment) than to a lower risk group, but the efficacy of this approach would need to be proven. On the other hand, if the specific causes of the link between dysfunctional families and more severe lung disease in CF are identified, then more specific effective therapies could be offered to these patients. This knowledge would also lead to improvements in outcome for others who are not of low SES but who may, nevertheless, be exposed to any of these risk factors. Second, because of its association with adverse health outcomes, low SES should be considered as a potential confounder in all clinical research trials, with appropriate adjustments made for it. Additionally, a better understanding of environmental risk factors and how they mediate adverse outcomes would provide insight into the pathogenesis of CF lung disease.
Footnotes
DECLARATIONS —
Competing interests None declared
Funding None
Ethical approval Not applicable
Guarantors AG and MR
Contributorship All authors contributed equally
Acknowledgements
The authors would like to thank all the members of the CF team at the Royal Brompton Hospital
References
- 1.Davis PB, Drumm M, Konstan MW. Cystic fibrosis – state of the art. Am J Respir Crit Care Med 1996;154:1229–56 [DOI] [PubMed] [Google Scholar]
- 2.The Cystic Fibrosis Genotype‐Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med 1993;329:1308–13 [DOI] [PubMed] [Google Scholar]
- 3.Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 2000;67(2):117–33 [DOI] [PubMed] [Google Scholar]
- 4.Dean M, Santis G. Heterogeneity in the severity of cystic fibrosis and the role of CFTR gene mutations. Hum Genet 1994;93:364–368 [DOI] [PubMed] [Google Scholar]
- 5.Adler NE, Boyce WT, Chesney MA, et al. Socioeconomic inequalities in health. JAMA 1993;269:3140–5 [PubMed] [Google Scholar]
- 6.Mackenbach JP, Kunst AE, Cavelaars AE, et al. Socioeconomic inequalities in morbidity and mortality in western Europe: the EU Working Group on Socioeconomic Inequalities in Health. Lancet 1997;349:1655–9 [DOI] [PubMed] [Google Scholar]
- 7.Britton J. Effects of social class, sex, and region of residence on age at death from cystic fibrosis. BMJ 1989;298:483–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schechter MS, Margolis PA. Relationship between socioeconomic status and disease severity in cystic fibrosis. J Pediatr 1998;132:260–4 [DOI] [PubMed] [Google Scholar]
- 9.Curtis JR, Burke W, Kassner A, et al. Absence of health insurance is associated with decreased life expectancy in patients with cystic fibrosis. Am J Respir Crit Care Med 1997;155:1921–4 [DOI] [PubMed] [Google Scholar]
- 10.Schechter MS, Shelton BJ, Margolis PA, et al. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med 2001;163:1331–7 [DOI] [PubMed] [Google Scholar]
- 11.Haire‐Joshu D, Morgan G, Fisher JEB. Determinants of cigarette smoking. Clin Chest Med 1991;4:711–24 [PubMed] [Google Scholar]
- 12.Kovesi T, Corey M, Levison H. Passive smoking and lung function in cystic fibrosis. Am Rev Respir Dis 1993;148:1266–71 [DOI] [PubMed] [Google Scholar]
- 13.Abman SH, Ogle JW, Butler‐Simon N, et al. Role of respiratory syncytial virus in early hospitalization for respiratory distress of young infants with cystic fibrosis J Pediatr 1988;113:826–30 [DOI] [PubMed] [Google Scholar]
- 14.Bor DH, Epstein PR. Pathogenesis of respiratory infection in the disadvantaged. Semin Respir Infect 1991;6:194–203 [PubMed] [Google Scholar]
- 15.Cohen S, Tyrrell DA, Smith AP. Psychological stress in humans and susceptibility to the common cold. N Engl J Med 1991;325:606–12 [DOI] [PubMed] [Google Scholar]
- 16.Dimissie K, Ernst P, Hanley JA, et al. Socioeconomic status and lung function among primary school children in Canada. Am J Respir Crit Care Med 1996;153:719–23 [DOI] [PubMed] [Google Scholar]
- 17.Henley LD, Hill ID. Errors, gaps, and misconceptions in the disease related knowledge of cystic fibrosis patients and their families. Pediatrics 1990;85:1008–14 [PubMed] [Google Scholar]
- 18.DiMatteo MR. Enhancing patient adherence to medical recommendations. JAMA 1994;271:79–82 [DOI] [PubMed] [Google Scholar]
- 19.Modi AC, Lim CS, Yu N, et al. A multi‐method assessment of treatment adherence for children with cystic fibrosis J Cystic Fibrosis 2006;5:177–85 [DOI] [PubMed] [Google Scholar]