

Abstract
GABA neurons of the cerebral cortex and other telencephalic structures are produced in the basal forebrain and migrate to their final destinations during the embryonic period. The embryonic basal forebrain is enriched in dopamine and its receptors, creating a favorable environment for dopamine to influence GABA neuron migration. However, whether dopamine receptor activation can influence GABA neuron migration is not known. We show that dopamine D1 receptor activation promotes and D2 receptor activation decreases GABA neuron migration from the medial and caudal ganglionic eminences to the cerebral cortex in slice preparations of embryonic mouse forebrain. Slice preparations from D1 or D2 receptor knock-out mouse embryos confirm the findings. In addition, D1 receptor electroporation into cells of the basal forebrain and pharmacological activation of the receptor promote migration of the electroporated cells to the cerebral cortex. Analysis of GABA neuron numbers in the cerebral wall of the dopamine receptor knock-out mouse embryos further confirmed the effects of dopamine receptor activation on GABA neuron migration. Finally, dopamine receptor activation mobilizes striatal neuronal cytoskeleton in a manner consistent with the effects on neuronal migration. These data show that impairing the physiological balance between D1 and D2 receptors can alter GABA neuron migration from the basal forebrain to the cerebral cortex. The intimate relationship between dopamine and GABA neuron development revealed here may offer novel insights into developmental disorders such as schizophrenia, attention deficit or autism, and fetal cocaine exposure, all of which are associated with dopamine and GABA imbalance.
Keywords: striatum, basal ganglia, ganglionic eminence, dopamine D1 receptor, dopamine D2 receptor, tangential neuronal migration
Introduction
GABA neurons constitute 20–30% of all neurons in the cerebral cortex and perform critical roles in modulating cortical functional output (Cherubini and Conti, 2001; Krimer and Goldman-Rakic, 2001). During development, GABA neurons modulate neocortical neuroepithelial cell proliferation, neuronal migration, and circuit formation (Owens and Kriegstein, 2002; Ben-Ari et al., 2004). Abnormalities in GABA neuron function occur in a variety of developmental disorders, including epilepsy, schizophrenia, anxiety, and autism (Benes, 2000; Treiman, 2001; Levitt et al., 2004). Cortical GABA neurons are produced in the medial and caudal ganglionic eminences of the basal forebrain and migrate to the cerebral wall during the embryonic period (Anderson et al., 1997; Lavdas et al., 1999; Wichterle et al., 2001). A variety of homeobox transcription factors, growth factors and their receptors, or chemoattractant/repulsive molecules regulate cortical GABA neuron migration and development (Rubenstein and Rakic, 1999; Zhu et al., 1999; Powell et al., 2001; Polleux et al., 2002; Alifragis et al., 2004; Flames et al., 2004). However, a direct link between these factors and developmental disorders involving GABA neuron dysfunction is not yet established, suggesting that some other upstream etiological factor(s) may contribute to GABA neuron dysfunction under those conditions.
A hallmark of developmental disorders such as schizophrenia, epilepsy, or autism is the involvement of multiple neurotransmitter systems, especially dopamine and GABA (Benes, 2000; Robinson et al., 2001; Treiman, 2001; Casanova et al., 2003; Levitt et al., 2004). Prenatal cocaine exposure also impairs dopamine (Wang et al., 1995; Friedman et al., 1996; Stanwood and Levitt, 2007) and GABA systems (Crandall et al., 2004). However, whether dopamine can influence GABA neuron development is not known. The embryonic basal forebrain, which is the source of cerebral cortical GABA neurons, is enriched in dopamine and its receptors (Ohtani et al., 2003), offering a favorable environment for dopamine and GABA interactions to occur during brain development.
We show that dopamine can modulate GABA neuron migration from the basal forebrain to the cerebral cortex in embryonic mice and that altering the physiological balance between D1 and D2 receptor activation can impair GABA neuron migration. These data suggest that dopamine imbalance may at least initially contribute to impairment of GABA neuron development in disorders such as schizophrenia or in prenatal cocaine exposure. Furthermore, our data show that dopamine imbalance can impair GABA neuron development even in regions such as the cerebral cortex that do not express high levels of dopamine or dopamine receptors early in embryonic development (Ohtani et al., 2003). Our data offer novel insights into an intimate and symbiotic relationship between dopamine and GABA in the developing brain, which may be pertinent for understanding the pathophysiology of an entire class of neurofunctional developmental disorders characterized by imbalance in multiple neurotransmitter systems.
Materials and Methods
Animals.
Timed-pregnant CD1 mice were purchased from Charles River Laboratories (Wilmington, MA). Colonies of dopamine D1 and D2 receptor knock-out mice (purchased from The Jackson Laboratory, Bar Harbor, ME) were maintained in our institutional animal facility. Female mice housed with a male for the previous 15–17 h were examined for the presence of vaginal plugs at 9:00 A.M. Presence of the plug was taken to indicate conception, and the day of plug discovery was designated embryonic day 0 (E0). Genotyping of the dopamine receptor knock-out mice was performed according to protocols available from The Jackson Laboratory.
Brain slice preparation and culture.
Neuronal migration from the basal forebrain to the cerebral wall was assayed in slice preparations of the embryonic telencephalon as described previously (Crandall et al., 2004). Briefly, E15 mice were removed one at a time from dams deeply anesthetized by an intraperitoneal injection of a mixture of ketamine (50 mg/kg) and xylazine (10 mg/kg). Age of the embryos was confirmed by crown-rump length (13–15 mm) and other external features (Theiler, 1972; Kaufman, 1992). The embryos were decapitated; the brains were removed and embedded in 8% agarose (type VII; Sigma, St. Louis. MO). Coronal sections of the brain were cut at a thickness of 250 μm on a vibratome. The sections were cultured individually on membrane filters in polycarbonate transwells (Costar, Corning, NY), placed in a six-well cell culture plate. Neurobasal medium (Invitrogen, Carlsbad, CA) containing 2% B-27 supplement, penicillin, streptomycin, and glutamine was added beneath the filter into each well. The slices were maintained in culture for 2 d. The medium was replaced at the end of the first 24 h.
Addition of dopamine or dopaminergic drugs.
Dopamine (1, 10, or 20 μm; Sigma) plus ascorbic acid (0.01%; Sigma), ascorbic acid alone (0.01%), dopamine D1 like receptor agonist SKF 81297 (6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide) (1 or 10 μm; Sigma), or D2 like receptor agonist quinpirole hydrochloride (10 or 20 μm; Sigma) was added to the medium 2–3 h after plating. Ascorbic acid was used as an antioxidant to retard oxidation of dopamine in the culture medium (Ohtani et al., 2003; Popolo et al., 2004). In other experiments, dopamine D1 like receptor antagonist SCH 23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride] (10 μm; Sigma) or D2 like receptor antagonist eticlopride (20 μm) was added to the culture medium 2–3 h after plating the cells, and, 2 h later, dopamine plus ascorbic acid or ascorbic acid alone were added.
DiI labeling of cells in the slice preparations.
Glass shards coated with DiI (Invitrogen) were prepared (Crandall et al., 2004) and inserted into the basal forebrain with the aid of a stereomicroscope immediately after plating each slice. We classified the slices into rostral or caudal groups based on anatomical landmarks (Fig. 1). A principal criterion was that the rostral slices contained both the lateral and medial ganglionic eminences (Fig. 1A), whereas a single, caudal ganglionic eminence could be identified in the caudal slices (Fig. 1B). Additionally, the rostral slices contained striatal and septal primordia (Fig. 1A), whereas rostral thalamic structures were visible in the caudal slices (Fig. 1B). Although the majority of the cells migrating to the cerebral wall originate in the medial and caudal ganglionic eminences (Wichterle et al., 1999, 2001; Parnavelas, 2000; Nery et al., 2002), at E15 some cells derived from the lateral ganglionic eminence also migrate to the cerebral wall (Anderson et al., 2001). It is not possible to reliably label only the cells derived from the lateral ganglionic eminence or the medial ganglionic eminence in the rostral slices because the migratory paths of the cells overlap within the basal forebrain. Therefore, in rostral slices, the DiI was inserted ∼200–400 μm ventral to the pallidal-striatal angle (Fig. 1A) so as to label cells originating in the lateral as well as medial ganglionic eminences. In the caudal slices, the migrating cells originate in the caudal ganglionic eminence, and the DiI crystals were inserted in the caudal ganglionic eminence, 200–400 μm ventral to the pallial-striatal angle in these slices (Fig. 1B).
Figure 1.

Neuronal migration assay using DiI labeling of migrating cells in forebrain slice preparations from E15 mice. A and B are bright-field images of representative rostral (A) and caudal (B) slices. DiI crystals (red clumps) were inserted approximately at the border between the lateral and medial ganglionic eminences (LGE and MGE, respectively) in rostral slices (A) and in the caudal ganglionic eminence (CGE) in caudal slices (B). In the rostral slices, the DiI-labeled cells originate in both the lateral and medial ganglionic eminences and migrate in multiple directions (red arrows), including toward the cerebral wall (CX). In the caudal slices, the DiI-labeled cells originate in the caudal ganglionic eminence and also migrate in multiple directions (red arrows), including toward the cerebral wall (CX). We counted the total number of DiI-labeled cells in the slices and calculated the percentage of cells entering the cerebral cortex (as a percentage of the total number of DiI-labeled cells in the slice). C–E are examples of slice preparations showing DiI-labeled cells (white arrows) that have migrated to the cerebral cortex (CX). The slices were exposed to medium without any drugs (control; C), medium containing the D1 receptor agonist SKF 81297 (1 μm; D), or the D2 receptor agonist quinpirole (20 μm; E) and maintained for 2 d in vitro. The DiI deposition site in the basal forebrain is visible (white asterisks). S, Septum; TH, thalamus, LV, lateral ventricle.
Analysis of cell migration.
After 2 d in culture, the slices were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, and viewed as whole mounts in a fluorescence microscope fitted with tetramethylrhodamine isothiocyanate filters (examples in Fig. 1C–E). The extent of the DiI injection site was delimited for each slice as a solidly intense fluorescent area. The position of DiI-labeled neurons was recorded with respect to the boundaries of the slice using an image analysis system (IPLab; Scanalytics, Rockville, MD) interfaced to the microscope. A single investigator blinded to the identity of the experimental group performed the analyses. An overlay grid of rectangles (each 640 × 480 μm), “grid boxes,” was placed on the image of the slice to measure the overall extent of neuronal migration. We calculated the percentage of DiI-labeled cells that entered the cerebral wall as a percentage of the total number of DiI-labeled cells in that slice. For this measurement, any DiI-labeled cell found dorsal and lateral to the pallial-striatal angle was considered to have reached the cortex.
Electroporation of D1 receptor constructs in slice preparations.
Wild-type mouse D1 receptor–enhanced green fluorescent protein (EGFP) (Araki et al., 2006) was inserted downstream of the second CAG promoter of pCAG–enhanced yellow fluorescent protein (EYFP)–CAG vector (Niwa et al., 1991) (kind gift from Dr. T. Saito, Kyoto University, Kyoto, Japan). The D1 receptor–EGFP construct used here induces expression of a functional D1 receptor as shown by agonist-induced upregulation of dopamine and cAMP-regulated phosphoprotein-32 and cAMP synthesis (Araki et al., 2006). However, the D1 receptor–EGFP construct did not produce strong EGFP fluorescence in the transfected cells (Araki et al., 2006). Therefore, in the present study, we used the pCAG–EYFP–CAG vector to enhance the fluorescence (with EYFP) to facilitate tracking the electroporated cells in our slice preparations. For electroporation, we placed a 1 mm platinum Petri dish electrode (positive pole; Protech International, San Antonio, TX) beneath the slice and injected 5 μg of DNA into the electroporation site using a micropipette (Narishige, Greenvale, NY). A 0.1 mm finely etched needle platinum electrode (gift from Edward Stern, Massachusetts General Hospital) was placed in the slice, and three to five electrical pulses were delivered (each pulse 5 ms width, 100 Hz, and 50V; ElectroSquarePorator T830; BTX, Hawthorne, NY).
As in the case of the DiI labeling experiments, the site of electroporation was ∼200–400 μm ventral to the pallial-striatal angle in the rostral and caudal slices (Fig. 2A). Thus, in the rostral slices, the electroporated cells originated in the lateral and medial ganglionic eminences, whereas in the caudal slices, the electroporated cells originated from the caudal ganglionic eminence (Fig. 2A). The electroporated cells do not express EYFP fluorescence for 12–24 h after electroporation. By 24–48 h, the EYFP expressing electroporated cells are widely scattered (Fig. 2B–F). As a result, the site of electroporation is not evident as a congregation of EYFP-positive cells. Therefore, unlike the DiI deposition sites (Fig. 1C–E, white asterisks), the electroporation sites are difficult to illustrate in photomicrographs. To ascertain that the site of electroporation was comparable with the site of DiI deposition (Fig. 1A,B), we included fast blue dye with the DNA constructs to verify our DNA injection and needle electrode placement sites in our preliminary experiments.
Figure 2.

D1 receptor gain-of-function assay was performed by electroporating D1 receptor–EYFP constructs into the basal forebrain of E15 mouse brain slices. A, The location of the electroporation sites (green asterisks) is shown in diagrams of representative rostral and caudal slices. As in the DiI-labeling experiments (Fig. 1), the constructs were electroporated at the border between the lateral and medial ganglionic eminences (LGE and MGE, respectively) in rostral slices and in the caudal ganglionic eminence (CGE) in caudal slices. The electroporated cells scatter widely within 24 h, making it difficult to illustrate a prominent EYFP-positive electroporation site. Green arrows in A indicate that EYFP-labeled cells migrate in multiple directions, including toward the cerebral wall (CX). B–F, Examples of E15 mouse brain slices electroporated with the EYFP-labeled constructs and maintained in vitro for 2 d. A fluorescence image (B) is superimposed on the bright-field image (C) to register position of labeled cells with anatomical landmarks. Images in D–F are such superimposed images. Labeled cells are seen near the site of electroporation in the lateral ganglionic eminence (LGE; B–D), medial ganglionic eminence (D), caudal ganglionic eminence (E), and in the differentiating striatum (STR; white arrows in B and C; black arrows in D and E). Some electroporated, EGFP-positive cells have migrated to the cortex over the 2 d period (black arrows in E). The shaded areas in A represent the lateral ventricles. V, Lateral ventricle; HIP, hippocampus.
The D1 receptor agonist SKF 81297 (1 μm; concentration found to be effective in previous slice preparation experiments) was added to the culture medium immediately after electroporation, and the slices were maintained in culture for 2 d. Control cultures did not receive any additives. After the 2 d culture period, electroporated EYFP-labeled cells were observed in the developing striatal fields (Fig. 2B–E), at the level of the lateral (Fig. 2B,C), medial (Fig. 2D), and caudal (Fig. 2E) ganglionic eminences as well as in the cortex (Fig. 2F). We analyzed the position and number of EYFP-labeled cells in the slices, as described above for DiI-labeling experiments.
GABA Immunohistochemistry.
E15 mice were perfused with 2% acrolein in 0.1 m phosphate buffer, pH 7.2, and the brains were fixed overnight at 4°C in the same fixative, cryoprotected in 30% sucrose, and sectioned at 50 μm thickness in the coronal plane on a sliding microtome. The sections were processed for immunohistochemistry using antibodies to GABA (rabbit polyclonal, 1:3000; Incstar, Stillwater, MN), biotinylated secondary antibody (Jackson ImmunoResearch, West Grove, PA), avidin–biotin kit (Vector Laboratories, Burlingame, CA), and nickel ammonium sulfate (0.02%; Sigma) enhancement of diaminobenzidine reaction product as described previously (Crandall et al., 2004).
Calculation of GABA-positive cell numbers.
GABA-positive cells were counted by a single investigator blinded to experimental treatment groups in 50-μm-thick sections through the presumptive medial prefrontal cortex. The counts were performed using a 40× objective lens in a sector that extended from the ventricular border to the pial surface of the cerebral wall. The counts were expressed as number of cells per 300 μm2. Subsequently, the number of GABA-positive cells was reanalyzed separately for the different laminas of the cerebral wall: ventricular zone (VZ)/subventricular zone (VZ), intermediate zone (IZ), subplate (SP)/cortical plate (CP), and the marginal zone (MZ).
Cytoplasmic dynein heavy chain and TuJ1 immunocytochemistry in striatal neurons exposed to D1 or D 2 receptor agonists in culture.
Dams carrying E15 mice were anesthetized, as described above. The embryos were removed and decapitated, and the brains were dissected in cold PBS. Basal forebrain samples containing neostriatal differentiating fields as well as the lateral, medial, and caudal ganglionic eminences were dissected from the overlying cerebral wall, washed with sterile PBS, and incubated with 0.25% trypsin at 37°C for 20 min. The samples were washed with culture medium, and the cells were gently dissociated using a sterile plastic pipette, passed through a 40 μm cell strainer, and resuspended in neurobasal medium (Invitrogen) containing 0.1 g/l penicillin/streptomycin and supplemented with B27 (Invitrogen) 2 mm l-glutamine, and 10% fetal bovine serum (Invitrogen). The cells were plated on 24-well poly-d-lysine and laminin BD Biocoat tissue culture plates (BD Biosciences, Bedford, MA) at a density of 300,000 cells per well. Cultures were maintained in a humidified incubator at 37°C under 5% CO2. On day 3 in vitro, cells were treated for 24 h with cytarabine (10 μm) to kill proliferating cells. On day 7 in vitro, cells were treated for 24 h with D1 receptor agonist SKF 81297 (1 μm) or D2 receptor agonist quinpirole hydrochloride (20 μm). Control cultures did not receive any additives. The cultures were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer for 15 min. Immunocytochemistry was performed using rabbit anti-cytoplasmic dynein heavy chain (CDHC) at 1:100 dilution (sc-9115; Santa Cruz Biotechnology, Santa Cruz, CA) or mouse anti-β-III tubulin at 1:500 (TuJ1 clone MAB5544; Chemicon, Temecula, CA) antibodies. Rabbit anti-CDHC signal was detected with biotinylated horse anti-rabbit at 1:500 (Jackson ImmunoResearch) and streptavidin–Alexa 546 at 1:500 (Invitrogen). β-III tubulin was detected with anti-mouse Alexa 488 at 1:500 (Invitrogen). Nuclei were labeled with DAPI.
Results
Although genes encoding five dopamine receptors have been cloned (Sibley and Monsma, 1992), based on mechanisms of action and availability of pharmacological ligands (Stoof and Kebabian, 1984), two major categories of dopamine receptors are commonly recognized: D1 receptor (D1 and D5; also called D1A and D1B, respectively, in rodents) and D2 receptor (D2, D3, and D4). When we refer to the D1 or D2 receptor in this study, we refer collectively to the D1 and D5 subtypes or D2, D3, and D4 subtypes, respectively. However, in the dopamine receptor knock-out mice used in this study, genes encoding either the D1 or the D2 subtype are deleted, leaving genes for the other receptor subtypes intact.
Dopamine D1 receptor activation increases and D2 receptor activation decreases basal forebrain to cerebral wall neuronal migration
We examined the effects of D1 or D2 receptor activation on neuronal migration from the basal forebrain to the cerebral wall in E15 mouse forebrain slice preparations. We added D1 receptor agonist SKF 81297 (1 or 10 μm) or D2 receptor agonist quinpirole (10 or 20 μm) to the culture medium immediately after application of the DiI. Control cultures did not contain any drugs. Our previous in vivo and in vitro studies (Ohtani et al., 2003; Popolo et al., 2004; Araki et al., 2006) confirmed the specificity and selectivity of both the agonists at the concentrations used here. Figure 1 shows examples of DiI-labeled slices from control (Fig. 1C), D1 agonist-treated (Fig. 1D), and D2 agonist-treated (Fig. 1E) experimental groups. Individual DiI-labeled cells at higher magnification are shown in Figure 3A.
Figure 3.
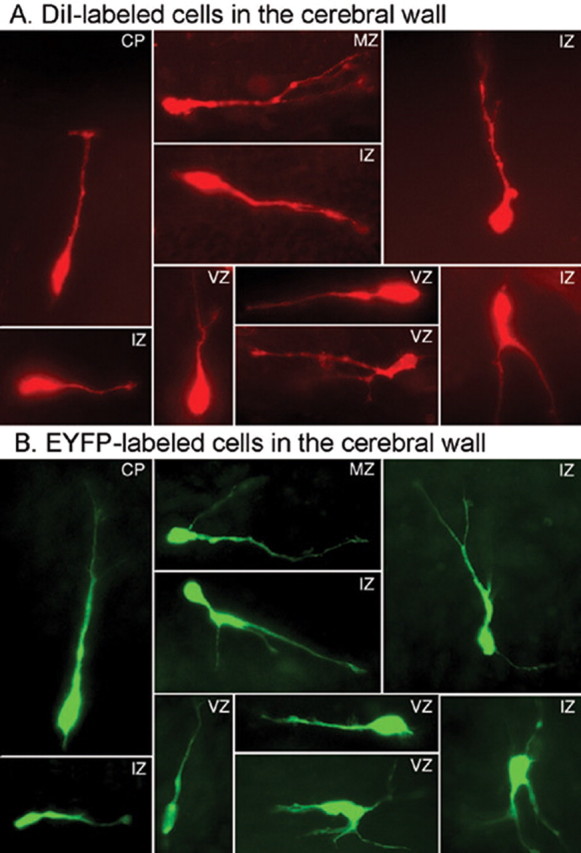
Morphology of migrating neurons. A, B, Cells with multiple morphological characteristics of migrating neurons were found entering the cerebral wall after DiI labeling (A) or electroporation of D1 receptor–EYFP constructs (B) in E15 forebrain slices. Examples are taken from different laminas of the cerebral wall of slices exposed to the D1 receptor agonist SKF 81297.
Initially, we analyzed the data separately for the rostral and caudal slices to determine whether the effects of D1 or D2 receptor activation on neuronal migration were different on cells originating in the lateral/medial ganglionic eminences (rostral slices) compared with the effects on cells derived from the caudal ganglionic eminence (caudal slices). The average percentage of DiI-labeled cells entering the cerebral wall was not different between the rostral and caudal slices after exposure to the D1 receptor agonist SKF 81297 [rostral (n = 5), 69.1 ± 5.1; caudal (n = 4), 65.6 ± 8.9; t = 0.37; p = 0.7] or the D2 receptor agonist quinpirole [rostral (n = 3), 33.9 ± 0.8; caudal (n = 4), 40.9 ± 3.4; t = 1.73; p = 0.17]. Therefore, in the following description, we present data combined from the rostral and caudal slices (i.e., from the lateral, medial, and caudal ganglionic eminences).
The D1 receptor agonist SKF 81297 (1 μm) significantly increased the percentage of DiI-labeled cells reaching the cerebral wall [∼20%; control (n = 16), 51.9 ± 3.6; SKF 81297 (n = 17), 62.7 ± 3.5; t = 2.18; p = 0.04], whereas the D2 receptor agonist quinpirole (20 μm) significantly decreased it by ∼38% [quinpirole at 20 μm (n = 14), 32.4 ± 3.5; control (n = 16), 51.9 ± 3.6; t = 4.01; p < 0.01). Quinpirole did not produce significant effects at 10 μm concentration [quinpirole at 10 μm (n = 13), 58.02 ± 4.8; control (n = 16), 51.9 ± 3.6; t = 0.274; p = 0.27). SKF 81297 also did not produce significant effects at 10 μm concentration [SKF 81297 at 10 μm (n = 9), 56.54 ± 2.7; control (n = 16), 51.9 ± 3.6; t = 0.83; p = 0.42). Therefore, we used 1 μm SKF 81297 and 20 μm quinpirole in of all our subsequent experiments.
Variability in the size of the DiI deposit can introduce variability in the numbers of DiI-labeled cells entering the cerebral wall. We took two measures to address the issue. First, as mentioned in Materials and Methods, we calculated the percentage of DiI-labeled cells that entered the cerebral wall as a percentage of the total number of DiI-labeled cells in a given slice, in all of our experiments. Therefore, any variability in the total number of DiI-labeled cells could not have contributed to variability in the relative numbers of DiI-labeled cells migrating to the cerebral wall in the different slices. Second, we compared the average total numbers of DiI-labeled cells in each slice in each of the experimental groups. We found that the average total DiI-labeled cell numbers were not significantly different among the different experimental groups [SKF 81297 (n = 17), 233.5 ± 47.7; control (no drugs; n = 16), 292.9 ± 47.0; t = 1.82; p = 0.18; quinpirole (n = 14), 256.9 ± 71.6 vs control, t = 1.14, p = 0.27]. Thus, variability in the total numbers of cells labeled in a slice did not produce artifacts in our data.
Next, we exposed the slices to the D1 receptor antagonist SCH 23390 [10 μm; concentration shown to be effective previously (Ohtani et al., 2003; Popolo et al., 2004; Araki et al., 2006)] for 2 h and then added dopamine (10 μm; see below for determination of effective concentration) to the medium in the presence of the antagonist. In this paradigm SCH 23390 blocks the D1 receptors, and dopamine can activate only D2 receptors. We found that the percentage of DiI-labeled cells reaching the cerebral wall was significantly decreased by ∼45% [SCH 23390 (n = 6), 23.4 ± 6.7; control (n = 16), 51.9 ± 3.6; t = 3.76; p < 0.01], confirming that blocking the D1 receptor and activating the D2 receptor decreases the neuronal migration. We performed the complementary experiment by adding the D2 receptor antagonist eticlopride [20 μm; concentration shown to be effective in previous studies (Ohtani et al., 2003; Popolo et al., 2004; Araki et al., 2006)] to the medium first and dopamine (10 μm) 2 h later. In this experiment, dopamine can activate only D1 receptors, and the percentage of DiI-labeled cells reaching the cerebral wall increased significantly by ∼18% [eticlopride (n = 4), 68.8 ± 7.1; control (n = 16), 51.9 ± 3.6; t = 2.12; p = 0.04]. Thus, blocking the D2 receptor and activating the D1 receptor increased the migration.
Dopamine increases basal forebrain to cerebral wall neuronal migration
Because dopamine is the naturally occurring ligand for D1 and D2 receptors, we examined whether dopamine per se influenced neuronal migration. We added dopamine (1, 10, or 20 μm) dissolved in the antioxidant ascorbic acid (0.01%) or ascorbic acid alone to the medium and placed DiI in the ganglionic eminence. Dopamine at the concentration used here activates its receptors in vivo and in vitro (Ohtani et al., 2003; Popolo et al., 2004; Araki et al., 2006). After a 48 h culture (change of medium and additives after the first 24 h), dopamine (10 μm) significantly increased by ∼15% the percentage of DiI-labeled cells reaching the cerebral wall [dopamine plus ascorbic acid (n = 17), 60.6 ± 1.5; ascorbic acid alone (n = 7), 45.5 ± 3.8; t = 2.87; p = 0.01]. Ascorbic acid alone did not produce significant effects [ascorbic acid (n = 7), 45.5 ± 3.8; control (no additives; n = 7), 51.9 ± 3.6; t = 0.95; p = 0.34]. Dopamine did not produce significant effects at 1 or 20 μm concentrations [1 μm (n = 15), 43.23 ± 3.2; 20 μm (n = 8), 44.27 ± 3.6; control (ascorbic acid; n = 7), 45.5 ± 3.8; 1 μm vs control, t = 1.70, p = 0.1; 20 μm vs control, t = 1.38, p = 0.18]. The 1 μm concentration may have been too low to activate the receptors, whereas at 20 μm, both D1 and D2 receptors may have been activated, offsetting the effects of each other.
These data indicate that dopamine increases neuronal migration to the cerebral wall, an effect that is similar to the effects of D1 receptor activation and opposite of D2 receptor activation. A likely explanation for D1 receptor-like effects of dopamine is that the density of D1 receptor binding sites is 10-fold higher than D2 receptor binding sites in the E15 mouse striatum (Ohtani et al., 2003). Therefore, dopamine at the concentration used here (10 μm) may activate the D1 receptors predominantly.
Gain of D1 receptor function increases neuronal migration from the basal forebrain to the cerebral wall
We electroporated D1 receptor–EYFP (D1R–EYFP) or EYFP-only constructs into cells of the basal forebrain in E15 slice preparations. The electroporated cells expressed EYFP fluorescence, were viable for at least 48 h in vitro, and were observed in the striatal fields, ganglionic eminences, and the cerebral cortex (Fig. 2B–F). Because the culture medium does not contain dopamine or any other D1 receptor ligand, we exposed the D1R–EYFP electroporated slices to the D1 receptor agonist SKF 81297 (1 μm). Control slices also received D1 receptor–EYFP electroporation but no pharmacological agents. There was a significant ∼30% increase in the average percentage of D1R–EYFP electroporated cells entering the cerebral wall in the slices exposed to SKF 81297 compared with control slices [control (n = 4), 47.4 ± 5.2; SKF 81297 (n = 3), 67.3 ± 4.2; t test = 2.82; p = 0.04]. These data demonstrate that activation of the electroporated D1 receptor is sufficient to augment neuronal migration.
Electroporation per se did not impair cell migration because migration of the EYFP-labeled cells to the cerebral wall was comparable with migration of DiI-labeled cells (compare Figs. 1, 2). In other words, the percentage of EYFP-labeled cells entering the cerebral wall in the control slices (i.e., without SKF 81297) was comparable with the percentage of labeled cells entering the cerebral wall in our DiI labeling studies under control conditions (i.e., no drugs added). Thus, the percentage of EYFP-labeled cells in the control slices was 47.4 ± 5.2; DiI-labeled cells in control slices was 51.9 ± 3.6 in our experiments using the D1 or D2 receptor agonists/antagonists and 52.5 ± 1.2 in slices from wild-type littermates of the dopamine receptor knock-out mice.
As in the case of the DiI-labeling experiments, we standardized the size of the electroporation site to minimize the variation in the number of EYFP-labeled cells per slice under the different conditions. There was no statistically significant difference in the total numbers of EYFP-labeled cells per slice [D1R-EYFP no drugs, 219.8 ± 32.6 (n = 4); D1R-EYFP plus SKF 81297, 236.7 ± 69.1 (n = 3); t = 0.24; p = 0.82). As was the case with the percentage of the EYFP-labeled cells entering the cerebral wall, the total numbers of EYFP-labeled cells per slice were comparable with the corresponding numbers obtained in our DiI-labeling experiments
We did not perform D2 receptor electroporation because we were concerned that the effects of gain of D2 receptor function may be less pronounced than that of the D1 receptor. In our studies with receptor agonists, the magnitude of the effects of activation of the endogenous D2 receptor was greater than that of the endogenous D1 receptor (38 vs 20%). Therefore, activation of the electroporated D2 receptor might not have decreased the migration further (compared with the activation of the endogenous receptors already present on the cells), making the D2 receptor electroporation potentially less informative than the D1 receptor electroporation.
Dopamine receptor activation influences migration of multiple cell types
Migrating neurons display different morphologies, each believed to correlate with specific stages or modes of migration (Nadarajah et al., 2002; Tabata and Nakajima, 2003; Bellion et al., 2005). Different morphologies may also correlate with neuronal neuropeptide content or functional subtype (Xu et al., 2004; Butt et al., 2005). Because neuropeptide expression patterns of GABA neurons are not fully established until postnatal stages (Xu et al., 2004), we examined different morphological types of DiI- or EYFP-labeled neurons in the cerebral wall. Cells with multiple morphologies, from bipolar to multipolar, were present under the different experimental conditions (Fig. 3). The laminar position of the migrating cells coupled with orientation of the long axis (horizontal or vertical) with respect to the ventricular surface may reflect modes of migration (e.g., ventricle or pia directed) (Nadarajah et al., 2001, 2002) or cell fate (progenitor or postmitotic) (Noctor et al., 2004). Our analysis did not reveal differences with respect to orientation of the cell soma in the different laminas of the cerebral wall: all orientations in all laminas were seen under the different conditions (Fig. 3). Therefore, dopamine receptor activation did not produce selective effects on particular cell types.
Dopamine receptor loss of function and basal forebrain to cerebral wall neuronal migration
We analyzed neuronal migration in slice preparations from wild-type and D1 or D2 receptor knock-out mice (crosses between heterozygous mice). We exposed the slices to dopamine (10 μm) plus ascorbic acid (0.01%) for 48 h. In the D1 receptor knock-out mouse, dopamine can activate only D2 receptors [and D5 receptors, which have low expression in the striatum (Vrana et al., 1995; Jung and Bennett, 1996; Surmeier et al., 1996)], and, in the D2 knock-out mouse, it can activate only D1 receptors [and D3, D4, or D5 receptors, low in the striatum (Vrana et al., 1995; Jung and Bennett, 1996; Surmeier et al., 1996)]. Because D1 receptor activation increases and D2 receptor activation decreases migration, we predicted that the percentage of DiI-labeled cells entering the cerebral wall would be decreased in the D1 receptor knock-out mice and increased in the D2 knock-out mice. This was indeed the case. We found an ∼51% decrease in the percentage of DiI-labeled cells entering the cerebral wall in the D1 knock-out mice (26.7 ± 2.8; n = 8) compared with wild-type (52.3 ± 2.3; n = 5) littermates (t = 6.32; p < 0.01). There was an ∼23% increase in neuronal migration in the D2 receptor knock-out (68.2 ± 2.6; n = 5) compared with wild-type (52.5 ± 1.2; n = 6) mice (t = 5.82; p < 0.001). These data confirm previous data using D1 or D2 receptor agonist/antagonist that activation of the D1 receptors increases and D2 receptors decreases basal forebrain to cerebral wall neuronal migration. Figure 4 shows examples of DiI-labeled slices from wild-type (Fig. 4A), D1 receptor knock-out (Fig. 4B), and D2 receptor knock-out (Fig. 4C) mice.
Figure 4.

A–C, Examples of slice preparations from wild-type (WT; A), D1 receptor knock-out (D1−/−; B) and D2 receptor knock-out (D2−/−; C) E15 mouse embryos showing DiI-labeled cells (white arrows) that have migrated from the basal forebrain to the cerebral cortex (Cortex). The slices were exposed to dopamine (10 μm) plus ascorbic acid (0.01%) and maintained for 2 d in vitro. The DiI deposition site in the basal forebrain is visible (white asterisks). There are fewer DiI-labeled cells in the cortex of the D1 receptor knock-out mouse and relatively greater numbers in the cortex of the D2 receptor knock-out mouse, indicating the opposite effects of the loss of D1 or D2 receptor on neuronal migration.
As in the case of the experiments using the D1 and D2 receptor agonists in the wild-type mice, we took two measures to address potential variation in the numbers of DiI-labeled cells per slice under the different conditions. First, we calculated the percentage of DiI-labeled cells that entered the cerebral wall as a percentage of the total number of DiI-labeled cells in a given slice. Therefore, any variability in the total number of DiI-labeled cells could not have contributed to variability in the relative numbers of DiI-labeled cells migrating to the cerebral wall in the different genotypes. Second, we compared the average total numbers of DiI-labeled cells in each slice in each genotype. There was no statistically significant difference in the total numbers of DiI-labeled cells per slice between the different groups [wild type, 237.5 ± 45 (n = 5); D1 knock-out, 260.9 ± 42.1 (n = 8); t = 0.37; p = 0.71; D2 knock-out, 299.8 ± 89.7 (n = 5); wild type, 281.0 ± 20.0 (n = 5); t = 1.41; p = 0.19].
Numerical density of GABA-positive cells in the cerebral wall of dopamine receptor knock-out mice
Because the majority of DiI-labeled neurons entering the cerebral wall are GABA neurons (Nery et al., 2002; Xu et al., 2004; Butt et al., 2005; Yozu et al., 2005) and because neuronal migration is affected in the D1 and D2 receptor knock-out mice, we predicted that the numbers of GABA-positive cells in the cerebral wall of E15 D1 or D2 receptor knock-out mice would be altered. We performed immunohistochemistry on sections of E15 wild-type (WT), D1 receptor knock-out (D1−/−), and D2 receptor knock-out (D2−/−) embryos to identify GABA-positive cells (Fig. 5) and calculated numerical density of GABA-positive cells in the presumptive medial prefrontal cortex (Table 1). We found a significant ∼25% decrease in GABA-positive cells in the D1 receptor knock-out mice and a significant ∼44% increase in the D2 receptor knock-out mice (Table 1). Early (E12–E14) versus late (E15) generated GABA neurons and GABA neurons derived from the lateral, medial, and caudal ganglionic eminences may follow different migration trajectories and arrive in different laminas of the embryonic cerebral wall (Anderson et al., 2001; Ang et al., 2003). The loss of D1 or D2 receptor may affect the different populations differently. Therefore, we analyzed GABA-positive cells separately in each lamina: VZ, SVZ, IZ, CP, SP, and MZ (Fig. 4, Table 1). The average GABA cell numerical density in the MZ or CP/SP was not affected significantly in D1 or D2 receptor knock-out mice (Table 1). However, in the IZ, GABA cell numerical density decreased by ∼48% in the D1 receptor knock-out mice and increased by ∼96% in the D2 receptor knock-out mice (Table 1). Similarly in the VZ/SVZ, the GABA cell numerical density significantly decreased by ∼53% in the D1 receptor knock-out mice and significantly increased by ∼143% in the D2 receptor knock-out mice (Table 1). Thus, the overall effects of the loss of the D1 or D2 receptor appear to be attributable to the significant changes in the IZ and VZ/SVZ.
Figure 5.

GABA-positive cells in the cerebral wall of E15 mouse embryos. A–C, GABA immunohistochemistry was performed in coronal sections through the medial prefrontal cortex of littermate E15 wild type (WT; A); D1 receptor knock-out (D1−/−; B) and D2 receptor knock-out (D2−/−; C) mice. GABA-positive cells (black arrows) are distributed in all laminas of the cerebral wall. Overall, there are fewer GABA-positive profiles in the D1−/− cerebral wall (B) and greater numbers in the D2−/− cerebral wall (C) compared with the wild type (A). GABA labeling shows variability among the different laminas and across genotypes (especially SP). Quantitative comparisons (see Table 1) showed significant differences among the three genotypes in the IZ, VZ, and SVZ.
Table 1.
GABA-positive cell numerical density in the E15 presumptive medial prefrontal cortex
| Experimental group | Mean ± SEM | t value* | p value |
|---|---|---|---|
| Entire cerebral wall (all laminas) | |||
| WT | 86.9 ± 5.9 | ||
| D1-KO | 64.8 ± 2.1 | 3.51 | 0.013 |
| D2-KO | 123.5 ± 1.9 | 5.87 | 0.001 |
| Marginal zone | |||
| WT | 34.6 ± 2.4 | ||
| D1-KO | 31.4 ± 0.8 | 1.26 | 0.25 (NS) |
| D2-KO | 37.4 ± 2.4 | 0.86 | 0.43 (NS) |
| Cortical plate + subplate | |||
| WT | 22.8 ± 2.4 | ||
| D1-KO | 18.2 ± 3.0 | 1.18 | 0.28 (NS) |
| D2-KO | 33.9 ± 6.8 | 1.62 | 0.16 (NS) |
| Intermediate zone | |||
| WT | 26.5 ± 3.0 | ||
| D1-KO | 13.8 ± 1.3 | 3.83 | 0.009 |
| D2-KO | 51.9 ± 3.3 | 5.68 | 0.001 |
| Ventricular zone + subventricular zone | |||
| WT | 3.0 ± 0.6 | ||
| D1-KO | 1.4 ± 0.3 | 2.44 | 0.02 |
| D2-KO | 7.3 ± 0.9 | 4.04 | 0.007 |
We calculated GABA-positive cell numbers in the entire cerebral wall as well as in each lamina between wild-type (WT), D1 receptor knock-out (D1-KO), and D2 receptor knock-out (D2-KO) embryos and compared the data between groups using a t test. GABA-positive cell numerical density in the E15 presumptive medial prefrontal cortex.
*, All comparisons are to the wild-type group; n = 4 embryos for each experimental group. NS, Not statistically significant, i.e., p > 0.05.
Dopamine receptor activation induces redistribution of cytoplasmic dynein motor protein
Migrating neurons undergo cytoskeletal reorganization concurrent with process extension or nuclear translocation (Bellion et al., 2005; Schaar and McConnell, 2005). A key mediator of the cytoskeletal reorganization is the motor protein CDHC. Changes in the intracellular distribution of CDHC are implicated in mediating neuronal migration (Morris et al., 1998; Morris, 2000). CDHC localizes to centrosomes near the nucleus in mitotically active neuroepithelial precursor cells and distributes along the neurites in differentiating or migrating neurons (Sasaki et al., 2000; Wynshaw-Boris and Gambello, 2001; Shu et al., 2004). We examined whether dopamine receptor activation altered the distribution of CDHC in primary neuronal cultures obtained from the ganglionic eminence/striatum of E15 mice. Because CDHC associates with neuronal class III β-tubulin (TuJ1), we also examined the distribution of the TuJ1 and its colocalization with CDHC. The cultures were exposed to the D1 receptor agonist SKF 81297 (1 μm) or the D2 receptor agonist quinpirole (20 μm), and CDHC and TuJ1 were labeled by immunocytochemistry. In control cultures, CDHC was localized predominantly to the cell body (Fig. 6B; higher-magnification view in BA), and TuJ1 was principally localized to the neurites (Fig. 6C). In cultures exposed to the D1 receptor agonist SKF 81297, CDHC localized to the soma, with extensions into the proximal segments of the neurites near the soma (Fig. 6F; higher magnification in FA). In these cultures, TuJ1 labeling was more intense than in controls, although it remained localized to the neurites (Fig. 6G), as in control cultures. CDHC and TuJ1 colocalization produced a prominent tuft-like pattern extending into the proximal segments of the neurites (Fig. 6H, arrows). In cultures exposed to the D2 receptor agonist quinpirole, the labeling pattern was strikingly different: the CDHC labeling was virtually exclusively localized to the cell body and did not extend into the neurites (Fig. 6J; higher magnification in JA). TuJ1 localized to neurites (Fig. 6K), as in control cultures (Fig. 6C), but unlike in SKF 81297-exposed cultures, it was not localized to the cytoplasm. CDHC–TuJ1 colocalization (Fig. 6L) did not reveal tuft-like localization that was seen in the SKF 81297-exposed cultures. Thus, D1 receptor activation increased TuJ1 labeling intensity and mobilized CDHC and TuJ1 to produce a tuft-like pattern extending into the proximal segments of the neurites, whereas D2 receptor activation produced a condensation of the labeling around the nucleus (Fig. 6M).
Figure 6.

Dopamine receptor activation and neuronal cytoskeleton. Striatal neurons from E15 mice were grown in culture for 5 d and exposed to the D1 receptor agonist SKF 81297 or the D2 receptor agonist quinpirole. Immunocytochemistry was performed to analyze distribution of the motor protein CDHC or the cytoskeletal protein neuronal β-III tubulin (TuJ1). A, E, I, Nuclei were labeled with the DNA stain DAPI. B, C, In control cultures (B), CDHC was distributed throughout the cell body and the neurites (higher-magnification view in BA), and TuJ1 was principally localized to the neurites (C). D, Merged image of CDHC and TuJ1 labeling revealed overlap between the two proteins mainly in the neurites. F, In cultures exposed to SKF 81297, CDHC was distributed mainly to the soma, with prominent extensions into the proximal segments of the neurites near the cell soma (higher magnification in FA). G, In these cultures, TuJ1 was distributed to the neurites, as in control cultures. However, TuJ1 labeling was more intense in the neurites in the SKF 81297-exposed cultures compared with controls (C vs G). H, Merged images of the SKF 81297-exposed cultures revealed prominent tuft-like colocalization of CDHC and TuJ1, near the nucleus and extending into the proximal segments of the neurites (arrows in H). J, In cultures exposed to quinpirole, CDHC labeling was condensed around the nucleus (high-power view in jA). K, TuJ1 was distributed to neurites, as in control cultures (C), but, unlike in SKF 81297-exposed cultures, it was not localized to the cytoplasm. L, Merged images of CDHC and TuJ1 labeling did not reveal the prominent tuft-like colocalization. M, In summary, D1 receptor activation mobilizes CDHC and TuJ1 to produce a tuft-like pattern extending into the proximal segments of the neurites, whereas D2 receptor activation produces a condensation of the labeling around the nucleus. Scale bars: 15 μm.
Discussion
Our data show that dopamine receptor activation can modulate neuronal migration from the basal forebrain to the cerebral wall. The effects were similar at rostral and caudal levels of the basal forebrain, suggesting comparable effects on migration of neurons originating in the medial and caudal ganglionic eminences. D1 receptor activation promoted the migration, whereas D2 receptor activation decreased it. We confirmed these findings by gain-of-function assay, in which D1 receptor was electroporated into striatal cells in vitro and activated with a pharmacological agonist. We also performed loss-of-function assays in which slice preparations from D1 or D2 receptor knock-out mice were used. Analysis of GABA neuron numbers in the cerebral wall of the dopamine receptor knock-out mice confirmed the effects of dopamine receptor activation on GABA neuron migration.
A hallmark of the effects of dopamine in the mature brain is the opposing actions of D1 and D2 receptors on a variety of functions (Stoof and Kebabian, 1984). This dichotomy is evident in the effects of dopamine on cell proliferation in the embryonic telencephalon (Ohtani et al., 2003; Popolo et al., 2004) and, as shown here, also in the effects of dopamine on GABA neuron migration. Thus, a physiological balance between D1 and D2 receptor expression and activation appears to be important for normal brain development. We do not know whether some neurons in the embryonic striatum express exclusively D1 receptors and others exclusively D2 receptors. It is likely that all neurons in the embryonic striatum express both D1 and D2 receptors but some are enriched in D1 and others in D2 receptors, as in the mature striatum (Gerfen, 1992; Surmeier et al., 1996; Aizman et al., 2000). It is tempting to speculate that neurons enriched in D1 receptors may be more likely to migrate away from the embryonic basal forebrain, whereas those enriched in D2 receptors may remain within it, offering a basis for specification of dorsal versus ventral fates for cells produced in the basal forebrain.
Our data raise the possibility that dopamine imbalance could underlie GABA dysfunction in developmental disorders such as schizophrenia, autism, or attention deficit because some degree of dysfunction in both dopamine and GABA systems is involved in these disorders (Benes, 2000; Robinson et al., 2001; Treiman, 2001; Casanova et al., 2003; Levitt et al., 2004). Fetal cocaine exposure also results in impairment of dopamine and GABA systems (Wang et al., 1995; Friedman et al., 1996; Crandall et al., 2004; Stanwood and Levitt, 2007). Thus, dopamine imbalance, GABA dysfunction, and developmental onset appear to be common factors in these conditions. Our data offer a novel perspective on the pathophysiology of these conditions by suggesting dopamine imbalance as a cause of impaired migration of GABA neurons. Another novel insight offered by our data is that dopamine imbalance can alter GABA neuron development in brain regions that may not be enriched in dopamine or its receptors. Specifically, during the period E12–E15 when GABA neuron migration from the basal forebrain to the cerebral wall is robust in mice, dopaminergic innervation and dopamine receptor expression are high in the basal forebrain and low in the cerebral wall (Ohtani et al., 2003; Popolo et al., 2004). However, our data show that imbalance in dopamine receptor activation occurring even earlier than E15 can alter cortical GABA neuron development. These observations underscore the significant role dopamine receptor activation in the embryonic basal forebrain can play for the development of GABA circuits throughout the forebrain, even in regions that may not be enriched in dopamine during development or at maturity.
We do not know whether the changes in cortical GABA cell numbers produced by imbalance in dopamine receptor function, such as those in the E15 D1 or D2 receptor knock-out mice, are permanent. However, recent evidence suggests that even a delay in the arrival of the GABA neurons in the cerebral wall, such as that which occurs in the D2 receptor knock-out mouse, can interfere with the normal development of cortical circuitry (Ben-Ari et al., 2004). Therefore, even a transient impairment of cortical GABA neuron development in the embryonic period can lead to lasting changes in cortical function, emphasizing the relevance of our data to an entire class of neuropsychiatric conditions characterized by developmental onset and dysfunction in both dopamine and GABA systems.
Previous studies had demonstrated a role for dopamine in cell proliferation and differentiation in the embryonic striatum and cerebral cortex (Zhang and Lidow, 2002; Ohtani et al., 2003; Popolo et al., 2004; Zhang et al., 2005). We were concerned that the changes in migration of DiI-labeled neurons produced by D1 and D2 receptor activation or by dopamine may be “secondary” to changes in cell proliferation produced by dopamine receptor activation (Ohtani et al., 2003; Popolo et al., 2004). In other words, the increase in the percentage of DiI-labeled cells reaching the cerebral wall after exposure of the slices to the D1 receptor agonist or dopamine may be attributable to a larger number of cells leaving the progenitor pool and becoming postmitotic in the ganglionic eminence as a result of D1 receptor activation (Ohtani et al., 2003; Popolo et al., 2004). The decrease in migration may be attributable to fewer cells becoming postmitotic as a result of D2 receptor activation. Thus, we could have mistaken changes in cell output for changes in cell migration. Two lines of evidence argue against those concerns. First, we expressed the number of DiI-labeled cells reaching the cerebral wall as a percentage of the total number of DiI-labeled cells in the entire slice. Therefore, our data were “corrected” for any changes in the total number of cells in the ganglionic region. Second, cells that migrate from the ganglionic eminence to the cerebral wall predominantly originate in the medial ganglionic eminence (Lavdas et al., 1999; Wichterle et al., 2001; Nery et al., 2002). The effects of dopamine on cell proliferation are evident in the basal forebrain principally in the lateral ganglionic eminence (Ohtani et al., 2003; Popolo et al., 2004). Finally, in our electroporation studies, many of the electroporated cells were in striatal differentiating fields, suggesting that they were postmitotic. Activation of the electroporated D1 receptor promoted migration of these cells. Thus, our data reveal a novel dimension for the effects of dopamine in the developing brain by demonstrating its role in GABA neuron migration, in addition to its role in proliferation of neuroepithelial cells in the telencephalon.
How dopamine can modulate neuronal migration is not known. Regardless of specific mechanisms involved, cytoskeletal reorganization is a prerequisite for neuronal migration or differentiation. Therefore, we sought a link between dopamine receptor activation and cytoskeletal changes in cultured striatal neurons. We show that the effects of dopamine receptor activation on neuronal migration are consistent with changes in neuronal cytoskeleton. Dopamine receptor activation induces redistribution of CDHC within cultured striatal neurons. CDHC is a motor protein implicated in movement of organelles during nuclear translocation (Sasaki et al., 2000). Activation of D1 or D2 receptors produces opposite effects on intracellular distribution of CDHC consistent with the opposite effects of these two receptors on neuronal migration. Activation of the D1 receptor-induced redistribution of CDHC and β-III tubulin to neurites, whereas activation of the D2 receptor resulted in a prominent perinuclear localization of CDHC and β-III tubulin. The patterns of localization of CDHC and β-III tubulin in response to D1 receptor activation are consistent with migrating or differentiating neurons, whereas the patterns in response to D2 receptor activation resemble undifferentiated or mitotically active cells (Sasaki et al., 2000; Wynshaw-Boris and Gambello, 2001; Shu et al., 2004). Thus, the effects of dopamine receptor activation on neuronal migration appear to have a basis at the level of the neuronal cytoskeleton. We recognize that the data on dopamine receptor-induced cytoskeletal changes are from differentiating neurons maintained in culture and not from migrating neurons per se. However, the conclusions from these experiments likely have a bearing on dopamine receptor-induced cytoskeletal changes associated with neuronal migration, especially because of the opposite effects of D1 and D2 receptor activation on the cytoskeleton as well as neuronal migration.
In conclusion, our data show that altering the physiological balance between dopamine D1 and D2 receptor activation can alter GABA neuron migration to the cerebral cortex in the embryonic brain. Because the embryonic basal forebrain is enriched in dopamine and its receptors and because the basal forebrain is the source of GABA neurons of multiple forebrain regions including the neocortex, hippocampus, striatum, nucleus accumbens, amygdala, and olfactory bulb (Parnavelas, 2000; Marin and Rubenstein, 2001), dopamine imbalance in the embryonic brain can impact GABA neuron development in multiple brain regions. Subtle but significant GABA dysfunction in multiple telencephalic regions is associated with a class of neurofunctional disorders exemplified by schizophrenia, attention deficit, or autism. Therefore, the intimate relationship between dopamine and GABA neuron development revealed here may be critical for gaining novel insights into the pathobiology of developmental neurofunctional disorders or fetal cocaine exposure, all of which involve dopamine and GABA neuron dysfunction.
Footnotes
This work was supported by United States Public Health Service Grants NS43426, NS45776, DA020796, and HD05515. We thank Dr. Edward Stern for fabricating the needle electrode and for his valuable advice regarding the electroporation experiments.
References
- Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- Alifragis P, Liapi A, Parnavelas JG. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their GABA phenotype. J Neurosci. 2004;24:5643–5648. doi: 10.1523/JNEUROSCI.1245-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JLR. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Ang ES, Jr, Haydar TF, Gluncic V, Rakic P. Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci. 2003;23:5805–5815. doi: 10.1523/JNEUROSCI.23-13-05805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki KY, Fujimura S, MacDonald ME, Bhide PG. Characterization of mouse striatal precursor cell lines expressing functional dopamine receptors. Dev Neurosci. 2006;28:518–527. doi: 10.1159/000095114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellion A, Baudoin JP, Alvarez C, Bornens M, Metin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J Neurosci. 2005;25:5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Represa A, Gozlan H. Interneurons set the tune of developing networks. Trends Neurosci. 2004;27:422–427. doi: 10.1016/j.tins.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. The Neuroscientist. 2003;9:496–507. doi: 10.1177/1073858403253552. [DOI] [PubMed] [Google Scholar]
- Cherubini E, Conti F. Generating diversity at GABAergic synapses. Trends Neurosci. 2001;24:155–162. doi: 10.1016/s0166-2236(00)01724-0. [DOI] [PubMed] [Google Scholar]
- Crandall JE, Hackett HE, Tobet SA, Kosofsky BE, Bhide PG. Cocaine exposure decreases GABA neuron migration from the ganglionic eminence to the cerebral cortex in embryonic mice. Cereb Cortex. 2004;14:665–675. doi: 10.1093/cercor/bhh027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flames N, Long JE, Garratt AN, Fischer TM, Gassmann M, Birchmeier C, Lai C, Rubenstein JL, Marin O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004;44:251–261. doi: 10.1016/j.neuron.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Friedman E, Yadin E, Wang HY. Effect of prenatal cocaine on dopamine receptor-G protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70:739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Jung AB, Bennett JP., Jr Development of striatal dopaminergic function. 1. Pre- and postnatal development of mRNAs and binding sites for striatal D1 (D1a) and D2 (D2a) receptors. Dev Brain Res. 1996;94:109–120. doi: 10.1016/0165-3806(96)00033-8. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. Ed 2. New York: Academic; 1992. The atlas of mouse development. [Google Scholar]
- Krimer LS, Goldman-Rakic PS. Prefrontal microcircuits: membrane properties and excitatory input of local, medium, and wide arbor interneurons. J Neurosci. 2001;21:3788–3796. doi: 10.1523/JNEUROSCI.21-11-03788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;99:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Morris NR. Nuclear migration. From fungi to the mammalian brain. J Cell Biol. 2000;148:1097–1101. doi: 10.1083/jcb.148.6.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM, Albrecht U, Reiner O, Eichele G, Yu-Lee LY. The lissencephaly gene product Lis1, a protein involved in neuronal migration, interacts with a nuclear movement protein, NudC. Curr Biol. 1998;8:603–606. doi: 10.1016/s0960-9822(98)70232-5. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Ventricle-directed migration in the developing cerebral cortex. Nat Neurosci. 2002;5:218–224. doi: 10.1038/nn813. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. J Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical neurones: new vistas. Trends Neurosci. 2000;23:126–131. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development. 2002;129:3147–3160. doi: 10.1242/dev.129.13.3147. [DOI] [PubMed] [Google Scholar]
- Popolo M, McCarthy DM, Bhide PG. Influence of dopamine on precursor cell proliferation and differentiation in the embryonic mouse telencephalon. Dev Neurosci. 2004;26:229–244. doi: 10.1159/000082140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Robinson PD, Schutz CK, Macciardi F, White BN, Holden JJ. Genetically determined low maternal serum dopamine beta-hydroxylase levels and the etiology of autism spectrum disorders. Am J Med Genet. 2001;100:30–36. doi: 10.1002/ajmg.1187. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Rakic P. Genetic control of cortical development. Cereb Cortex. 1999;9:521–523. doi: 10.1093/cercor/9.6.521. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci USA. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to Regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine D1 receptor activity and subcellular distribution. J Neurosci. 2007;27:152–157. doi: 10.1523/JNEUROSCI.4591-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoof JC, Kebabian JW. Two dopamine receptors: biochemistry, physiology and pharmacology. Life Sci. 1984;35:2281–2296. doi: 10.1016/0024-3205(84)90519-8. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiler K. Berlin: Springer; 1972. The house mouse. Development and normal stages from fertilization to 4 weeks of age. [Google Scholar]
- Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42(Suppl 3):8–12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x. [DOI] [PubMed] [Google Scholar]
- Vrana SL, Kluttz BW, Vrana KE. Application of quantitative RT-PCR to the analysis of dopamine receptor mRNA levels in rat striatum. Brain Res Mol Brain Res. 1995;34:127–134. doi: 10.1016/0169-328x(95)00152-i. [DOI] [PubMed] [Google Scholar]
- Wang HY, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J Pharmacol Exp Ther. 1995;273:492–498. [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A. Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci. 1999;2:461–466. doi: 10.1038/8131. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Wynshaw-Boris A, Gambello MJ. LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 2001;15:639–651. doi: 10.1101/gad.886801. [DOI] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yozu M, Tabata H, Nakajima K. The caudal migratory stream: a novel migratory stream of interneurons derived from the caudal ganglionic eminence in the developing mouse forebrain. J Neurosci. 2005;25:7268–7277. doi: 10.1523/JNEUROSCI.2072-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lidow MS. D1 dopamine receptor regulation of cell cycle in FGF- and EGF-supported primary cultures of embryonic cerebral cortical precursor cells. Int J Dev Neurosci. 2002;20:593–606. doi: 10.1016/s0736-5748(02)00104-1. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bai J, Undie AS, Bergson C, Lidow MS. D1 dopamine receptor regulation of the levels of the cell-cycle-controlling proteins, cyclin D, P27 and Raf-1, in cerebral cortical precursor cells is mediated through cAMP-independent pathways. Cereb Cortex. 2005;15:74–84. doi: 10.1093/cercor/bhh110. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Li HS, Zhou L, Wu JY, Rao Y. Cellular and molecular guidance of GABAergic neuronal migration from an extracortical origin to the neocortex. Neuron. 1999;23:473–485. doi: 10.1016/s0896-6273(00)80801-6. [DOI] [PubMed] [Google Scholar]