

Abstract
Stress hormones significantly impact dendritic cell (DC) activation and function, typically in a suppressive fashion. However, a social stressor termed social disruption (SDR) has been shown to induce an increase in inflammatory responses and a state of glucocorticoid resistance in splenic CD11b+ monocytes. These experiments were designed to determine the effects of SDR on DC activation, toll-like receptor-induced cytokine secretion and glucocorticoid sensitivity. Compared to cells obtained from control animals, splenic DCs from SDR mice displayed increased levels of MHC I, CD80 and CD44, indicative of an activated phenotype. In addition, DCs from SDR mice produced comparatively higher TNF-α, IL-6 and IL-10 in response to in vitro stimulation with LPS and CpG DNA. Increased amounts of TNF-α and IL-6 were also evident in SDR-DC cultures stimulated with poly(I:C). Furthermore, as shown previously in CD11b+ monocytes, the CD11c+ DCs obtained from SDR mice were glucocorticoid resistant. Taken together, the data suggest that social stress, in the absence of any immune challenge, activates DCs, increases DC cytokine secretion in response to toll specific stimuli and renders DCs glucocorticoid resistant.
Keywords: Dendritic Cell, Innate Immunity, Social Stress, Psychoneuroimmunology
Introduction
Dendritic cells (DCs) are a critical bridge between the innate and adaptive immune responses. Their acquisition and presentation of antigen via MHC and their co-stimulatory signals, as well as their production of immunomodulatory cytokines are essential for antigen-specific T-cell activation and clonal expansion (Banchereau et al., 2000; Lenschow et al., 1996). Prior to activation, immature DCs express a variety of receptors, including toll-like receptors (TLRs), cytokine receptors, and hormone receptors. These receptors ensure that DCs are highly sensitive and reactive to localized microenvironmental cues (Maestroni, 2006; Mazzoni and Segal, 2004; Pulendran, 2005; Sabatte et al., 2007). In the absence of exogenous antigen, immature DCs can respond to changes in their environment (e.g. shifts in cytokine or hormone secretion, direct contact with infiltrating or activated immune cells) and become activated via signaling through one or more of its various receptors (Munz et al., 2005; Sabatte et al., 2007). Once a signal is received, DCs can either become immunogenic or tolerogenic, depending on maturation status and the nature of environmental stimulus, which will lead to alterations in both innate and adaptive immunity (Bros et al., 2007; Grohmann et al., 2001; Lutz and Schuler, 2002). Upon antigen encounter, activated DCs rapidly mature and can then effectively prime T cells and direct differentiation and clonal expansion.
Exposure to stressful stimuli (e.g. environmental, physical, social or psychological stress) can cause changes in the local microenvironment of DCs due to the release of potent neuro-immune mediators including cytokines, glucocorticoids and catecholamines. As a consequence, the stress response has a significant impact on overall immunity (Stratakis and Chrousos, 1995). More often than not, the effects of stress hormones have been reported to be immunosuppressive. For example, DCs exposed to corticosterone are poor APCs because of impaired MHC-antigen complex formation (Elftman et al., 2007; Truckenmiller et al., 2005). Exposure of immature DCs to glucocorticoids during activation and maturation cause them to become tolerogenic as a result of reduced co-stimulatory molecule expression and decreased proinflammatory cytokine secretion (Moser et al., 1995). In addition, the number of DCs and the level of TLR expression on plasmacytoid DCs, are significantly decreased in dexamethasone (Dex)-treated cultures (Abe 2006). Glucocorticoids also impair maturation and function of DCs when TLR expression and binding are increased (which is normally a trigger for DC maturation) (Rozkova et al., 2006).
However, some types of stressors can selectively enhance specific components of both innate and adaptive immunity. Acute stress, induced by short term, low intensity stressors, activates the innate and adaptive immune system. Activation results in increased inflammatory cytokine gene expression, maturation and trafficking of DCs, increased macrophage number, and increased T cell recruitment and activation (Dhabhar and McEwen, 1997; Viswanathan et al., 2005). Acute psychological stress has also been shown to enhance DC activation resulting in more efficient primary and memory Ag-specific T cell immune responses (Saint-Mezard et al., 2003). A social stressor termed social disruption stress (SDR), results in functional glucocorticoid resistance in splenic CD11b+ monocyte populations, and enhances inflammatory responses in mice (Stark et al., 2001). In addition, when compared to cells from non-stressed home cage control mice, CD11b+ cells from SDR mice are more sensitive to toll specific stimuli and produce significantly higher levels of cytokines (TNF-α and IL-6) after stimulation in culture with LPS (Avitsur et al., 2005; Avitsur et al., 2002; Stark et al., 2002). CD11b+ macrophages from SDR mice have enhanced microbicidal activity against E. coli (Bailey et al., 2007). Because assessing the influence of SDR on CD11b+ monocytes/macrophages has been the focus to this point, little is known with regard to the effects of SDR on CD11c+ DCs. The following study was designed to determine whether the experience of repeated social defeat during SDR will promote DC activation and enhance DC function. This study demonstrates that SDR alone can activate DCs, prime DCs for enhanced cytokine secretion in response to toll specific stimuli, and induce a state of glucocorticoid resistance in DCs.
Methods
Animals
Male C57BL/6 mice between the ages of 6–8 weeks were purchased from Charles River Laboratories (Wilmington, MA) and allowed to acclimate to the animal facility for 1 week prior to any procedures. Mice were housed in groups of 3 per cage and were maintained on a 12 h light/dark schedule with the lights on at 0600 h. Aggressive intruders were individually housed. Food and water were available ad libitum, and all procedures with animals were approved by the Ohio State University Laboratory Animal Care and Use committee.
Social disruption
Social disruption or SDR was employed as previously described (Avitsur et al., 2001). Briefly, aggressive intruder male mice were introduced into cages of established cohorts of three mice for 2h between 1630 and 1830 for six consecutive days. During the SDR period, the aggressor attacked and defeated all of the resident mice within 20 minutes of introduction into the cage. If fighting did not begin within the first 5 minutes after the intruder was introduced, the aggressor was removed and a new aggressor was introduced. At the end of the 2 hr SDR period, the aggressor was returned to its cage and the residents were left undisturbed until the next day. Eighteen hours after the 6th cycle of SDR, the resident mice were sacrificed and tissues were harvested for experimental use. At this time, mice were re-assessed for the extent of wounding as a consequence of fighting. Scoring was based on a 3 point scale, with 0 indicating no wounds, 1 indicating 1-3 superficial skin wounds, 2 indicating 4-5 superficial skin wounds, and 3 indicating > 5 superficial skin wounds. In no case did wounding exceed superficial skin wounds. Animals were also monitored/assessed for infection of existing wounds and no infection was observed through the course of the experiments. It should be noted that aggressive/intruder mice were not used for experimental analysis.
Preparation of Cells
Mice were housed in groups of 3 and cagemate spleens were pooled in order to obtain enough DCs for experiments performed. Spleens from each group (SDR or HCC) of animals were pooled in threes in ice-cold Hanks' balanced salt solution (HBSS) and mechanically disrupted to obtain single cell suspensions. Red blood cells were lysed by adding 2 ml of room temperature lysis buffer (0.16 M NH4Cl, 10 mM KHCO3, and 0.13mM EDTA) for 2 min, followed by one wash with HBSS/10% heat-inactivated fetal bovine serum (FBS). Each cell pellet was resuspended in HBSS, filtered and washed a final time in HBSS. Cells were counted and samples were resuspended (2.5 × 106 cells/ml) in supplemented RPMI medium (10% heat-inactivated FBS, 0.075% sodium bicarbonate, 10 mM HEPES buffer, 100 U/ml penicillin G, 100 mg/ml streptomycin sulfate, 1.5 mM L-glutamine, and 0.0035% 2-mercaptoethanol).
Enrichment of CD11c+ cells
Dendritic cells were selected from cell suspensions using Pan DC MicroBeads (Miltenyi Biotec, Auburn, CA) according to the manufacturer's protocol. MicroBeads are superparamagnetic particles of approximately 50 nanometers in diameter that are biodegradable and decompose when cells are cultured. Binding of the antibody portion of the MicroBead to the cell of interest does not typically activate isolated cells, influence their function, or affect their viability (Badell et al., 2000; Lyons et al., 2007). Therefore, no bead detachment is required post-selection and positively isolated cells can be used immediately for experimental purposes. In addition, studies have shown that positive selection of cells yields a more pure population than negatively selected cells (Lyons et al., 2007).
Briefly, 100μl Pan DC microbeads were added to 108 total cells in 400μl MACs buffer, and samples were vortexed and incubated for 15 min at 4°C. Labeled cells were then positively selected by magnetic separation using MS MACs columns and a MACs separator (Miltenyi Biotec). The negative fraction containing CD11c- cells was collected in a tube, and the column was washed three times with 500μl of MACs buffer. After the column was removed from the MACs separator, the positive fraction containing CD11c+ cells was eluted and collected in 1 ml of MACs buffer. Flow cytometric analysis showed greater than 95% purity.
Flow cytometric analysis
Single cell suspensions derived from HCC or SDR spleen tissue (1 ×106 cells per sample) were incubated with 1 μg each of fluorescently-labeled monoclonal antibodies (or the appropriate isotype controls) after a 20-min blocking step with Fc receptor block. Antibody labeling was performed at 4°C for 30 min. The cells were then washed twice in PBS containing 1% FBS and 0.09% NaN3 and fixed with 1% paraformaldehyde. All antibodies were obtained from BD PharMingen (San Diego, CA), including PE-labeled anti-CD11c, APC-labeled anti-CD11b, and FITC-labeled anti-CD80, anti-CD86, anti-MHC I (H-2Db, clone KH95), anti-MHC II (I-A/I-E, clone 2G9) or anti-CD44 (Pharmingen). Lymphocytes were gated based on forward versus side scatter and a total of 10,000 events were analyzed on a FACSCalibur flow cytometer using Cell Quest and Cell Quest Pro analysis software (Becton-Dickenson, San Jose, CA).
Cytokine measurement by Cytometric Bead Array (CBA)
Supernatants were harvested at 18 hours post culture from 96-well plate cultures of CD11c+ cells (2 × 105cells/well) stimulated with culture medium alone, LPS from Escherichia coli (1 μg/ml) (serotype O111:B4; Sigma-Aldrich, St. Louis, MO, USA) poly(I:C) (100μg/ml) (Invitrogen,, San Diego, CA) or CpG DNA (4 μg/ml) (Cell Sciences, Canton, MA). A dose response for each mitogen was performed to determine optimal in vitro concentrations. In addition, a time course analysis was done to determine the optimal time for cytokine measurement. The relative levels of secreted cytokines (TNF-α, IL-12p70, IL-10, IFN-γ, MIP-1α and IL-6) were detected simultaneously and with high sensitivity using the mouse Inflammatory Cytokine CBA kit according to the manufacturer's instructions (BD PharMingen, San Diego, CA). Briefly, 50 μl of each sample was mixed with 50 μl of mixed capture beads and 50 μl of the Inflammation PE detection reagent. The samples were incubated at room temperature for 3 h in the dark. After incubation with the PE detection reagent, the samples were washed once and resuspended in 300 μl of wash buffer before acquisition on the FACSCalibur (BD Biosciences, Sunnyvale, CA). Data were analyzed using CBA software (BD PharMingen). Using the mixed cytokine standard provided in each kit, standard curves were generated for each cytokine. The concentration for each cytokine in cell supernatants was determined by extrapolation from the corresponding standard curve. The range of detection was 20–5000 pg/ml for each cytokine measured by CBA. To address intra-assay performance of the CBA Mouse Inflammation cytokine assay, ten replicates of each of three different levels of IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70 were tested. To address inter-assay performance of the CBA Mouse Inflammation kit, three different levels of IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70 were tested in four experiments. The intra-assay average percentage CV and inter-assay percentage CV for each cytokine was as follows: IL-6 was 4.6% and 9.6% respectively, IL-10 (5.6% and 9.7%), MCP-1 (4.6% and 8.7%), IFN-g (4.0% and 6.3%), TNF-a (3.7% and 8.0%), IL-12p70 (8.0% and 5.7%). It should be noted that no significant differences in IFN-γ, MIP-1α and IL-12p70 were noted between groups with any mitogen tested in any experiment.
Cell viability assay
Cells were sorted and separated as stated above. CD11c+ DCs (2 × 105 cells per well) were plated in triplicate in a 96-well plate and stimulated with RPMI media or 1μg/ml LPS (Sigma) and increasing concentrations of corticosterone at a dose range of 0.005-5μM to determine glucocorticoid sensitivity. Corticosterone was obtained commercially from Sigma and dissolved in ethanol; the final concentration of ethanol in the culture medium was 0.2%. After plating samples in triplicate, culture plates were incubated at 37°C in 5% CO2. After 48 hours in culture, the Cell Titer 96 aqueous nonradioactive proliferation assay (Promega; Madison, WI) was used to determine cell viability. The tetrazolium substrate solution (20 μl) was added to each well and the plates were incubated at 37°C in 5% CO2 for 3 h, and the resulting color changes were quantified by obtaining optical density (OD) readings at 490 nm on an ELISA plate reader. To account for differences in background activity of cells, the mean OD of three wells containing RPMI with 0.2% ethanol were subtracted for a given treatment from each of the corresponding LPS-stimulated values.
Statistical Analysis
Results are reported as mean +/- SEM. The data were analyzed by a Student's t test or a three way or mixed parameter ANOVA with a post hoc t test with Bonferroni correction. Groups were considered statistically significant at p<0.05.
Results
CD11c+ cells from SDR mice display an activated profile
Previous studies from our laboratory have shown that SDR increases the number and function of CD11b+ monocytes in splenomegalic animals. In addition, the CD11b+ cells have been determined to be glucocorticoid insensitive (Bailey et al., 2007; Stark et al., 2001). This study was designed to further explore the effects of SDR on innate immunity with a focus on DCs. The first set of studies was designed to determine the effect of SDR on DC activation. Activation status and maturation of DCs were determined by flow cytometric analysis of cell surface marker expression. The levels of MHC I and MHC II as well as CD44, CD80 and CD86 were determined on purified CD11c+ DCs from the spleens of SDR or HCC mice. DCs from SDR mice displayed significantly higher levels of CD80 (t(4) = 5.32, p<0.01) and MHC I (t(4) = 5.14, p<0.01) on the cell surface and increased staining intensity on a per cell basis when compared to HCC CD11c+ cells (Figure 1A-D). In sharp contrast to CD80, the percentage of cells expressing CD86 and the intensity of CD86 expression on a per cell basis was significantly decreased (t(4) = 6.88, p<0.01) in SDR DCs (Figure 1E-F). There were no comparative differences in the level or intensity of MHC II expression, which is constitutively expressed on DCs; expression was high in both SDR and HCC groups (data not shown). DCs from the spleens of SDR mice also had significantly elevated levels of CD44 on the cell surface (t(4) = 6.04, p<0.01) and a shift in intensity (Figure G-H). Changes in the level of expression of MHC, CD80, CD86 and CD44 are not only indicative of DC activation after exposure to SDR, but also suggest that SDR DCs may be more efficient at presenting antigen to T cells than HCC DCs.
Figure 1. DCs from SDR mice display an increase in activation markers.
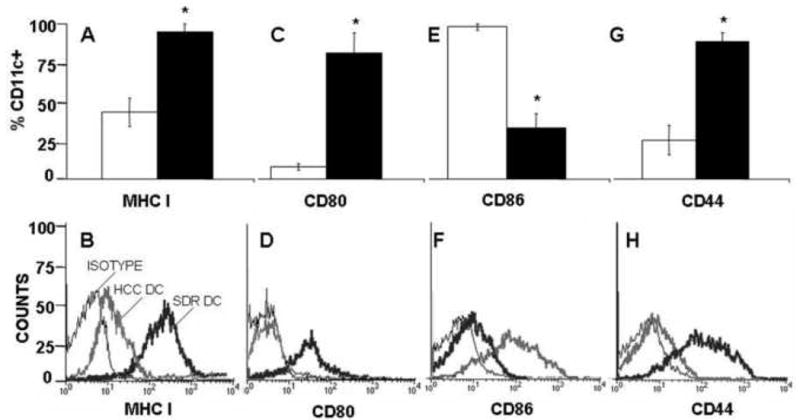
18 hours after the completion of the final cycle of SDR, CD11c+ cells isolated from the spleens of HCC(□) or SDR (■) mice were stained with anti-CD11c (A-H) and anti-MHC I (A-B), or anti-CD80 (C-D), or anti-CD86 (E-F) or anti-CD44 (G-H) to determine the activation status. Data are presented as the percent of CD11c/MHCI double positive cells (A), CD11c/CD80 double positive cells (C), CD11c/CD86 double positive cells (E) and CD11c/CD44 double positive cells (G) from purified CD11c+ spleen cells from HCC or SDR DC mice. The second panel represents the fluorescence intensity on a per cell basis on CD11c+ cells for MHC I (B), CD80 (D), CD86 (F) or CD44 (H). Asterisks (*) indicate that the difference between groups is statistically significant (p < 0.01) in repeated experiments. Results are the combined data of three separate experiments. (n= 12 mice per group, per experiment).
Flow analysis also revealed that approximately 80% of the CD11c+ population was also CD11b+ (data not shown), indicating a conventional dendritic cell phenotype. Few CD11c+ cells were B220+, and the number of CD11c+ cells expressing B220 was relatively equal in both groups, showing that SDR did not change the proportion of plasmacytoid DCs in the spleen. Similar results were obtained in the peripheral lymph nodes (data not shown). As in previous studies utilizing the SDR model to study the effects of stress on the immune response, mice exposed to SDR were splenomegalic, with increased cellularity in the spleen. However, unlike CD11b+ monocytes, the number of CD11c+ DCs was not significantly altered by SDR (data not shown).
SDR DCs secrete more cytokines in response to toll specific stimuli
In order to determine the effect of SDR on the response of DCs to toll specific stimuli, inflammatory cytokine production was examined. CD11c+ cells were isolated from the spleens of SDR and HCC mice and were cultured with LPS (1μg/ml), poly (I:C) (100μg/ml) or CpG DNA (4μg/ml); these are ligands for TLR4, TLR3, and TLR9, respectively. Following stimulation with each toll specific ligand, the cytokine response in the supernatants of purified SDR and HCC DCs was measured by cytometric bead array at 18 hours post culture. When left untreated in culture medium alone, both HCC and SDR DC cell cultures had no detectable levels of TNF-α (Figure 2A-C), IL-6 or IL-10 in culture (data not shown). In response to each toll specific stimuli, DC cell cultures from SDR mice had significantly higher levels of TNF-α and IL-6 and, in some cases, IL-10 at 18h (Figures 2-5). However, only TNF-α secretion by SDR DCs was significantly increased at 4h in response to LPS (F(1,8) = 41.03, p<0.001), Poly I:C (F(1, 8) = 62.42, p<0.001) and CpG DNA (F(1,8) = 99.33, p<0.001) (Figures 2A-2C).
Figure 2. TNF-α secretion is more sensitive to mitogen stimulation in SDR DCs.
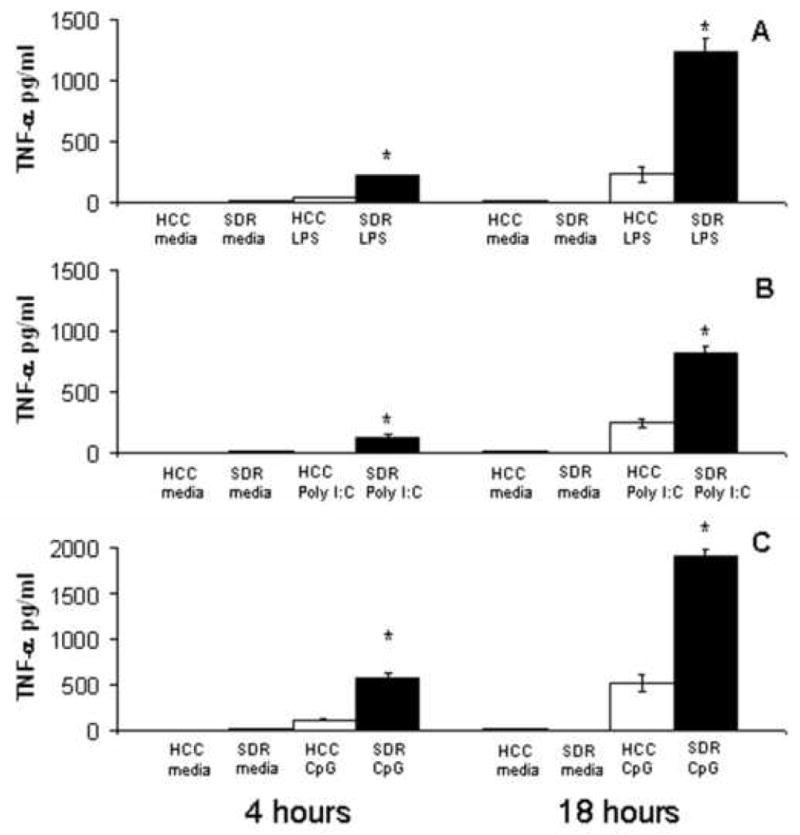
CD11c+ cells from HCC (□) or SDR (■) mice were stimulated in culture with media alone, 1μg/mL LPS, 100μg/mL Poly(I:C) or 4μg/mL CpG DNA. At 4h and 18h post culture, supernatants were analyzed by cytometric bead array. Data are presented as the amount of TNF-α in the supernatants of HCC or SDR DC cultures. Asterisks (*) indicate that the difference between groups is statistically significant (*p < 0.001) in repeated experiments. Results are representative of three experiments. (n= 12 mice per group, per experiment).
Figure 5. Cytokine secretion in response to CpG DNA stimulation is significantly increased in SDR DCs.
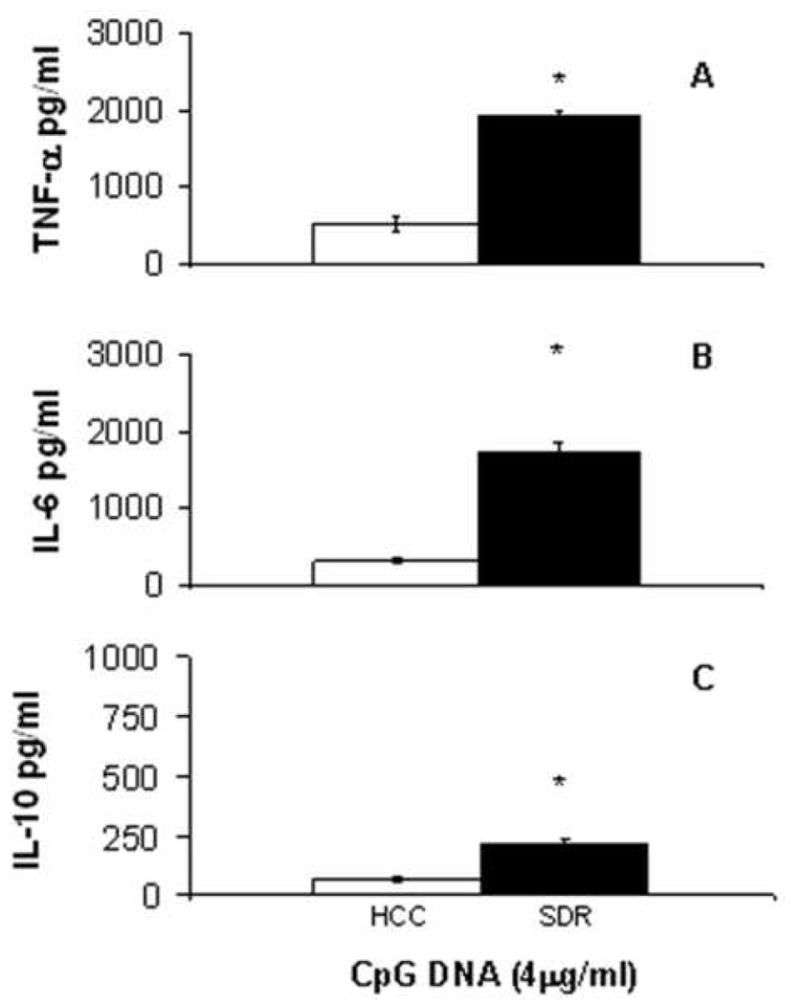
CD11c+ cells from HCC (□) or SDR (■) mice were stimulated in culture with 4μg/mL CpG DNA. At 18h post culture supernatants were analyzed by cytometric bead array. Data are presented as the amount of TNF-α (A), IL-6 (B) and IL-10 (C) in the supernatants of HCC or SDR DC cultures. Asterisks (*) indicate that the difference between groups is statistically significant (p < 0.01) in repeated experiments. Results are representative of three experiments. (n= 12 mice per group, per experiment).
With LPS stimulation, there were significant increases of TNF-α (t(4) = 7.42, p<0.01) and IL-6 (t(4) = 4.67, p<0.01) production by SDR DCs compared to HCC DCs (Figure 3A-3B). IL-10 secretion by SDR DCs was also significantly elevated (t(4) = 3.34, p<0.03) in cultures stimulated with LPS (Figure 3C).
Figure 3. Cytokine secretion in response to LPS stimulation is significantly increased in SDR DCs.
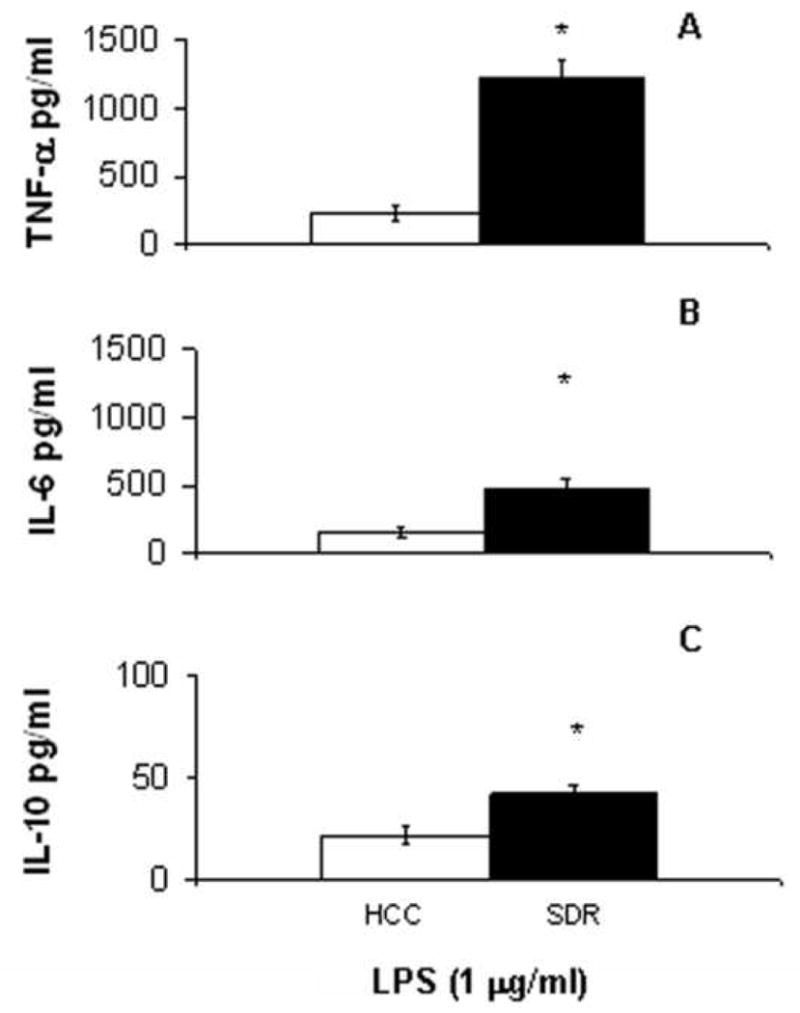
CD11c+ cells from HCC (□) or SDR (■) mice were stimulated in culture with 1 μg/mL LPS. At 18h post culture supernatants were analyzed by cytometric bead array. Data are presented as the amount of TNF-α (A), IL-6 (B) and IL-10 (C) in the supernatants of HCC or SDR DC cultures. Asterisks (*) indicate that the difference between groups is statistically significant (*p < 0.01, **p < 0.03) in repeated experiments. Results are representative of three experiments. (n= 12 mice per group, per experiment).
With poly(I:C) stimulation, there was a significant increase (t(4) = 8.85, p<0.01) in TNF-α production by SDR DCs (Figure 4A). IL-6 secretion by SDR DCs was also significantly elevated (t(4) = 6.73, p<0.01) with poly(I:C) stimulation (Figure 4B). However, no significant differences in IL-10 secretion from SDR DCs and HCC DCs were found after stimulation with poly(I:C) (Figure 4C).
Figure 4. Cytokine secretion in response to poly(I:C) stimulation is significantly increased in SDR DCs.
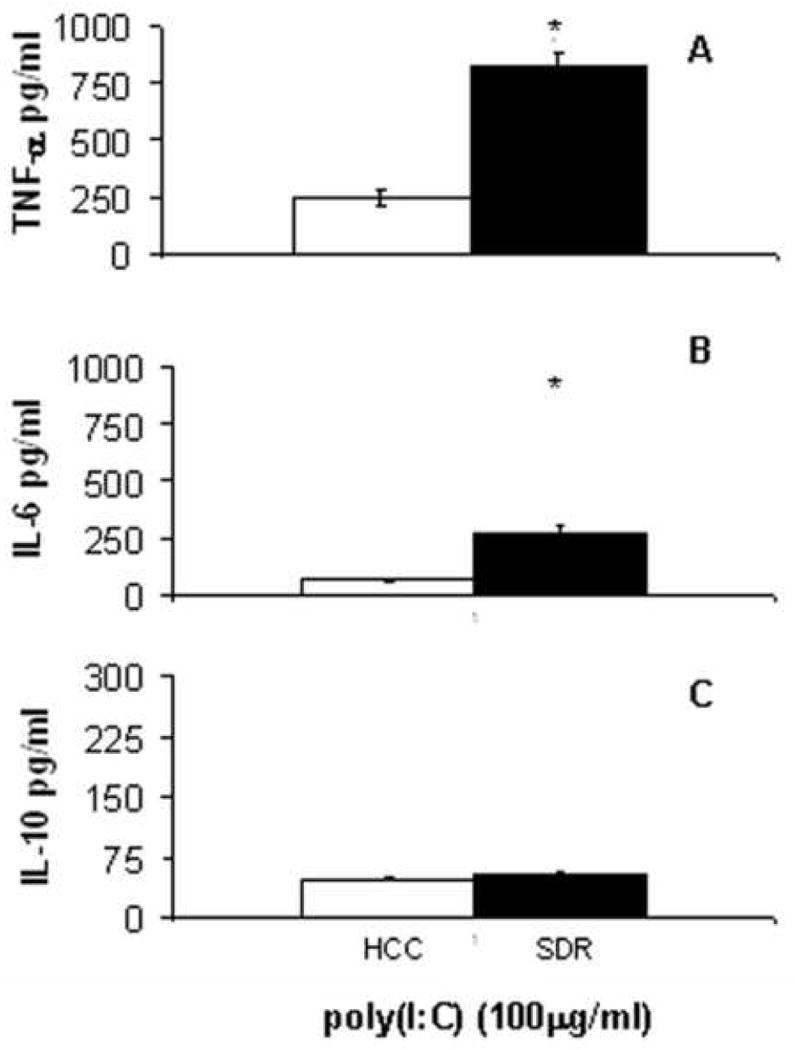
CD11c+ cells from HCC (□) or SDR (■) mice were stimulated in culture with 100μg/mL Poly(I:C). At 18h post culture supernatants were analyzed by cytometric bead array. Data are presented as the amount of TNF-α (A), IL-6 (B) and IL-10 (C) in the supernatants of HCC or SDR DC cultures. Asterisks (*) indicate that the difference between groups is statistically significant (p < 0.01) in repeated experiments. Results are representative of three experiments. (n= 12 mice per group, per experiment).
After stimulation with CpG DNA, there was a significant increase (t(4) = 11.59, p<0.01) of TNF-α by SDR DCs compared to HCC DCs (Figure 5A). IL-6 (t(4) = 10.58, p<0.01) and IL-10 (t(4) = 6.65, p<0.01) secretion by SDR DCs, when compared to HCC DCs, was also significantly elevated 18 hours post culture with CpG DNA (Figure 5B-5C).
CD11c+ cells from SDR mice are glucocorticoid resistant
As mentioned previously, SDR induces GC resistance in CD11b+ cells (Stark et al., 2001). We hypothesized that if GC resistance was induced in the CD11c+ DC population, it might provide a mechanism for enhanced activation and cytokine production in response to TLR stimulation. Therefore, GC sensitivity of the CD11C+ cell population was assessed. Cells from SDR and HCC mice were plated with 2ug/mL LPS and increasing concentrations of corticosterone (0.005–5 μM). The data showed that the 48-hour viability of CD11c+ cells from control animals decreased as the concentration of corticosterone increased. In contrast, CD11c+ cells from SDR mice showed a statistically significant decrease in sensitivity to glucocorticoid-induced cell death (F(5,50) = 33.60, p < 0.001). At the highest corticosterone concentrations, 1-5μM, approximately 50% of the SDR DCs survived compared to approximately 10% of the HCC DCs (Figure 6). In addition, significant differences were evident at the (0.05-1μM) concentrations of corticosterone.
Figure 6. SDR DCs are glucocorticoid resistant.
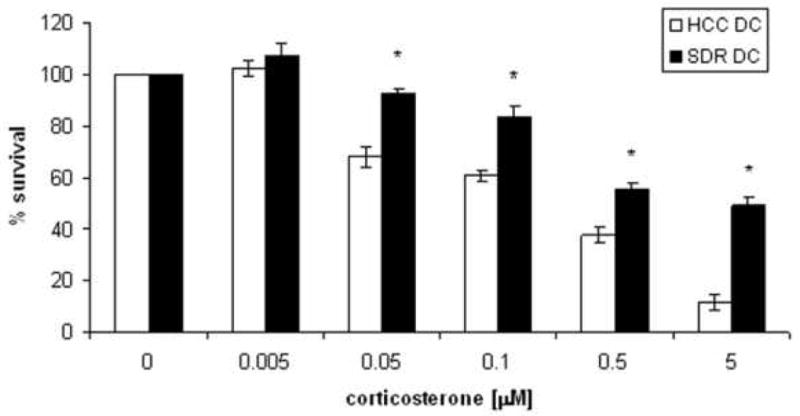
CD11c+ dendritic cells from mice exposed to SDR or from non-stressed control mice (HCC) were stimulated with 1μg/ml LPS and increasing doses of (0.005-5μM) corticosterone. White bars (□) represent CD11c+ cells from HCC, while black bars (■) represent CD11c+ cells from SDR mice. These are the combined data of two separate experiments. (n=12 mice per group, per experiment) (*p<0.001).
Discussion
These data indicate that the response to social stress significantly alters CD11c+ DCs in subordinate animals. In the absence of exogenous antigen, DCs exposed to SDR were activated, primed and glucocorticoid resistant (similar to previous reports of SDR-induced changes in CD11b + populations (Avitsur et al., 2005; Bailey et al., 2007). An increase in DC cell surface expression of MHCI, CD80 and CD44 was indicative of activation and increased DC cytokine production in response to toll specific stimuli indicated priming, both important events that will eventually lead to more efficient and more effective elimination of pathogens in vivo.
The upregulation of co-stimulatory molecules (e.g. CD80, CD86, CD40) on the surface of activated DCs is a critical event in the maturation of a DC. T cells stimulated in the absence of such signals from DCs become anergic (Lenschow et al., 1996). Additional surface markers induced upon activation of a DC, such as CD44, can lead to enhanced migration to sites of inflammation. CD44 also serves as an adhesion molecule that both attracts DCs to sites of inflammation and promotes interactions between DCs and T cells (Termeer et al., 2003; Termeer et al., 2001; Weiss et al., 1997). SDR induced the increased expression of MHCI, CD80 and CD44 on DCs, indicating that DCs from SDR mice would be strong APCs and drive a robust adaptive immune response to infection. Ongoing studies in our laboratory show that social stress increases the frequency of antigen specific CD8+ T cells in response to viral infection (Sheridan et. al, unpublished observations). A possible mechanism for such SDR specific increases in antigen specific immunity may be due to enhanced activation and priming of DCs in SDR mice.
Increased cytokine production by DCs and increased frequencies of co-stimulatory molecules on the surface of DCs will promote activation of naïve T cells; however, the type of co-stimulatory molecules on the DC surface can influence the nature of the T cell response (Guermonprez et al., 2002). SDR preferentially decreases CD86 and increases CD80 on the surface of DCs. CD86 is constitutively expressed early on the surface of DCs, whereas CD80 is upregulated later during activation and maturation of the DC. Both are capable of binding CD28 on the T cell surface and effectively activate the T cell when present simultaneously with MHC and antigen (Greenwald et al., 2005). While both CD80 and CD86 are important co-stimulatory molecules and both capable of enhancing T cell activation, each can have different functional implications after binding to their common ligands CD-28 and CTLA-4. CD80, not CD86, has been shown to be an important inducer of CD25 expression on CD8+ T cells, thereby promoting CD8+ T cell activation. (Pejawar-Gaddy and Alexander-Miller, 2006). CD80 has also been shown to effectively downregulate and control CTL responses upon binding CTLA-4, whereas CD86 has been shown to be more important in generating naïve Th1 and Th2 responses (Lang et al., 2002). Social stress alone was able to activate DCs and cause a shift in the CD86 to CD80 ratio which would predicate an enhanced CD8+ T cell response, and would support our observations of enhanced CD8+T cell responses to viral infection both in vitro and in vivo after SDR (Mays et al., unpublished observation). Additionally, regulatory T cells, potent suppressors of T cell function, are differentially modified by CD86 and CD80. Antibodies directed against CD86 enhanced suppression by CD4+CD25+ Treg cells. In contrast, blocking CD80 favored proliferative responses by hindering Treg suppression (Zheng et al., 2004). These results indicate that CD80, upon ligation of CTLA-4, would support T reg suppression of T cell responses. We are currently investigating the effects of SDR on the number and function of CD4+CD25+ T reg cells.
SDR enhanced cytokine secretion by DCs in culture. TNF-α secretion was present at 4h in SDR DC cultures with each TLR-ligand indicating an increase in sensitivity to TLR stimuli. Additionally, more TNF-α, IL-6 and in some cases, IL-10 were secreted by SDR DCs at 18h when compared to HCC DCs. Alterations in cytokine production coupled with DC activation has significant implications for the overall immune response to infection. This is primarily because cytokines produced by antigen presenting cells (APCs) in the local microenvironment in which T cells are matured, play a major role in directing the immune response to a given pathogen (de Jong et al., 2005; Kapsenberg, 2003; Nakamura et al., 1997; Steinman and Hemmi, 2006). Alterations of the APC itself, that specifically increases in the number of TLRs or the sensitivity of TLR signaling in DCs will likely result in more efficient DC maturation, migration, cytokine secretion and antigen presentation to T cells (Lehner et al., 2007; Watts et al., 2007).
In addition to SDR-induced increases in DC activation and cytokine production, CD11c+ DCs from SDR were glucocorticoid resistant. Under baseline conditions, DCs are highly sensitive to glucocorticoids. Studies in vivo and in vitro have shown that exposure of DCs to physiologic and pharmacologic levels of corticosterone can decrease survival, impair antigen presentation, interfere with migration and shift cytokine secretion (Elftman et al., 2007; Mainali and Tew, 2004; Moser et al., 1995; Saint-Mezard et al., 2003; Truckenmiller et al., 2006; Truckenmiller et al., 2005). These studies suggest that social stress, which activates the HPA axis and subsequently increases glucocorticoid production, could have deleterious effects on the exposed animals. However, SDR-induced glucocorticoid resistance in CD11c+ DCs may be responsible, in part, for increased cell surface expression of co-stimulatory molecules and increased cytokine production upon TLR ligation in the presence of increased glucocorticoids. Mechanisms driving differentiation, activation and glucocorticoid sensitivity of monocytes are currently underway.
Circulating cytokines are sufficient to induce monocyte differentiation and activation. Previous reports indicate increased plasma levels of IL-6 and IL-1β in SDR mice. Along with M-CSF, IL-6 promotes monocyte differentiation into macrophages rather than DCs (Chomarat et al., 2000). This may be, in part, an explanation for the increase in macrophage number and not DCs in the spleen after SDR. A possible factor influencing DC activation in the SDR model is the increased levels of IL-1β in the plasma, spleen and liver of SDR mice (Engler et. al. 2008). Elevation of IL-1β in the plasma and tissues of SDR mice may precede DC activation as IL-1β has been shown to have immunostimulatory effects on dendritic cells with downstream influences on T cell-mediated inflammation. Other factors associated with SDR are currently under investigation such as the influence of SDR-induced changes in norepinephrine release and glucocorticoid receptor binding on DC activation.
The stress response is a natural and unavoidable aspect of life for all organisms. A stressor can be physical, environmental, psychological or social. The physiological adaptation to stress is critical to the survival of a species. Although it tends to carry a negative connotation, stress can be beneficial. The “fight or flight” response is a direct consequence of physiological changes in response to stress. Stress, under some conditions, has been shown to enhance competition, immune function and ultimately, survival. Social stress has been shown in previous studies, and in this study, to activate the innate immune system and augment immune responses to infection. Determining the effects of social stress on the immune response will further our understanding of how stress impacts immunity, and pave the way to determining and developing new therapeutic targets and interventions for inflammatory and infectious diseases.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the technical assistance of Amy Hufnagle, Jon Wells and Jacqueline Verity and critical reading by Shannon Barnes. This work was supported by NIH grants NIMH/RO1MH046801-17 and NIDCR/T32DE014320-7 to JFS and DAP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avitsur R, Kavelaars A, Heijnen C, Sheridan JF. Social stress and the regulation of tumor necrosis factor-alpha secretion. Brain Behav Immun. 2005;19:311–317. doi: 10.1016/j.bbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Dhabhar FS, Sheridan JF. Social stress alters splenocyte phenotype and function. J Neuroimmunol. 2002;132:66–71. doi: 10.1016/s0165-5728(02)00310-7. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- Badell E, Oeuvray C, Moreno A, Soe S, van Rooijen N, Bouzidi A, Druilhe P. Human malaria in immunocompromised mice: an in vivo model to study defense mechanisms against Plasmodium falciparum. J Exp Med. 2000;192:1653–1660. doi: 10.1084/jem.192.11.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Engler H, Powell ND, Padgett DA, Sheridan JF. Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1180–1190. doi: 10.1152/ajpregu.00307.2007. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- Bros M, Jahrling F, Renzing A, Wiechmann N, Dang NA, Sutter A, Ross R, Knop J, Sudowe S, Reske-Kunz AB. A newly established murine immature dendritic cell line can be differentiated into a mature state, but exerts tolerogenic function upon maturation in the presence of glucocorticoid. Blood. 2007;109:3820–3829. doi: 10.1182/blood-2006-07-035576. [DOI] [PubMed] [Google Scholar]
- Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1:510–514. doi: 10.1038/82763. [DOI] [PubMed] [Google Scholar]
- de Jong EC, Smits HH, Kapsenberg ML. Dendritic cell-mediated T cell polarization. Springer Semin Immunopathol. 2005;26:289–307. doi: 10.1007/s00281-004-0167-1. [DOI] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Stiner-Jones LM, Quan N, Sheridan JF. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology. 2008;33:108–117. doi: 10.1016/j.psyneuen.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11:286–306. doi: 10.1006/brbi.1997.0508. [DOI] [PubMed] [Google Scholar]
- Elftman MD, Norbury CC, Bonneau RH, Truckenmiller ME. Corticosterone impairs dendritic cell maturation and function. Immunology. 2007;122:279–290. doi: 10.1111/j.1365-2567.2007.02637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–532. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Grohmann U, Fallarino F, Bianchi R, Belladonna ML, Vacca C, Orabona C, Uyttenhove C, Fioretti MC, Puccetti P. IL-6 inhibits the tolerogenic function of CD8 alpha+ dendritic cells expressing indoleamine 2,3-dioxygenase. J Immunol. 2001;167:708–714. doi: 10.4049/jimmunol.167.2.708. [DOI] [PubMed] [Google Scholar]
- Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Lang TJ, Nguyen P, Peach R, Gause WC, Via CS. In vivo CD86 blockade inhibits CD4+ T cell activation, whereas CD80 blockade potentiates CD8+ T cell activation and CTL effector function. J Immunol. 2002;168:3786–3792. doi: 10.4049/jimmunol.168.8.3786. [DOI] [PubMed] [Google Scholar]
- Lehner M, Morhart P, Stilper A, Petermann D, Weller P, Stachel D, Holter W. Efficient chemokine-dependent migration and primary and secondary IL-12 secretion by human dendritic cells stimulated through Toll-like receptors. J Immunother (1997) 2007;30:312–322. doi: 10.1097/01.cji.0000211345.11707.46. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23:445–449. doi: 10.1016/s1471-4906(02)02281-0. [DOI] [PubMed] [Google Scholar]
- Lyons PA, Koukoulaki M, Hatton A, Doggett K, Woffendin HB, Chaudhry AN, Smith KG. Microarray analysis of human leucocyte subsets: the advantages of positive selection and rapid purification. BMC Genomics. 2007;8:64. doi: 10.1186/1471-2164-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestroni GJ. Sympathetic nervous system influence on the innate immune response. Ann N Y Acad Sci. 2006;1069:195–207. doi: 10.1196/annals.1351.017. [DOI] [PubMed] [Google Scholar]
- Mainali ES, Tew JG. Dexamethasone selectively inhibits differentiation of cord blood stem cell derived-dendritic cell (DC) precursors into immature DCs. Cell Immunol. 2004;232:127–136. doi: 10.1016/j.cellimm.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Mazzoni A, Segal DM. Controlling the Toll road to dendritic cell polarization. J Leukoc Biol. 2004;75:721–730. doi: 10.1189/jlb.1003482. [DOI] [PubMed] [Google Scholar]
- Moser M, De Smedt T, Sornasse T, Tielemans F, Chentoufi AA, Muraille E, Van Mechelen M, Urbain J, Leo O. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur J Immunol. 1995;25:2818–2824. doi: 10.1002/eji.1830251016. [DOI] [PubMed] [Google Scholar]
- Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med. 2005;202:203–207. doi: 10.1084/jem.20050810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Kamogawa Y, Bottomly K, Flavell RA. Polarization of IL-4- and IFN-gamma-producing CD4+ T cells following activation of naive CD4+ T cells. J Immunol. 1997;158:1085–1094. [PubMed] [Google Scholar]
- Pejawar-Gaddy S, Alexander-Miller MA. Ligation of CD80 is critical for high-level CD25 expression on CD8+ T lymphocytes. J Immunol. 2006;177:4495–4502. doi: 10.4049/jimmunol.177.7.4495. [DOI] [PubMed] [Google Scholar]
- Pulendran B. Variegation of the immune response with dendritic cells and pathogen recognition receptors. J Immunol. 2005;174:2457–2465. doi: 10.4049/jimmunol.174.5.2457. [DOI] [PubMed] [Google Scholar]
- Rozkova D, Horvath R, Bartunkova J, Spisek R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin Immunol. 2006;120:260–271. doi: 10.1016/j.clim.2006.04.567. [DOI] [PubMed] [Google Scholar]
- Sabatte J, Maggini J, Nahmod K, Amaral MM, Martinez D, Salamone G, Ceballos A, Giordano M, Vermeulen M, Geffner J. Interplay of pathogens, cytokines and other stress signals in the regulation of dendritic cell function. Cytokine Growth Factor Rev. 2007;18:5–17. doi: 10.1016/j.cytogfr.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Saint-Mezard P, Chavagnac C, Bosset S, Ionescu M, Peyron E, Kaiserlian D, Nicolas JF, Berard F. Psychological stress exerts an adjuvant effect on skin dendritic cell functions in vivo. J Immunol. 2003;171:4073–4080. doi: 10.4049/jimmunol.171.8.4073. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF. Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. J Neuroimmunol. 2002;124:9–15. doi: 10.1016/s0165-5728(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1799–1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Chrousos GP. Neuroendocrinology and pathophysiology of the stress system. Ann N Y Acad Sci. 1995;771:1–18. doi: 10.1111/j.1749-6632.1995.tb44666.x. [DOI] [PubMed] [Google Scholar]
- Termeer C, Averbeck M, Hara H, Eibel H, Herrlich P, Sleeman J, Simon JC. Targeting dendritic cells with CD44 monoclonal antibodies selectively inhibits the proliferation of naive CD4+ T-helper cells by induction of FAS-independent T-cell apoptosis. Immunology. 2003;109:32–40. doi: 10.1046/j.1365-2567.2003.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termeer C, Johannsen H, Braun T, Renkl A, Ahrens T, Denfeld RW, Lappin MB, Weiss JM, Simon JC. The role of CD44 during CD40 ligand-induced dendritic cell clustering and maturation. J Leukoc Biol. 2001;70:715–722. [PubMed] [Google Scholar]
- Truckenmiller ME, Bonneau RH, Norbury CC. Stress presents a problem for dendritic cells: corticosterone and the fate of MHC class I antigen processing and presentation. Brain Behav Immun. 2006;20:210–218. doi: 10.1016/j.bbi.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Truckenmiller ME, Princiotta MF, Norbury CC, Bonneau RH. Corticosterone impairs MHC class I antigen presentation by dendritic cells via reduction of peptide generation. J Neuroimmunol. 2005;160:48–60. doi: 10.1016/j.jneuroim.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Viswanathan K, Daugherty C, Dhabhar FS. Stress as an endogenous adjuvant: augmentation of the immunization phase of cell-mediated immunity. Int Immunol. 2005;17:1059–1069. doi: 10.1093/intimm/dxh286. [DOI] [PubMed] [Google Scholar]
- Watts C, Zaru R, Prescott AR, Wallin RP, West MA. Proximal effects of Toll-like receptor activation in dendritic cells. Curr Opin Immunol. 2007;19:73–78. doi: 10.1016/j.coi.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Sleeman J, Renkl AC, Dittmar H, Termeer CC, Taxis S, Howells N, Hofmann M, Kohler G, Schopf E, Ponta H, Herrlich P, Simon JC. An essential role for CD44 variant isoforms in epidermal Langerhans cell and blood dendritic cell function. J Cell Biol. 1997;137:1137–1147. doi: 10.1083/jcb.137.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. 2004;172:2778–2784. doi: 10.4049/jimmunol.172.5.2778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.