

Abstract
αvβ8 integrin expression is restricted primarily to kidney, brain, and placenta. Targeted αv or β8 deletion is embryonic lethal due to defective placenta and brain angiogenesis, precluding investigation of kidney αvβ8 function. We find that kidney β8 is localized to glomerular mesangial cells, and expression is decreased in mouse models of glomerulosclerosis, suggesting that β8 regulates normal mesangial cell differentiation. To interrogate β8 signaling pathways, yeast two-hybrid and co-precipitation studies demonstrated β8 interaction with Rho guanine nucleotide dissociation inhibitor-1 (GDI). Selective β8 stimulation enhanced β8-GDI interaction as well as Rac1 (but not RhoA) activation and lamellipodia formation. Mesangial cells from itgb8−/− mice backcrossed to a genetic background that permitted survival, or gdi−/− mice, which develop glomerulosclerosis, demonstrated RhoA (but not Rac1) activity and α-smooth muscle actin assembly, which characterizes mesangial cell myofibroblast transformation in renal disease. To determine whether Rac1 directly modulates RhoA-associated myofibroblast differentiation, mesangial cells were transduced with inhibitory Rac peptide fused to human immunodeficiency virus-Tat, resulting in enhanced α-smooth muscle actin organization. We conclude that the β8 cytosolic tail in mesangial cells organizes a signaling complex that culminates in Rac1 activation to mediate wild-type differentiation, whereas decreased β8 activation shifts mesangial cells toward a RhoA-dependent myofibroblast phenotype.
Integrins are a family of α and β heterodimeric extracellular matrix receptors that mediate vital cell functions, including adhesion, migration, proliferation, and survival. Eighteen α and eight β subunits assemble to form 24 heterodimers. The β8 subunit is a 769-amino acid polypeptide that partners exclusively with αv (1). Although β8 expression was initially reported to be restricted to brain, placenta, and kidney (1), subsequent reports have documented its expression in lung and eyelid (2, 3). β8 gene deletion causes embryonic lethality due to impaired placenta and brain angiogenesis (4), which is similar to the phenotype observed in αv−/− mice (5), indicating that β8 is the major αv partner during development (5, 6). Kidney β8 localization and function have not been described.
Similar to other cell surface receptors, ligand binding to integrins leads to generation of intracellular signals and cytoskeletal rearrangement, i.e. outside-in signaling. The major site for soluble signaling molecule and cytoskeleton binding is the β-subunit cytosolic tail, and the α-subunit generally serves a regulatory function (7), although two groups have recently shown that α-subunits may also direct signal transduction pathways (8, 9). A unique feature of integrins is that independently generated intracellular signals can induce conformational changes to extracellular integrin domains, which enhances ligand affinity and/or extracellular matrix assembly, i.e. inside-out signaling. Neither outside-in nor inside-out signaling pathways have been described for αvβ8. The β8 cytoplasmic tail is 66 amino acids in length (10, 11) and is predicted to contain an α-helix that extends from residues 730−744 (psiphred) but no definable protein interaction domains (SMART, Motifscan) or sequence homology with other integrins (BLAST). β-Integrin domain swapping experiments showed that the β8 cytosolic tail does not directly affect cell adhesion (10), further suggesting that β8 signaling is distinct from other integrins.
The cytosolic tail of other β-integrin subunits has been shown to regulate the Ras superfamily of small molecular weight GTP-binding proteins (G-proteins). In particular, the Rho subfamily, which is commonly represented by three members, Rho, Rac, and Cdc42, is required for integrin-dependent cytoskeletal assembly (12). RhoA activation establishes stable, integrin-based focal adhesions at the cell periphery and is characterized in vitro by focal adhesion kinase activation. Rac1 mediates several aspects of integrin-dependent cell migration, including formation of membrane ruffles and lamellipodia at the leading edge of migrating cells and focal complexes at more internal sites. Integrin-associated Cdc42 function is required for development of actin-rich filopodia at the leading edge of migrating cells. Because β8 mediates nascent adhesion formation in migration (10, 13) and the β8 cytosolic tail lacks consensus focal adhesion kinase or talin binding sequences (14), we hypothesized that β8 preferentially activates Rac or Cdc42 signaling.
G-proteins act as binary switches, which cycle between the GDP-bound inactive state and the GTP-bound active conformation. Rho family G-proteins are regulated by direct binding to GTP exchange factors (GEFs),4 GTPase-activating proteins (GAPs), and Rho guanine nucleotide dissociation inhibitors (GDIs). Rho family G-proteins are also regulated by subcellular compartmentalization, as activation requires that lipid-modified G-proteins translocate to membrane microdomains for GTP loading and localization to receptors and effectors. It has been suggested that GDI bi-directionally regulates G-protein localization by chaperoning Rho family G-proteins to selective membrane signaling domains, which permits GEF-regulated GDP-GTP exchange as well as by removal of GDP-bound G-proteins from membrane sites and sequestration within the cytosol (15-18).
We find that under physiologic conditions, kidney β8 integrin is localized to glomerular mesangial cells (MCs), and animal models of glomerulosclerosis are associated with decreased MCβ8 expression. In vitro, MCβ8 stimulation leads to β8-GDI interaction, Rac1 activation, and suppression of RhoA-regulated, pathologic features. The data suggest that the β8 cytosolic tail provides specificity to G-protein signaling and regulates MC phenotype by spatially coordinating GDI-bound Rac1 to discrete domains containing Rac-GEFs and effector molecules.
MATERIALS AND METHODS
Reagents
Rabbit anti-human β8 integrin antisera has been previously characterized (19). Mouse monoclonal anti-β1, β3, and β5 antibodies and rabbit polyclonal anti-desmin antibody were purchased from Chemicon International (Temecula, CA). Monoclonal Tac antibodies were harvested from 7G7B6 mouse hybridoma cell line (ATCC, Manassas, VA) media. Rabbit polyclonal RhoA antibodies were obtained from Santa Cruz Bio-technology (Santa Cruz, CA). Mouse monoclonal anti-Rac1 and anti-Cdc42 antibodies and peroxidase-conjugated goat anti-mouse and anti-rabbit antibodies were purchased from BD Biosciences. Mouse monoclonal anti-α-smooth muscle actin (α-SMA) antibodies were purchased from Sigma. Fluorescein isothiocyanate-conjugated anti-mouse IgG and Texas Red-conjugated streptavidin were purchased from Vector Laboratories (Burlingame, CA). Alexa 568-conjugated phalloidin was purchased from Molecular Probes (Eugene, OR). DNA oligonucleotides were purchased from Operon Biotechnologies (Huntsville, AL). Purified vitronectin was purchased from Promega (Madison, WI); poly-l-lysine was from Sigma. Biotin-conjugated HIV-Tat, Rac (17-32)-HIV Tat, and Cdc42 (17-32)-HIV Tat fusion proteins were synthesized by New England Peptide (Gardner, MA). GST-PAK and GST-rhotekin fusion proteins were gifts from Dr. J. Collard (Netherlands Cancer Institute) and Dr. M. A. Schwartz (University of Virginia), respectively. Human interleukin-2 receptor (IL-2R)/β1, IL-2R/β3, and β1-integrin cDNAs were gifts from Dr. S. E. LaFlamme (Albany Medical College).
Animal Models
ROP-Os/+ mice, which were purchased from The Jackson Laboratory (Bar Harbor, ME), have a radiation-induced inversion mutation on chromosome 8, which results in a 50−75% decrease in nephron number and oligosyndactyly. ROP-Os/+ mice develop proteinuria and renal lesions, which resemble focal and segmental glomerulosclerosis over 3−6 months and ultimately die from renal failure (20). Mice overexpressing a non-infectious HIV transgene lacking gag and pol genes develop glomerular disease, which is indistinguishable from human HIV-associated nephropathy (HIVAN) (21). Characteristic features include the nephrotic syndrome, focal and segmental glomerulosclerosis, microcystic tubular dilatation, and progression to end-stage renal failure. All protocols were approved by the Institutional Animal Care and Use Committees at Case Western Reserve University.
In Situ Hybridization
Non-radioactive in situ hybridization was performed on mouse kidneys, which were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned to 5-μm thickness as described (22). To generate riboprobes, a 584-bp β8 integrin cDNA fragment from a highly conserved sequence between species was generated by PCR using 5′-ATGCACAATAATATAGAAAAA-3′ (nt 475−495) and 5′-TCCTTGTACCAATGAAACTG-3′ (nt 1039−1058) oligonucleotide primers from full-length β8 cDNA template (1) (a gift from Dr. S. L. Nishimura, University of California, San Francisco). The PCR product was bidirectionally subcloned into pCRII vector (Invitrogen). Digoxigenin-labeled non-hybridizing (sense) and hybridizing (antisense) riboprobes were synthesized using T7 RNA polymerase (Roche Applied Science) and quantified by spectrophotometry. In situ hybridizations were performed using equal concentrations of sense and antisense probe (42 °C, overnight). Hybridized probe was detected by labeling with an alkaline phosphatase-conjugated, anti-digoxigenin antibody.
Northern Blot Analysis
Established methods were followed for Northern blotting (13). Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA), and 20 μg per lane was fractionated on a denaturing 1.0% agarose, 0.67% formaldehyde gel, transferred to nylon membranes, and cross-linked by UV light exposure. To assess β8 integrin mRNA levels, full-length human β8 cDNA (1) was used as a template to generate probes, which were labeled with [α-32P]dCTP to specific activity ≥1.0 × 108 cpm/μg DNA (RTS Random Prime DNA labeling system, Invitrogen). Hybridization and high stringency washes were conducted according to previously described methods (13). Blots were stripped and re-hybridized with a random-primed oligonucleotide probe derived from chicken β-actin cDNA template (BD Biosciences) as a control for housekeeping gene expression.
Quantitative Reverse Transcription (RT)-PCR
Real-time quantitative RT-PCR analysis was employed to determine mRNA content in mouse kidney using LightCycler and SYBR Green technology (Roche Applied Science). Each analysis included mouse kidney samples of unknown mRNA concentration and, to confirm amplification specificity, random-primed RNA in the absence of RT or RNA template as negative controls. Total RNA (3 μg) was first treated with DNase I and then reverse-transcribed using the Thermoscript RT-PCR System (Invitrogen) in a 20-μl volume. Two-μl cDNA products were PCR-amplified in buffer containing 2 μl of LightCycler-Fast-Start DNA Master SYBR Green I mix (Roche Applied Science), 18 μl of hybridization buffer, 5 μm gene-specific primers, and 3 mm MgCl2. β8-Specific primers were 5′-ATGCACAATAATATAGAAAAA-3′ (nt 475−495) and 5′-TCCTTGTACCAATGAAACTG-3′ (nt 1039−1058). To quantify and validate RNA integrity, real-time PCR for β-actin internal standard was also performed using 5′-ATCTGGCACCACACCTTCTACAATGAGCTGCG-3′ (nt 333−364) and 5′-CGTCATACTCCTGCTTGCTGATCCACATCTGC-3′ (nt 1139−1169) primers (BD Biosciences Clontech, Palo Alto, CA). Thermocycling conditions were 95 °C for 10 min (initial denaturation), then 45 cycles at 95 °C for 10 s (denaturation), 52 °C for 20 s (annealing), and 72 °C for 30 s (extension). PCR products from all primer pairs were subjected to melting curve analysis and then analyzed by agarose gel electrophoresis to confirm amplification of a single product of the predicted size. LightCycler software was used to establish amplification cycle thresholds (CT), which demarcates the cycle number when sample fluorescence is above background and within the initial logarithmic phase. For each set of probes and primer pairs, serial dilutions validated that control and experimental gene amplification efficiencies were similar. Transcript quantification was determined by the comparative CT (ΔΔCT) method (23). Data are expressed as relative β8 mRNA abundance, which is defined as the β8-integrin level normalized to β-actin transcript content within the same sample.
Standard RT-PCR reactions for 30 cycles were employed to detect β-integrin expression in CHO-B2/v7 cells using primers 5′-ATGCACAATAATATAGAAAAA-3′ (nt 475−495) and 5′-TCCTTGTACCAATGAAACTG-3′ (nt 1039−1058) for β8, ATTGGCCTTGCCGCCCTGCTCATCTG-3′ (nt 2197−2222) and 5′-CAGGCTGATAATGATCTGAGGATGAC-3′ (nt 2377−2402) for β3, and 5′-GCTGTGGTCGGTAGCATCCTCCTTG-3′ (nt 2176−2200) and 5′-CTCCAGCCCCTCGGAGAAGGAAACA-3′ (nt 2401−2425) for β5.
Cell Line Isolation and Culture
Mouse kidney glomeruli were isolated by microdissection (24), and tubules were isolated by previously described Percoll gradient centrifugation methods (25). Rat MCs and human MCs from discarded nephrectomies were harvested according to established sieving methods (26). MCs from mice with targeted deletion of β8 (itgb8−/−) (4) or RhoGDI-1 (gdi−/−) (27) genes were harvested from microdissected glomeruli as described (24) and distinguished from other glomerular cells by desmin expression upon immunocytochemical analysis. itgb8−/− mice on out-bred C57BL/6J-129/Sv genetic background were crossed with CD1 background mice, yielding progeny in reduced numbers that survived 3−4 weeks, which was sufficient for MC culture but inadequate to assess renal phenotype. Human renal proximal tubule cells (a gift from Dr. L. Racusen, Johns Hopkins) were derived from human proximal tubule (28-30), HEK293 cells were from ATCC (Manassas, VA), and U373 human astrocytoma cells were a gift from Dr. D. Kunze (Case Western Reserve University). CHO and CHO-B2/v7 cell lines (31) were gifts from Dr. E. Ruoslahti (Burnham Institute). All cell lines were cultured in Dulbecco's modified Eagle's medium/F-12 (Invitrogen) containing 10% fetal bovine serum (Hyclone, Logan, UT) supplemented with 1% penicillin G-streptomycin sulfate-amphotericin (Sigma).
Plasmid Transfections
Plasmids were transformed into competent DH-5α bacterial strain according to the manufacturer's protocol (Invitrogen), extracted using a Maxiprep Kit (Qiagen, Valencia, CA), and amplified by culture in Luria-Bertani broth containing appropriate antibiotics. cDNAs were transiently transfected into CHO-B2/v7 or HEK293 cells according to previously described methods (32, 33). Briefly, cells were plated in 6-well dishes (0.25 × 106 cells/well) and cultured overnight in Dulbecco's modified Eagle's medium/F-12 plus 10% fetal bovine serum to achieve 80% confluence. The transfection mixture, which contained 2.0 μg of plasmid DNA and 6 μl of FuGENE 6 transfection reagent (Roche Applied Science) in 100 μl serum-free Dulbecco's modified Eagle's medium (Invitrogen), was mixed for 20 min at room temperature and then added to each well with complete medium for 24 h. Cells were evaluated for protein expression by immunoprecipitation (see the methods below) 24−48 h post-transfection.
Stable β8 integrin transfectants were generated from HEK293 cells as previously described (13). Briefly, cells were cultured to 50% confluence in 10-cm dishes, then transfected with 2 μg of human β8 integrin cDNA (1) subcloned into pcDNA1-Neo vector using FuGENE 6 reagent. Cells were then cultured in complete media containing G418 (Sigma). Individual G418-resistant clones were isolated by limiting dilution, subcultured, and assayed for β8 integrin expression by biotin surface labeling and immunoprecipitation according to published methods (13). Stable transfectants with persistent β8 integrin overexpression were utilized after three to five passages.
Immunoprecipitation and Immunoblot Analysis
Cells were lysed in Nonidet P-40 (1%, v/v), sodium deoxycholate (0.25%, w/v), 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin (34). Aliquots of equal protein content were then immunoprecipitated with specified anti-integrin antibodies, resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and blotted with peroxidase-conjugated, secondary antibodies according to previously described methods (13). In some experiments cells were initially surface-biotinylated, then lysates were immunoprecipitated with β8 antisera, and immunoblots were probed with peroxidase-conjugated streptavidin, as described (13).
Yeast Two-hybrid Screening
Methods have previously been reported (22) and have been modified as described below. The two-hybrid bait construct was generated by PCR amplification of the cDNA encoding the C-terminal 60-amino acid sequence of human integrin β8 (1) with 5′-CAGGTGGAATTCCAATGGAATAGT-3′ (nt 2116−2139) and 5′-AGAATAGGATCCTCTAGATG-3′ (vector sequence 3′ to stop codon) primers. The resulting fragment was inserted into pAS2−1 vector (MATCHMAKER Two-Hybrid System 3; BD Sciences Clontech) in-frame with the GAL4 DNA binding domain to generate pASβ8. Two-hybrid analysis was performed in the Saccharomyces cerevisiae strain AH109 (MATα trp1−901 leu2−3112 ura3−52 his3−200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ). No transcriptional activation by bait fusion protein alone was detected, as measured by expression of three reporters, lacZ, ADE2, and HIS3 (see Table 1). To select protein partners interacting with β8 cytosolic tail, AH109 cells were co-transformed with pASβ8 and Human Kidney Matchmaker cDNA Library (Clontech) in pACT2 vector, which contained the GAL4 transcriptional activation domain fused to inserts. Transformants were initially selected on Leu−/Trp−/His− media in the presence of 5 mm 3-aminotriazole. Selected clones were further tested for growth on Leu−/Trp−/Ade− and Leu−/Trp−/His−/Ade− media by replica plating. Positive clones were further tested by β-galactosidase expression and sequenced. To exclude nonspecific protein interaction with β8 fusion protein, control WTIP constructs (22) were inserted into the activation domain plasmid, co-transformed with pASβ8, and analyzed for reporter expression. Using a similar strategy, GDI specificity was tested for interaction with WTIP and human β3-integrin negative controls. The sequence encoding the entire β3 cytoplasmic tail was amplified from the IL-2R/β3 construct using oligonucleotide primers 5′-CAGGCTGATAATGATCTGAGGATGAC-3′ containing an NcoI site and 5′-ATTGGCCTTGCCGCCCTGCTCATCTG-3′ containing a BamHI site, cloned into pAS2−1. To ensure specificity and direct bait-prey interaction, individual plasmid prey constructs were co-transformed in AH109 with pASβ8 to recapitulate results obtained during the library screening process.
TABLE 1.
RhoGDI-1 specifically interacts with β8-integrin cytosolic domain in yeast
| GAL4 DNA binding domain fusion protein | GAL4 transcriptional activation domain fusion protein | Reporter Gene |
||
|---|---|---|---|---|
| HIS3 | ADE2 | lacZ | ||
| β8 cytosolic domain | GAL4AD | − | − | White |
| β8 cytosolic domain | RhoGDI-1 | + | + | Blue |
| β3 cytosolic domain | RhoGDI-1 | − | − | White |
| β8 cytosolic domain | ΔN-WTIPa | − | − | White |
| GAL4 DNA binding domain | RhoGDI-1 | − | − | White |
| Full-length WTIP | RhoGDI-1 | − | − | White |
| ΔN-WTIP | RhoGDI-1 | − | − | White |
Wilms tumor-interacting protein with N-terminal deletion.
Generation of IL-2R/β8 Chimeric Receptors
To selectively stimulate β8 integrin signaling pathways, a chimeric receptor composed of the IL-2R transmembrane and ectodomains fused to human β8 integrin intracellular domain was constructed (35, 36) using gene splicing by overlap extension (gene SOEing) methods (37). The IL-2R portion of the chimera was generated by PCR using the human IL-2R cDNA template (38) (a gift from Dr. W. C. Greene, Duke University) with 5′-CGCGAATTCCGCCACCATGGATTCATACCTGCTGATG-3′ (primer 1, the EcoRI site is underlined, nt 1−21 from IL-2 receptor cDNA) and 5′-CTTAATTTTATTACTATTCCAGAGCCCACTCAGGAGGAGG-3′ (primer 2, nt 759−777 were from IL-2 receptor cDNA, and nt 2131−2151 were from β8 cDNA). The β8 chimera portion was generated from full-length human β8 cDNA template (1) using 5′-CCTCCTCCTGAGTGGGCTCTGGAATAGTAATAAAATTAAG-3′ (primer 3, nt 759−777 were from IL-2 receptor cDNA, and nt 2131−2151 were from β8 cDNA) and 5′-CGCGGATCCCGAAGTTGCACCTGAAAGTTTC-3′ (primer 4, the BamHI site is underlined, nt 2287−2307 were from β8 cDNA). The transcribed products were engineered to contain overlapping sequences to permit annealing as well as 5′-EcoRI and 3′-BamHI restriction sites. Annealed fragments were then PCR-amplified using primers 1−4 to generate chimeric IL2R-β8. Negative control IL-2R extracellular and transmembrane domain-only (IL-2RΔ) constructs were generated by PCR amplification using 5′-CGCGAATTCCGCCACCATGGATTCATACCTGCTGATG-3′ (primer 1) and 5′-CGCGGATCCCGCCACCGAGCCCACTCAGGAGGAGG-3′ (nt 759−777) primers. β8 cytosolic domain-only (β8-cd) negative control was generated using PCR primers 5′-CGCGAATTCCGCCACCATGGATTGGAATAGTAATAAAATTAAG-3′ (nt 2130−2151) and 5′-CGCGGATCCCGAAGTTGCACCTGAAAGTTTC-3′ (primer 4). IL2R-β8, IL-2RΔ, and β8-cd PCR products were cloned into 5′-EcoR1-and 3′-BamH1-digested pEGFP-N2 vector (BD Sciences Clontech) to generate green fluorescent protein-tagged constructs. HEK293 cells, which do not express the IL-2R (data not shown), were transiently transfected with chimeric receptor, IL-2RΔ, or β8-cd constructs. Receptors were clustered with anti-IL-2R (Tac) monoclonal antibodies according to established methods (35, 36, 39). Adherent cells were incubated directly with Tac (1 μg/ml, 60 min, 37 °C). For cell suspension experiments, Tac was fixed to GammaBind-Sepharose beads (Amersham Biosciences). Beads were precleared with 2% bovine serum albumin, and 20 μl of packed bead volume was suspended in 20 μl of phosphate-buffered saline, to which 5 μg of Tac or isotype control antibodies were bound. Cells from confluent 10-cm dishes were lifted, suspended in 100 μl Dulbecco's modified Eagle's medium, and incubated with 40 μl beads (30 min, 37 °C).
G-protein Activity Assays
RhoA and Rac1/Cdc42 activities were determined as described previously (40). αvβ8 integrin was stimulated by plating β8-transfected CHO-B2/v7 cells on vitronectin (10 μg/ml) in serum-free media or by incubating HEK293 cells expressing IL2R-β8 with Tac antibodies. Cells were lysed in a buffer containing 50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 10 mm MgCl2, 150 mm NaCl, 0.5% sodium deoxycholate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin on ice for 30 min. Cell lysates were immediately incubated with glutathione-Sepharose 4B beads coupled to GST-rhotekin (to capture GTP-Rho) or GST-PAK binding domain (to capture GTP-Rac or GTP-Cdc42) for 45 min at 4 °C. Beads were then washed and re-suspended in SDS sample buffer. GTP-bound RhoA, Rac1, or Cdc42 were analyzed by immunoblotting with anti-RhoA, anti-Rac1, or anti-Cdc42 antibodies, respectively. Total G-protein levels were determined by immunoblot analysis of whole cell lysates.
Immunocytochemistry
Cells were evaluated by previously described immunocytochemical methods (30, 41). All cells were plated on glass coverslips, fixed in paraformaldehyde (4%, 10 min, room temperature), blocked, and permeabilized with 5% bovine serum albumin in 0.2% Triton X-100. Actin was labeled with Alexa 568-conjugated phalloidin (1:40, room temperature) and by incubation with anti-α-SMA antibodies (1:100, 1 h, room temperature) followed by fluorescein isothiocyanate-conjugated secondary antibody (1:300, 1 h, room temperature). In some experiments cells were transfected with green fluorescent protein cDNA to assess transfection efficiency and transduced with biotin epitope-tagged peptide composed of HIV-Tat fused to Rac1 amino acid residues 17−32 (Tat-Rac-(17-32)), Cdc42 residues 17−32 (Tat-Cdc42 (17-32)), or control biotinylated HIV-Tat (40 μg/ml, 90 min) according to published methods (42). Biotinylated HIV-Tat-Rac was detected with Texas Red-conjugated streptavidin (1:300, 1 h, room temperature). Coverslips were mounted in 4′,6-diamidino-2-phenylindole-containing Vectashield medium (Vector Laboratories) and viewed with a Nikon epifluorescence microscope (Tokyo, Japan) equipped with filters for red, green, and blue wavelength light emission. Images were generated with a Spot Digital System camera (Diagnostic Instruments, Sterling Heights, MI) and Image Pro software (Media Cybernetics, Silver Spring, MD). For quantitation of α-SMA stress fibers, cells containing more than 10 fibers, which extended for greater than half of the cell diameter, were considered positive.
Data Presentation
All data are representative of three to four experiments per condition. Graphical results are presented as the mean ± S.E. unless otherwise indicated.
RESULTS
Kidney β8 Integrin Is Localized to MCs
Our original interest in the β8 integrin stemmed from investigation of Fas (CD95)-directed pathway activation in renal tubular epithelial cells. Using a hybridization array approach, we found that Fas stimulation up-regulated β8 expression in cultured proximal tubule epithelial cells (13). To characterize β8 expression in vivo, mouse kidney sections were probed for β8 mRNA expression by in situ hybridization, since suitable antibodies for immunohistochemical studies were not available. Kidney β8 was unexpectedly expressed in a predominant, glomerular mesangial pattern (Fig. 1, A and C). Mouse kidney glomerular and tubular mRNA expression was also assessed by RT-PCR and confirmed that β8 is expressed primarily in glomeruli (Fig. 1D). β8 protein content was determined in cultured MC and tubule cell lines by immunoprecipitation of lysates from biotin surface-labeled cells, which revealed robust expression in rat MCs, and to a lesser extent in the HRPT human proximal tubule cell line (Fig. 1E, upper panel), consistent with previously published data (13). HEK293 cells do not express endogenous β8 mRNA or protein (Fig. 1E, upper panel), in agreement with previous reports (43). For this reason HEK293 cells were stably transfected with β8 cDNA and used in subsequent experiments. In control studies β1 protein expression was noted to be similar between the three cell lines (Fig. 1E, lower panel).
FIGURE 1. β8 integrin is expressed in MCs.
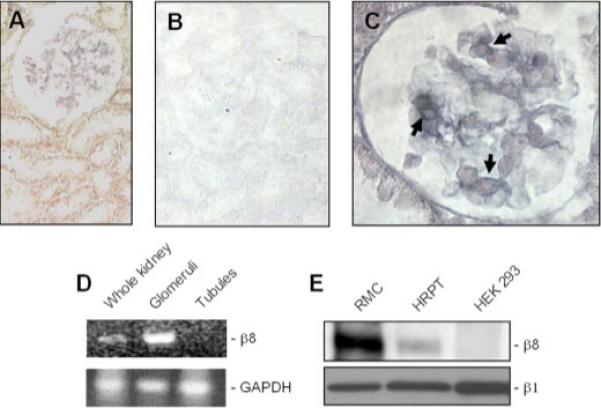
In situ hybridization with an alkaline phosphatase detection system (stains blue) using β8 integrin riboprobes in mouse kidney sections is shown. A, glomeruli with mesangial staining pattern and the absence of tubule cell labeling (×400 magnification). Alkaline phosphatase activity due to riboprobe trapping is noted within the interstitial space and along tubular basement membranes. B, to assess background labeling, control hybridization was conducted with a sense probe, which yielded no alkaline phosphatase activity (×400 magnification). C, cytoplasmic staining of cells with a mesangial distribution (shown by arrows, ×1000 magnification) in a single glomerulus. D, mouse kidney glomeruli and tubules were fractionated by microdissection and Percoll gradient centrifugation, respectively. β8 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH ) mRNA were amplified by RT-PCR (30 cycles). E, rat mesangial cells in primary culture (RMC ), human renal proximal tubule cells (HRPT), and HEK293 human embryonic kidney tubule cells were surface-biotinylated, and lysates with equal protein content were immunoprecipitated with rabbit β8 integrin anti-sera and probed by immunoblot analysis with peroxidase-conjugated streptavidin (upper panel ). Parallel lysates from the same cell lines (20 μg of protein per lane were probed for β1 integrin expression by immunoblot analysis (lower panel ).
Kidney β8 Integrin Expression Is Reduced in Mouse Models of Renal Disease
To elucidate β8 function in kidney, mouse models of glomerulosclerosis were examined for β8 expression. Northern blots from wild-type and ROP-Os/+ kidney (20) demonstrated progressively decreased β8 expression over time in the ROP-Os/+ group (Fig. 2A). Kidney β8 protein levels were similarly decreased in ROP-Os/+ compared with wild-type mice (Fig. 2B). In contrast, β1 integrin immunoblots revealed no difference between ROP-Os/+ and wild-type kidneys (Fig. 2B). To determine whether this observation is generalized to other models, β8 mRNA expression was examined by real-time PCR in a mouse model of HIVAN (21) as well as in ROP-Os/+ mice. These experiments demonstrated decreased β8 mRNA content in HIVAN and ROP-Os/+ compared with age-matched control kidneys (Fig. 2C). We conclude from these studies that glomerular β8 expression is suppressed in models of primary (Os) and secondary (HIVAN) forms of glomerular disease.
FIGURE 2. Kidney β8 integrin expression is reduced in mouse models of renal disease.
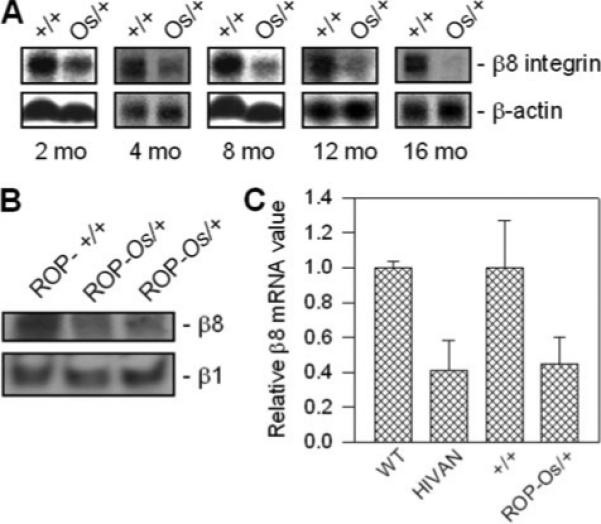
A, total RNA was harvested from wild type (+/+) and ROP-Os/+ (Os/+) mouse kidneys at the indicated time points, resolved by agarose gel electrophoresis (20 μg/lane), and Northern-blotted for β8-integrin mRNA expression. β-Actin mRNA expression is shown as a loading control. B, kidney cortex lysates (20 μg of protein per lane) from ROP+/+ (left lane, 9 months) and ROP-Os/+ (middle lane, 9 months; right lane, 3 months) mice were probed forβ8 and β1 integrin expression by immunoblot analysis. C, kidney total RNA from a mouse model of HIVAN and age-matched (16 week) wild-type (WT) control mice and ROP-Os/+ and age-matched (12 week) +/+ mice were analyzed for β8-integrin and β-actin mRNA expression by real-time PCR. Data are expressed as mean β8 transcript levels (±S.E.) normalized to β-actin from three mice in each group.
β8 Interacts with RhoGDI-1 (GDI)
Upon ligation with extracellular matrix proteins, integrins undergo conformational changes that permit β-subunit cytoplasmic tails to associate with intracellular signaling molecules (44). However, unlike other β-integrins, the β8 tail contains no predicted protein interaction or signaling domains (11). To identify cell signaling pathways regulated by β8, we employed a yeast two-hybrid screen with the β8 cytoplasmic domain as bait (Table 1). Screening of 2.5 × 106 transformants yielded 18 positive clones, 2 of which corresponded to the C terminus of GDI. Directed yeast two-hybrid assays with β3 or β8 integrin cytoplasmic tail sequences in pAS2−1 and C-terminal GDI sequence in pACT2 confirmed the β8-GDI interaction, whereas GDI did not interact with β3 (Table 1). The β8-GDI interaction is of particular interest because gdi−/− mice develop a renal phenotype characterized by glomerulosclerosis and proteinuria (27). Yeast two-hybrid screening also identified Band 4.1B as a β8 interactor, which confirms recently published data from McCarty et al. (43).
Fig. 3A demonstrates that in rat MCs in primary culture, the U373 human astrocytoma cell line, and HEK293 cells stably expressing β8, RhoGDI-1 co-precipitates with β8, confirming the yeast two-hybrid findings. In co-precipitation experiments using HEK293 cells overexpressing chimeric receptors, with the IL-2 receptor extracellular domain fused to β1, β3, or β8 cytoplasmic tails, GDI interacted exclusively with IL-2R/β8 (Fig. 3B), suggesting that the integrin-GDI interaction is specific for β8. To address whether ligand occupancy drives β8-GDI interaction, rat MCs were plated on vitronectin ligand or poly-l-lysine, which permits cell attachment by an integrin-independent mechanism. β8-GDI association was assessed by immunoprecipitating GDI and blotting for β8. Fig. 3C shows that lysates from vitronectin-stimulated MCs demonstrated robust β8-GDI interaction, whereas co-precipitation was not observed in lysates from poly-l-lysine-treated cells, indicating that ligand stimulation recruits GDI to bind the β8 cytosolic tail.
FIGURE 3. β8 stimulation regulates interaction with GDI.
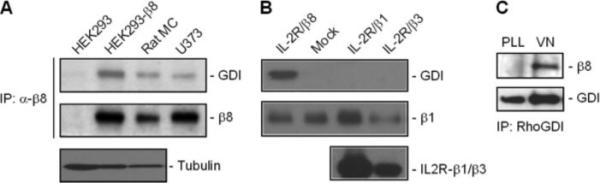
A, untransfected HEK293 cells or HEK293 cells stably transfected with β8 cDNA (HEK293-β8), rat MCs, and U373 astrocytoma cells were grown to confluence in serum-containing media. Whole cell lysates were immunoprecipitated (IP) with β8 antisera and immunoblotted with anti-RhoGDI-1 IgG (upper panel). Blots were stripped and re-probed with anti-β8 IgG (middle panel). Parallel lysates from the HEK293 cell lines and rat MCs were probed for tubulin expression by immunoblot analysis as a loading control. B, HEK293 cells were transiently transfected with empty vector (mock) or chimeric receptors for IL-2 receptor extracellular domain fused to β1-, β3-, or β8- integrin cytosolic domains (IL-2R/β1, IL-2R/β3, and IL-2R/β8, respectively). Each transfected cell line was maintained in serum-free media for 24 h before incubation with clustering anti-IL-2 receptor (Tac) antibody-coated beads (1 μg/ml, 30 min, 37 °C). Cells were then lysed and immunoprecipitated with Tac antibodies, resolved by SDS-PAGE, and then immunoblotted with anti-RhoGDI-1 antibodies (upper panel). Parallel HEK293 cell lysates were immunoblotted with anti-β1 integrin antibodies (middle panel). To verify chimeric receptor expression in IL-2R/β1- and IL-2R/β3-transfected controls, parallel cell lysates were immunoblotted with anti-IL-2R (Tac) antibodies (lower panel). C, rat MCs were added to 10-cm plates that were coated with poly-l-lysine (PLL, 10μg/ml, 30 min) or vitronectin (VN, 10 μg/ml, 30 min) and blocked with bovine serum albumin (3 ml, 2 mg/ml). Lysates were immunoprecipitated with anti-RhoGDI-1 IgG and immunoblotted with anti-β8 integrin IgG (upper panel). The lower panel represents stripped blot re-probed for GDI expression.
β8 Ligation Stimulates Rac1 and Suppresses RhoA Activation
Because GDI and integrins regulate Rho family G-protein signaling, we next tested for β8-dependent G-protein activation. In these experiments we used fibronectin binding-deficient CHO-B2/v7 cells, which have been stably transfected to express αv, because this cell line does not express β-integrins that partner with αv (31). This was verified by evaluating CHO-B2/v7 cells for β3- and β5-subunit expression by RT-PCR. Fig. 4A demonstrates that neither β3 nor β5 was detectable. CHO-B2/v7 cells were then transiently transfected with β8 integrin cDNA and plated on vitronectin ligand to selectively activate αvβ8 or on poly-l-lysine negative control and probed for Rac1 activity. Fig. 4B shows that Rac1 activation was increased in αvβ8-expressing CHO-B2/v7 cells incubated with vitronectin but not poly-l-lysine. CHO-B2/v7 cells transfected with empty vector did not activate Rac1 (not shown).
FIGURE 4. β8 ligation stimulates Rac1 activity.
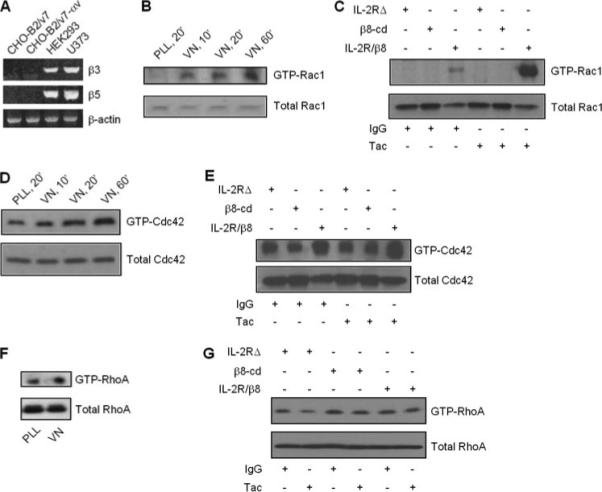
A, β3 and β5 integrin subunit expression in CHO cells expressing αv (CHO-B2/v7) was determined by RT-PCR as described under “Materials and Methods.” B and D, CHO-B2/v7 cells were transiently transfected with β8 cDNA and plated on vitronectin (VN) or poly-l-lysine (PLL) as described for Fig. 3 for the indicated times. C, E, and G, as an alternative method of β8 activation, HEK293 cells were stably transfected with chimeric receptor constructs composed of IL-2 receptor extracellular domain fused to β8 cytoplasmic domain (IL-2R/β8), negative control IL-2R extracellular domain only (IL-2RΔ), or negative control β8 cytosolic domain only (β8-cd). Transfected cells were maintained in serum-free media for 24 h before incubation with clustering Tac or isotype control antibody-coated beads. F, GTPase activity was determined in β8-transfected CHO-B2/v7 cells plated on vitronectin (VN, 10 μg/ml, 30 min) or negative control poly-l-lysine (PLL, 10 μg/ml, 30 min). G-protein activity was determined by pull-down assays, whereby whole cell lysates were first incubated with GST beads bound to PAK1 binding domain (B–E ) or rhotekin (F and G). GTP-bound G-proteins were detected by immunoblotting for Rac1 (B and C ), Cdc42 (D and E), or RhoA (F and G). Corresponding whole cell lysates were probed for expression of individual G-proteins by immunoblot analysis (B–G, lower panels).
CHO-B2/v7 cells express small amounts of β1-integrin, which couples with many α-integrin partners, including αv (Ref. 31 and data not shown). Therefore, to verify that Rac signaling is specific to β8 stimulation, Rac1 activity was also assessed in HEK293 cells expressing IL-2R extracellular domain-β8 intracellular domain chimeric receptors (IL-2R/β8), which permits integrin clustering and activation of intracellular signals by anti-IL-2R Tac antibody incubation (39). Fig. 4C demonstrates that Rac1 was robustly stimulated by receptor clustering with Tac, whereas Rac1 activity was undetectable in cells expressing truncated IL-2RΔ or β8-cd after Tac or isotype control IgG incubation. Irrelevant IgG exposure to IL-2R/β8-expressing cells resulted in modest Rac1 activation, perhaps due to overexpression and spontaneous clustering (45). It is doubtful that serum-free media contains significant IL-2. Less robust Cdc42 activity was observed in response to vitronectin (Fig. 4D) or chimeric receptor clustering (Fig. 4E), whereas neither vitronectin nor chimeric receptor stimulation of β8 activated RhoA (Fig. 4, F and G).
To further address the role of β8-regulated G-protein signaling in MCs, poly-l-lysine- and vitronectin-stimulated Rac1 and RhoA activities were determined in itgb8+/+ and itgb8−/− MC lines (Fig. 5A). Fig. 5B demonstrates Rac1 activation by vitronectin in wild-type MCs. In contrast, Rac1 activity was undetectable after plating itgb8−/− MCs on poly-l-lysine or vitronectin. RhoA activity was extremely low in wild-type MCs exposed to poly-l-lysine or vitronectin (some activity was observed with longer film exposures) and constitutively activated in itgb8−/− MCs. These data are consistent with Fig. 4 results and demonstrate that β8 up-regulates Rac1 and suppresses RhoA activation. In data not shown, MC stimulation with serum (10% fetal calf serum, 5 min) to activate Rho and Rac pathways did not affect β8 expression, as determined by RT-PCR.
FIGURE 5. β8 expression down-regulates RhoA activity.
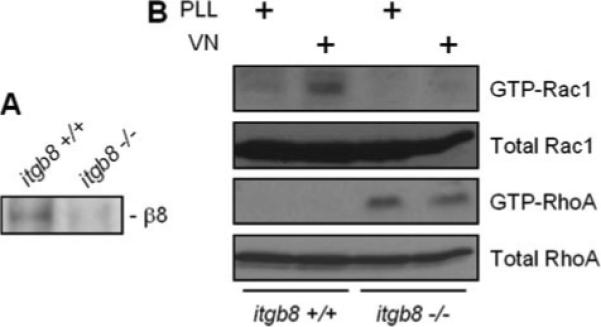
MCs from itgb8+/+ and itgb8−/− mice were harvested and maintained in primary culture. A, to verify itgb8 gene deletion, whole cell lysates were probed for β8 integrin protein expression by immunoblot analysis. B, itgb8+/+ and itgb8−/− MCs were incubated with poly-l-lysine (PLL, 10 μg/ml, 30 min) or vitronectin (VN, 10 μg/ml, 30 min) and then assayed for Rac1 and RhoA activities, as described in Fig. 4.
β8 Activation I Associated with Rac1 Release from GDI
For Rac1 activation to occur, GDP-bound Rac1 must first be released from GDI, which may be facilitated by a GDF before interaction with a RacGEF for GTP loading. β8 ligand binding or clustering regulates Rac1 signaling as well as GDI interaction with the β8 cytosolic tail, consistent with mechanisms whereby the β8 tail could function as a GDF or GEF. As an initial test to distinguish between these possibilities, CHO-B2/v7 cells expressing chimeric IL-2R/β8 or IL-2R/β3 receptors were clustered with Tac antibodies. Integrin cytosolic tail interaction with GDI or Rac1 was then assessed by immunoprecipitation. As seen in Fig. 6, integrin clustering resulted in GDI interaction with β8, but not β3, in agreement with Fig. 3 data. Moreover, β8 did not interact with Rac1, consistent with GDF, rather than RacGEF activity.
FIGURE 6. β8 activation is associated with Rac1 release from GDI.
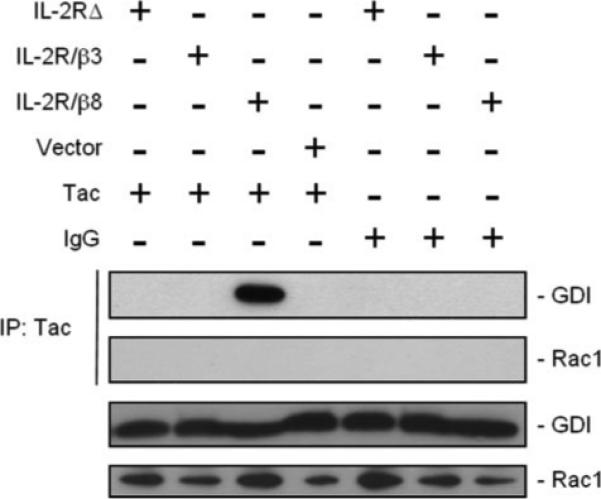
HEK293 cells expressing IL-2RΔ, IL-2R/β8, IL-2R/β3, or empty vector were incubated with clustering Tac or control antibodies. Chimeric integrins were immunoprecipitated (IP) from cell lysates with Tac IgG, resolved by SDS-PAGE, and then probed for GDI or Rac1 by immunoblot analysis (upper two panels). Fractions of cell lysate before immunoprecipitation were immunoblotted with anti-GDI and anti-Rac1 antibodies (lower two panels) to assess expression.
β8 Regulation of G-protein-dependent Cell Morphology
Characteristic in vitro manifestations of Rho family G-protein activation are actin-rich lamellipodia (Rac1), filopodia (Cdc42), and stress fiber formation (RhoA) (12, 46). To test for β8 regulation of these morphologic features, HEK293 cells expressing IL-2RΔ or IL-2R/β8 were stimulated with Tac antibodies and then labeled for F-actin with phalloidin. As seen in Fig. 7A, IL-2RΔ-stimulated cells were small, with little cytoplasm or actin assembly. In contrast, IL-2R/β8-expressing cells developed marked morphologic changes, with cytoplasmic spreading, broad lamellipodia, rare filopodia, and the absence of stress fibers (Fig. 7B). Taken together with data from Figs. 4 and 5, we conclude that the β8 cytosolic tail stimulates Rac1 and Cdc42 and suppresses RhoA activation.
FIGURE 7. β8 regulates Rac-dependent morphologic features.
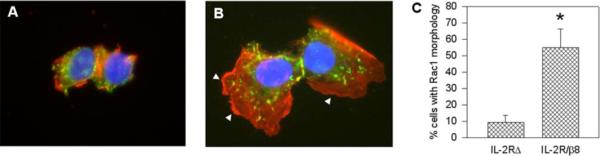
HEK293 cells were transiently transfected with IL-2RΔ (A) or chimeric IL-2R/β8 (B) receptors, which contain a green fluorescent protein tag at the C terminus to permit identification of transfected cells. Both groups were incubated with clustering IL-2R (Tac) antibodies (1 μg/ml, 60 min, 37 °C). Actin was labeled with Alexa 568-phalloidin (red) and 4′,6-diamidino-2-phenylindole-stained nuclei are in blue. Representative ×1000 images are shown. Arrowheads demarcate lamellipodia. C, quantitation of IL-2RΔ or IL-2R/β8 receptor-transfected cells demonstrating Rac1-dependent morphologic features. In each condition, transfected cells were examined for spreading and actin-rich lamellipodia; cells prominently exhibiting both features compared with surrounding untransfected cells were considered positive. Results are expressed as the mean ± S.E. *, p<0.05 compared with IL-2RΔ-expressing cells by Student's t test.
Rac1 Regulates MC Myofibroblast Phenotype
A cardinal feature of myofibroblast differentiation, which characterizes MC pathology, is α-SMA-containing stress fibers. α-SMA expression and assembly are regulated by RhoA in mesenchymal cells, including MCs (47, 48). Opposing Rac1 and RhoA signaling in response to β8 stimulation, therefore, suggests that Rac1 may regulate MC-myofibroblast transformation by inhibiting RhoA (44, 49-51). The next set of experiments was designed to test whether Rac1 directly modulates RhoA-dependent myofibroblast differentiation.
G-protein functions are dependent upon interaction with effector molecules, such as the serine-threonine kinase PAK and the Wiscott-Aldrich protein (WASP). As a tool to interrogate the effect of Rac1 or Cdc42 upon RhoA-regulated cell phenotype, a peptide corresponding to Rac or Cdc42, which blocks the interaction with PAK, was fused with the HIV Tat protein (52-54) and incubated with human MCs in primary culture. α-SMA assembly was employed as the assay for functional RhoA activation. At incubation times ranging from 8 to 36 h, compared with cells treated with Tat alone, Tat-Rac (17-32)-treated cells and to a lesser extent Cdc42 (17-32) cells displayed a more spread morphology and increased α-SMA organization (Fig. 8). The data are consistent with Rac1 suppression of RhoA-dependent myofibroblast differentiation.
FIGURE 8. Rac1 regulates MC myofibroblast phenotype.
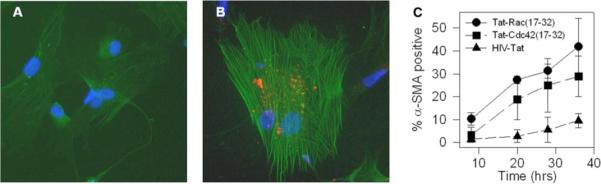
Human MC in primary culture were left in growth media for 1.5 h (A) or incubated with biotin-conjugated Tat-Rac (17-32) (B, 40 μg/ml, 1.5 h). Tat-peptide transduction efficiency range was 75−100%. Cells were fixed in paraformaldehyde, mounted with 4′,6-diamidino-2-phenylindole-containing media to label nuclei (blue), then incubated with Texas Red-streptavidin and counterstained with anti-α-SMA antibodies followed by fluorescein isothiocyanate-conjugated secondary antibody. C, the percentage of MCs demonstrating α-SMA assembly was calculated as described under “Materials and Methods” from 20 random fields per coverslip by an observer blinded to experimental conditions. Comparisons were then made between a population of MCs that took up the Tat-Rac (17-32), Tat-Cdc42 (17-32), or HIV-Tat peptides. Data represent the mean ± S.E. from four separate experiments.
Implications of MC Rho Family GTPase Signaling by GDI
MCs derived from gdi−/− mice demonstrate enhanced RhoA, but not Rac1 activation (not shown), consistent with divergent GDI regulation of Rho family G-proteins (44). To test for regulation of cell morphology by GDI, quiescent mouse gdi−/− and gdi+/+ MCs were stained for α-SMA or F-actin by immunocytochemical methods. As seen in Fig. 9B, serum-starved gdi−/− cells demonstrated robust α-SMA assembly into stress fibers, consistent with associated RhoA activation, whereas wild-type cells displayed few α-SMA stress fibers (Fig. 9A). Differences in phalloidin staining were also observed between gdi−/− and gdi+/+ MCs (Figs. 9, C and D), indicating that stress fibers were derived from α-SMA and β-actin. In addition, lamellipodia were less prominent in gdi−/− compared with gdi+/+ MC.
FIGURE 9. Implications of MC Rho family GTPase signaling by GDI.
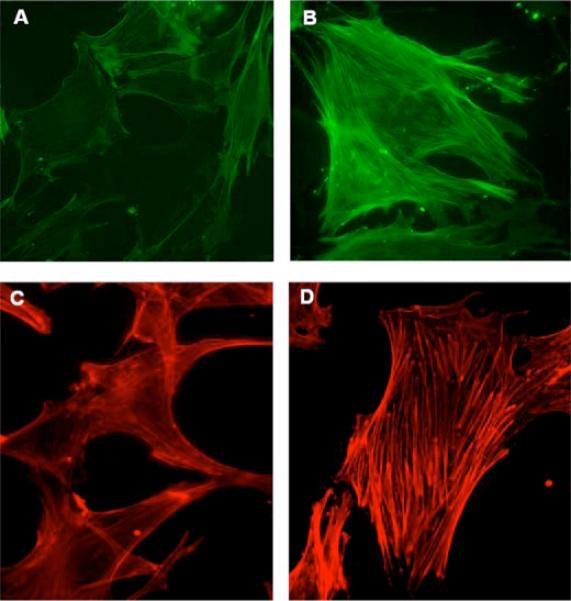
MCs harvested by microdissection from wild-type (A and C) and gdi−/− (B and D) mice were maintained in primary culture. Cells were changed to media containing 0% serum for 24 h before fixation in paraformaldehyde. Cells were labeled with anti-α-SMA antibodies followed by fluorescein isothiocyanate-conjugated secondary antibody (A and B) or Alexa 568-conjugated phalloidin (C and D). Representative ×400 fluorescence images are shown.
Taken together the data support the hypothesis that β8 interacts with GDI to promote wild-type MC phenotype via Rac1-dependent suppression of RhoA. In MCs with targeted deletion of itgb8 or gdi, ligand-mediated Rac1 activity is lost, which permits RhoA-dependent myofibroblast features to predominate.
DISCUSSION
Cell-matrix interaction and associated signal transduction pathways are regulated by multiple mechanisms, including cell-specific expression of different α-β integrin heterodimers. Until now, the only well established MC β-integrin subunit was β1, which partners with α1, and to a lesser extent with α2, α3, and α6 (55). The original β8 cDNA cloning data demonstrated abundant mRNA expression in kidney (1), but the current report represents the first description of kidney β8 localization, which is most prominent in the MCs. We also found that kidney β8 mRNA expression was markedly diminished in two different models of progressive glomerular disease, suggesting that β8 may specify the normal MC differentiation state and that loss of β8 expression may contribute to glomerulosclerosis pathogenesis. Diminished β8 expression is unlikely to be due to scarring and loss of MCs, since kidney β1 integrin expression persisted in mouse models of glomerulosclerosis.
MC expression of both β1 and β8 permits switching between integrin pools to achieve specific responses, such as intracellular signaling, cytoskeleton remodeling, and motility, to dynamic extracellular cues (56, 57). Unlike β1 integrins, which recognize many matrix proteins, αvβ8 is less promiscuous, with mammalian ligands including vitronectin, latent TGFβ, and perhaps laminin-1 and type IV collagen (10, 58, 59). Type IV collagen isoforms, fibronectin and laminins 8 and 9, reside in normal glomerular mesangium (60), whereas vitronectin, laminin-1, and latent TGFβ do not (61-64). We, therefore, deduce that under normal circumstances, MC αvβ8 may bind type IV collagen in vivo. Only collagen IV isoforms containing α1 or α2 chains are expressed in mesangial matrix, and it has not yet been established that αvβ8 binds to these specific isoforms. However, mesangial matrix components have not been exhaustively identified, so native MC αvβ8 ligands may still be unknown. In vivo, MCs are centrally located in glomeruli and surrounded by extracellular (mesangial) matrix. Kikkawa et al. (65) recently demonstrated that MC projections also adhere to glomerular basement membrane through α3β1 integrin binding to laminin α5 chains (65). Therefore, MC αvβ8 may also be spatially regulated to interact with a cadre of distinct mesangial or glomerular basement membrane matrix proteins.
An intriguing possibility is that β8 could regulate TGFβ signaling, which has been implicated in myofibroblast differentiation (66). Recent reports have demonstrated that the latency-associated peptide portion of the latent TGFβ complex may be a β8 ligand (59). Latent TGFβ is not abundantly expressed in normal glomeruli, but it is induced in animal models of glomerular disease and secreted by MCs (63, 64, 67). Unlike latency-associated peptide ligation to αvβ6, which induces a conformational change in the integrin that leads to direct TGFβ1 activation (68), αvβ8-regulated TGFβ1 activity requires concomitant latency-associated peptide cleavage by the membrane-associated metalloproteinase, MT1-MMP (59). MT1-MMP is also inducible in MCs but generally only in pathologic conditions (69). We, therefore, speculate that in disease states, latent TGFβ may displace the natural extracellular matrix ligand for β8 to initiate glomerular injury, which includes down-regulation of β8 expression and bioactive TGFβ release within metalloproteinase-rich microenvironments.
Our studies represent the first characterization of a β8-regulated signal transduction pathway, namely activation of the small molecular weight G-protein, Rac1, and to a lesser degree, Cdc42. The canonical integrin signaling pathway is initiated by focal adhesion kinase followed by downstream activation of Rho family members, Rho, Rac, and Cdc42. However, focal adhesion kinase is not predicted to bind β8, and it was not identified as a β8 interactor by yeast two-hybrid assays. Furthermore, although other integrins have been shown to activate Rac1, including IL-2 receptor β1 and β3 integrin chimeras (39), Rac1 activation in association with integrin-GDI binding is unique to β8. That β8 stimulation activated Rac1, but not RhoA, is consistent with antagonism between Rac and Rho pathways in other systems, including β1 integrin-mediated adhesion to fibronectin (44). It was originally demonstrated that Rac1 can activate RhoA in fibroblasts, although weakly and with delayed kinetics (12, 46). Several subsequent reports in epithelial and mesenchymal cells have shown that Rac1 down-regulates RhoA activity (70-73), which was corroborated by our data. RhoA activation induces stress fiber and focal adhesion formation (44), whereas activated Rac1 prevents these processes (44, 49) through PAK-dependent inhibition of myosin heavy chain phosphorylation (50) and myosin light chain kinase activity (51). Stress fibers are predominantly composed of β-actin. However, MCs also express α-SMA, which assembles into stress fibers only during myofibroblast differentiation in pathologic states.
Taken together, the in vivo and in vitro studies support a model (Fig. 10) of constitutive MC αvβ8 activation by mesangial matrix or glomerular basement membrane ligands which stimulates Rac1-bound GDI shuttling to β8-containing microdomains to facilitate GDI release of Rac1 for membrane insertion in proximity to appropriate GEFs and other effectors. This scheme is consistent with recent reports describing spatial regulation of G-proteins to sites of activation by GDI in mammalian (16) as well as plant cells (74). Downstream, Rac1-directed signals maintain the normal differentiation state by suppression of RhoA-regulated α-SMA stress fiber formation. In the context of glomerular disease, MC β8 expression is diminished, which leads to altered Rac1 targeting, decreased Rac1 activity, and stimulation of RhoA-dependent α-SMA assembly. The model, therefore, predicts that the shift from Rac1 to RhoA signaling drives MC pathophysiology.
FIGURE 10. Schematic model of β8-integrin regulation of Rho family G-proteins in MC differentiation.
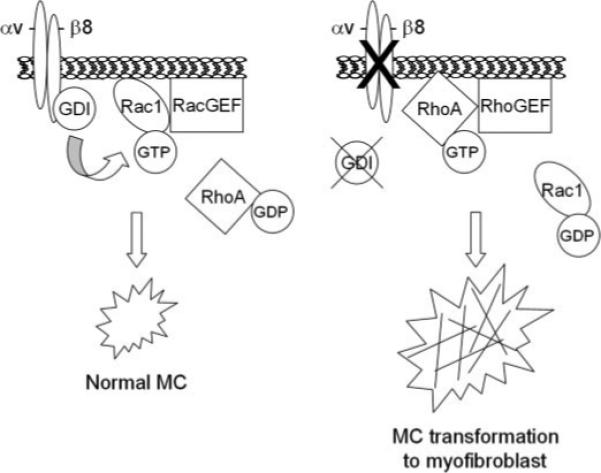
The figure on the left represents the normal state, wherein MC differentiation is regulated by αvβ8-dependent docking of RhoGDI-1 with the β8 cytoplasmic tail, which stimulates release of Rac1 from GDI for Rac-GEF interaction. The figure on the right represents the pathologic state, modeled by either decreased β8 or GDI expression, resulting in up-regulated RhoA-dependent α-SMA assembly, which defines pathologic, myofibroblast differentiation.
The unique finding that GDI interacts with the β8 cytosolic tail in the context of G-protein activation is not easily rationalized with data demonstrating that G-protein-GDI interaction is unnecessary for Rac1 and Cdc42 activation (75, 76) or that the sole function of GDI is to modulate G-protein activity by cytosolic sequestration. Based upon a recent report by Moissoglu et al. (77) demonstrating that GDI inhibits Rac1 membrane targeting, one unifying explanation is that β8 may enhance Rac1 membrane association and activation by sequestering free GDI, i.e. GDI not complexed with Rac1 (78, 79). Alternatively, GDI down-regulation of Rac1 and Cdc42 may be peculiar to systems employing overexpression of constitutively active G-proteins and, therefore, may not reflect endogenous G-protein function (18, 80).
In addition to the well described role of GDI as a negative regulator of G-protein activation, GDI interaction with Rac1 and Cdc42 has been associated with G-protein activation by mechanisms such as GDI-regulated inhibition of GTPase activity (15, 81-83), shielding G-proteins from protease cleavage (84) or trafficking G-proteins to appropriate membrane domains (85). To accommodate dual roles for GDI in G-protein activation and inactivation, it has been postulated that GDI dynamically regulates G-protein signaling by chaperoning G-proteins from cytosol to membrane activation domains and by removal of G-proteins from membrane sites and retention as an inactive cytosolic complex (18). Because β8 ligation was associated with GDI binding and Rac1 activation, we hypothesized that the integrin could acts as a GEF or GDF (86-90). Because Rac1 was not detected in the complex with activated β8, our data are more consistent with β8 functioning as a GDF rather than a GEF.
β8 stimulation by multiple strategies resulted in Rac1 but not RhoA activation. Because GDI binds all three classes of small molecular weight G-proteins (Rac1, RhoA, Cdc42), we speculate that if β8 has GDF activity, it may discriminately regulate G-protein pathways. Microinjection of fibroblasts with radixin, the first described GDF, resulted in activation of RhoA, but not Rac1 (87), thereby establishing a precedent for selective G-protein regulation by GDFs. Specificity of GDI release may also be regulated by post-translational modification and conformational changes of GDI. For example, PAK phosphorylation of Ser-101 and Ser-174 GDI residues caused release and activation of Rac1 but not RhoA (91). By analogy, β8-dependent PAK activation could represent an additional mechanism for preferential Rac1 activation. Finally, a recent model proposes that integrins are juxtaposed with lipid rafts containing specific G-proteins and effectors, and signal amplification is achieved by integrin prevention of lipid raft microdomain internalization (85). This study, therefore, suggests that the β8-GDI complex could be targeted to lipid domains, which are enriched for Rac1 rather than RhoA effectors (87, 91).
Data from gdi−/− mice (27) support biologic relevance of the β8-GDI interaction in MCs. Despite ubiquitous GDI expression in normal mice, a limited number of gdi−/− phenotypes was observed. The most profound was renal dysfunction, which included massive proteinuria and premature death due to renal failure. Histologic examination revealed glomerulosclerosis in a mesangial distribution. MC morphology was not addressed in detail, although gdi gene deletion in other mesenchymal cells enhanced stress fiber formation (92). MC-to-myofibroblast transition, characterized by α-SMA stress fiber formation, is a recognized glomerular disease feature. Evidence that β8 and GDI may be partners within a complex that regulates MC differentiation include the following. (a) β8 and GDI co-precipitate, (b) kidney expression of β8 is most prominent in glomerular mesangium, and gdi−/− mice have a mesangial phenotype, and (c) itgb8−/− and gdi−/− MCs exhibited pathologic, myofibroblast features including enhanced RhoA activity and RhoA-dependent α-SMA stress fiber organization.
In conclusion, the β8 integrin is localized to kidney glomerular MCs in vivo and in vitro, and animal models of glomerulosclerosis are associated with decreased MC β8 expression. In vitro, β8 stimulation leads to β8-GDI interaction, Rac1 and Cdc42 (but not RhoA) activation, and suppression of pathologic MC features. Taken together the data suggest that under basal, physiologic conditions, the β8 cytosolic tail provides specificity to G-protein signaling and regulates MC phenotype, perhaps by docking and sequestering GDI, to permit Rac1 targeting to discrete signaling domains. A corollary effect of β8-dependent Rac1 activation is concomitant RhoA suppression. Diminished MC β8 expression may impair Rac1 targeting and activation, thereby permitting MCs to develop a RhoA-regulated myofibroblast phenotype.
Footnotes
This work was supported by National Institutes of Health Grants P50 DK054178, R01 DK064719, R01 CA092259, R01 CA096533, and R01 DK061395. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: GEF, GTP exchange factor; HIVAN, HIV-associated nephropathy; IL-2R, human interleukin-2 receptor; WTIP, Wilms tumor-interacting protein; GDI, guanine nucleotide dissociation inhibitor-1; MC, mesangial cells; α-SMA, anti-α-smooth muscle actin; HIV, human immunodeficiency virus; GST, glutathione S-transferase; nt, nucleotides; RT, reverse transcription; CT, cycle threshold; HEK cells, human embryonic kidney cells; CHO, Chinese hamster ovary; β8-cd, β8 cytosolic domain only; TGFβ, transforming growth factor β; PAK, p21-activated kinase; GDF, GDI displacement factor.
REFERENCES
- 1.Moyle M, Napier MA, McLean JW. J. Biol. Chem. 1991;266:19650–19658. [PubMed] [Google Scholar]
- 2.Cambier S, Mu DZ, O'Connell D, Boylen K, Travis W, Liu WH, Broaddus VC, Nishimura SL. Cancer Res. 2000;60:7084–7093. [PubMed] [Google Scholar]
- 3.Stepp MA. Dev. Dyn. 1999;214:216–228. doi: 10.1002/(SICI)1097-0177(199903)214:3<216::AID-AJA5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Zhu JW, Motejlek K, Wang DN, Zang KL, Schmidt A, Reichardt LF. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, Shani M, Dvorak HF, Wolburg H, Bader BL, Dvorak AM, Hynes RO. Mol. Cell. Biol. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 7.Humphries MJ, McEwan PA, Barton SJ, Buckley PA, Bella J, Mould AP. Trends Biochem. Sci. 2003;28:313–320. doi: 10.1016/s0968-0004(03)00112-9. [DOI] [PubMed] [Google Scholar]
- 8.Mattila E, Pellinen T, Nevo J, Vuoriluoto K, Arjonen A, Ivaska J. Nat. Cell Biol. 2005;7:78–85. doi: 10.1038/ncb1209. [DOI] [PubMed] [Google Scholar]
- 9.Nishiya N, Kiosses WB, Han J, Ginsberg MH. Nat. Cell Biol. 2005;7:343–352. doi: 10.1038/ncb1234. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura SL, Sheppard D, Pytela R. J. Biol. Chem. 1994;269:28708–28715. [PubMed] [Google Scholar]
- 11.Stefansson A, Armulik A, Nilsson I, von Heijne G, Johansson S. J. Biol. Chem. 2004;279:21200–21205. doi: 10.1074/jbc.M400771200. [DOI] [PubMed] [Google Scholar]
- 12.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 13.Jarad G, Wang B, Khan S, DeVore J, Miao H, Wu K, Nishimura SL, Wible BA, Konieczkowski M, Sedor JR, Schelling JR. J. Biol. Chem. 2002;277:47826–47833. doi: 10.1074/jbc.M204901200. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Mol. Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 15.Hancock JF, Hall A. EMBO J. 1993;12:1915–1921. doi: 10.1002/j.1460-2075.1993.tb05840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA. Nat. Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- 17.Dransart E, Morin A, Cherfils J, Olofsson B. J. Biol. Chem. 2005;280:4674–4683. doi: 10.1074/jbc.M409741200. [DOI] [PubMed] [Google Scholar]
- 18.Dovas A, Couchman JR. Biochem.J. 2005;390:1–9. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pytela R, Pierschbacher MD, Argraves S, Suzuki S, Ruoslahti E. Methods Enzymol. 1987;144:475–489. doi: 10.1016/0076-6879(87)44196-7. [DOI] [PubMed] [Google Scholar]
- 20.Zalups RK. Am. J. Physiol. 1993;264:F53–F60. doi: 10.1152/ajprenal.1993.264.1.F53. [DOI] [PubMed] [Google Scholar]
- 21.Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, Eckhaus M, Bryant JL, Notkins AL, Klotman PE. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1577–1581. doi: 10.1073/pnas.89.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srichai MB, Konieczkowski M, Padiyar A, Konieczkowski DJ, Mukherjee A, Hayden PS, Kamat S, El Meanawy MA, Khan S, Mundel P, Lee SB, Bruggeman LA, Schelling JR, Sedor JR. J. Biol. Chem. 2004;279:14398–14408. doi: 10.1074/jbc.M314155200. [DOI] [PubMed] [Google Scholar]
- 23.Applied Biosystems . Applied Biosystems User Bulletin #2. Applied Biosystems; Foster City, CA: 2001. [Google Scholar]
- 24.Gandhi PN, Gibson RM, Tong XF, Miyoshi J, Takai Y, Konieczkowski M, Sedor JR, Wilson-Delfosse AL. Biochem. J. 2004;378:409–419. doi: 10.1042/BJ20030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schelling JR, Hanson AS, Marzec R, Linas SL. J. Clin. Investig. 1992;90:2472–2480. doi: 10.1172/JCI116139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werber HI, Emancipator SN, Tykocinski ML, Sedor JR. J. Immunol. 1987;138:3207–3212. [PubMed] [Google Scholar]
- 27.Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Doi T, Mizoguchi A, Matsuura N, Niho Y, Nishimune Y, Nishikawa S, Takai Y. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 28.Racusen LC, Monteil C, Sgrignoli A, Lucskay M, Marouillat S, Rhim JGS, Morin JP. J. Lab. Clin. Med. 1997;129:318–329. doi: 10.1016/s0022-2143(97)90180-3. [DOI] [PubMed] [Google Scholar]
- 29.Schelling JR, Nkemere N, Kopp JB, Cleveland RP. Lab. Investig. 1998;78:813–824. [PubMed] [Google Scholar]
- 30.Khan S, Cleveland RP, Koch CJ, Schelling JR. Lab. Investig. 1999;79:1089–1099. [PubMed] [Google Scholar]
- 31.Zhang Z, Morla AO, Vuori K, Bauer JS, Juliano RL, Ruoslahti E. J. Cell Biol. 1993;122:235–242. doi: 10.1083/jcb.122.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan S, Koepke A, Jarad G, Schlessman K, Cleveland RP, Wang BC, Konieczkowski M, Schelling JR. Kidney Int. 2001;60:65–76. doi: 10.1046/j.1523-1755.2001.00771.x. [DOI] [PubMed] [Google Scholar]
- 33.Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, Konieczkowski M, Sedor JR, Schelling JR. J. Biol. Chem. 2004;279:26280–26286. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]
- 34.Loo DT, Kanner SB, Aruffo A. J. Biol. Chem. 1998;273:23304–23312. doi: 10.1074/jbc.273.36.23304. [DOI] [PubMed] [Google Scholar]
- 35.LaFlamme SE, Akiyama SK, Yamada KM. J. Cell Biol. 1992;117:437–447. doi: 10.1083/jcb.117.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YP, O'Toole TE, Shipley T, Forsyth J, LaFlamme SE, Yamada KM, Shattil SJ, Ginsberg MH. J. Biol. Chem. 1994;269:18307–18310. [PubMed] [Google Scholar]
- 37.Horton RM, Cai ZL, Ho SN, Pease LR. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 38.Nikaido T, Shimizu A, Ishida N, Sabe H, Teshigawara K, Maeda M, Uchiyama T, Yodoi J, Honjo T. Nature. 1984;311:631–635. doi: 10.1038/311631a0. [DOI] [PubMed] [Google Scholar]
- 39.Berrier AL, Martinez R, Bokoch GM, LaFlamme SE. J. Cell Sci. 2002;115:4285–4291. doi: 10.1242/jcs.00109. [DOI] [PubMed] [Google Scholar]
- 40.Miao H, Nickel CH, Cantley LG, Bruggeman LA, Bennardo LN, Wang B. J. Cell Biol. 2003;162:1281–1292. doi: 10.1083/jcb.200304018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu KL, Khan S, Lakhe-Reddy S, Wang LM, Jarad G, Miller RT, Konieczkowski M, Brown AM, Sedor JR, Schelling JR. Am. J. Physiol. Renal Physiol. 2003;284:829–839. doi: 10.1152/ajprenal.00314.2002. [DOI] [PubMed] [Google Scholar]
- 42.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 43.McCarty JH, Cook AA, Hynes RO. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13479–13483. doi: 10.1073/pnas.0506068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burridge K, Wennerberg K. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 45.Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH, Wallach D. J. Biol. Chem. 1995;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 46.Nobes CD, Hall A. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 47.Kreisberg JI, Ghosh-Choudhury N, Radnik RA, Schwartz MA. Am. J. Physiol. 1997;273:F283–F288. doi: 10.1152/ajprenal.1997.273.2.F283. [DOI] [PubMed] [Google Scholar]
- 48.Patel K, Harding P, Haney LB, Glass WF. J. Cell. Physiol. 2003;195:435–445. doi: 10.1002/jcp.10267. [DOI] [PubMed] [Google Scholar]
- 49.Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, Lim L. Mol. Cell. Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Nat. Cell Biol. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- 51.Sanders LC, Matsumura F, Bokoch GM, De Lanerolle P. Science. 1999;283:2083–2085. doi: 10.1126/science.283.5410.2083. [DOI] [PubMed] [Google Scholar]
- 52.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vastrik I, Eickholt BJ, Walsh FS, Ridley A, Doherty P. Curr. Biol. 1999;9:991–998. doi: 10.1016/s0960-9822(99)80447-3. [DOI] [PubMed] [Google Scholar]
- 54.Kiosses WB, Hood J, Yang S, Gerritsen ME, Cheresh DA, Alderson N, Schwartz MA. Circ. Res. 2002;90:697–702. doi: 10.1161/01.res.0000014227.76102.5d. [DOI] [PubMed] [Google Scholar]
- 55.Kreidberg JA, Symons JM. Am. J. Physiol. 2000;279:F233–F242. doi: 10.1152/ajprenal.2000.279.2.F233. [DOI] [PubMed] [Google Scholar]
- 56.Danen EH, van Rheenen J, Franken W, Huveneers S, Sonneveld P, Jalink K, Sonnenberg A. J. Cell Biol. 2005;169:515–526. doi: 10.1083/jcb.200412081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou H, Kramer RH. J. Biol. Chem. 2005;280:10624–10635. doi: 10.1074/jbc.M411900200. [DOI] [PubMed] [Google Scholar]
- 58.Venstrom K, Reichardt L. Mol. Biol. Cell. 1995;6:419–431. doi: 10.1091/mbc.6.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. J. Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansen K, Abrass CK. Kidney Int. 2003;64:110–118. doi: 10.1046/j.1523-1755.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- 61.Schvartz I, Seger D, Shaltiel S. Int. J. Biochem. Cell Biol. 1999;31:539–544. doi: 10.1016/s1357-2725(99)00005-9. [DOI] [PubMed] [Google Scholar]
- 62.Hansen KM, Berfield AK, Spicer D, Abrass CK. Matrix Biol. 1998;17:117–130. doi: 10.1016/s0945-053x(98)90025-7. [DOI] [PubMed] [Google Scholar]
- 63.Tamaki K, Okuda S, Miyazono K, Nakayama M, Fujishima M. Lab. Investig. 1995;73:81–89. [PubMed] [Google Scholar]
- 64.Hugo C, Shankland SJ, Pichler RH, Couser WG, Johnson RJ. Kidney Int. 1998;53:302–311. doi: 10.1046/j.1523-1755.1998.00774.x. [DOI] [PubMed] [Google Scholar]
- 65.Kikkawa Y, Virtanen I, Miner JH. J. Cell Biol. 2003;161:187–196. doi: 10.1083/jcb.200211121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. J. Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaname S, Uchida S, Ogata E, Kurokawa K. Kidney Int. 1992;42:1319–1327. doi: 10.1038/ki.1992.423. [DOI] [PubMed] [Google Scholar]
- 68.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 69.Turck J, Pollock AS, Lee LK, Marti HP, Lovett DH. J. Biol. Chem. 1996;271:15074–15083. doi: 10.1074/jbc.271.25.15074. [DOI] [PubMed] [Google Scholar]
- 70.Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. J. Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zondag GC, Evers EE, ten Klooster JP, Janssen L, van der Kammen RA, Collard JG. J. Cell Biol. 2000;149:775–782. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nimnual AS, Taylor LJ, Bar-Sagi D. Nat. Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 73.Meng WX, Numazaki M, Takeuchi K, Uchibori Y, Ando-Akatsuka Y, Tominaga M, Tominaga T. EMBO J. 2004;23:760–771. doi: 10.1038/sj.emboj.7600095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carol RJ, Takeda S, Linstead P, Durrant MC, Kakesova H, Derbyshire P, Drea S, Zarsky V, Dolan L. Nature. 2005;438:1013–1016. doi: 10.1038/nature04198. [DOI] [PubMed] [Google Scholar]
- 75.Gibson RM, Wilson-Delfosse AL. Biochem. J. 2001;359:285–294. doi: 10.1042/0264-6021:3590285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akakura S, Sukhwinder SA, Spataro M, Akakura R, Kim JI, Albert ML, Birge RB. Exp. Cell Res. 2004;292:403–416. doi: 10.1016/j.yexcr.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 77.Moissoglu K, Slepchenko BM, Meller N, Horwitz AF, Schwartz MA. Mol. Biol. Cell. 2006;17:2770–2779. doi: 10.1091/mbc.E06-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allenspach EJ, Cullinan P, Tong J, Tang Q, Tesciuba AG, Cannon JL, Takahashi SM, Morgan R, Burkhardt JK, Sperling AI. Immunity. 2001;15:739–750. doi: 10.1016/s1074-7613(01)00224-2. [DOI] [PubMed] [Google Scholar]
- 79.DerMardirossian C, Bokoch GM. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Lin Q, Fuji RN, Yang WN, Cerione RA. Curr. Biol. 2003;13:1469–1479. doi: 10.1016/s0960-9822(03)00613-4. [DOI] [PubMed] [Google Scholar]
- 81.Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Nature. 1991;353:668–670. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- 82.Hart MJ, Maru Y, Leonard D, Witte ON, Evans T, Cerione RA. Science. 1992;258:812–815. doi: 10.1126/science.1439791. [DOI] [PubMed] [Google Scholar]
- 83.Chuang TH, Xu X, Knaus UG, Hart MJ, Bokoch GM. J. Biol. Chem. 1993;268:775–778. [PubMed] [Google Scholar]
- 84.Zhang B, Zhang Y, Dagher MC, Shacter E. Cancer Res. 2005;65:6054–6062. doi: 10.1158/0008-5472.CAN-05-0175. [DOI] [PubMed] [Google Scholar]
- 85.Grande-Garcia A, Echarri A, Del Pozo MA. Biochem. Soc. Trans. 2005;33:609–613. doi: 10.1042/BST0330609. [DOI] [PubMed] [Google Scholar]
- 86.Chuang TH, Bohl BP, Bokoch GM. J. Biol. Chem. 1993;268:26206–26211. [PubMed] [Google Scholar]
- 87.Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. J. Biol. Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- 88.Yamashita T, Tohyama M. Nat. Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- 89.Sivars U, Aivazian D, Pfeffer SR. Nature. 2003;425:856–859. doi: 10.1038/nature02057. [DOI] [PubMed] [Google Scholar]
- 90.Sakisaka T, Meerlo T, Matteson J, Plutner H, Balch WE. EMBO J. 2002;21:6125–6135. doi: 10.1093/emboj/cdf603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DerMardirossian C, Schnelzer A, Bokoch GM. Mol. Cell. 2004;15:117–127. doi: 10.1016/j.molcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 92.Miura Y, Kikuchi A, Musha T, Kuroda S, Yaku H, Sasaki T, Takai Y. J. Biol. Chem. 1993;268:510–515. [PubMed] [Google Scholar]