

Abstract
The Thermomyces lanuginosa lipase has been extensively studied in industrial and biotechnological research because of its potential for triacylglycerol transformation. This protein is known to catalyze both hydrolysis at high water contents and transesterification in quasi-anhydrous conditions. Here, we investigated the Thermomyces lanuginosa lipase structure in solution in the presence of a tributyrin aggregate using 30 ns molecular-dynamics simulations. The water content of the active-site groove was modified between the runs to focus on the protein-water molecule interactions and their implications for protein structure and protein-lipid interactions. The simulations confirmed the high plasticity of the lid fragment and showed that lipid molecules also bind to a secondary pocket beside the lid. Together, these results strongly suggest that the lid plays a role in the anchoring of the protein to the aggregate. The simulations also revealed the existence of a polar channel that connects the active-site groove to the outside solvent. At the inner extremity of this channel, a tyrosine makes hydrogen bonds with residues interacting with the catalytic triad. This system could function as a pipe (polar channel) controlled by a valve (the tyrosine) that could regulate the water content of the active site.
Introduction
Lipases (EC 3.1.1.3) are a class of enzymes that are able to catalyze different reactions, such as hydrolysis and transesterification of triglycerides (TGs). They have been characterized for various species and present differences despite a well-conserved α/β hydrolase fold (1). Some lipases, such as the mammal α-pancreatic lipase, are associated with another protein, the colipase. The role of the colipase is not yet well understood, but it seems to be responsible for the anchoring of the lipase/colipase complex to cell membranes or micelles (2–4) and is necessary for the enzyme to work properly in the presence of bile salts (5). Other members of the family, such as the Thermomyces lanuginosa lipase (TLL), are active without any cofactor. Lipases can catalyze several reactions in different compounds. They can mediate TG hydrolysis in di- or monoglycerides and free fatty acids, but can also drive transesterification or interesterification (6). Transesterification consists of the exchange of acyl chains between two TGs. As consumers' demand for healthy food is increasing, the use of such enzymes for TG modification is crucial for the food industry. Moreover, these proteins show good potential for use in industrial and biotechnological research and applications (6).
The water content and activity of the catalytic environment greatly influences the reaction rates (7). For most lipases, a low water content (<5%) is needed for optimal transesterification (7). In all cases, the enzymatic activity increases drastically when the lipid concentration reaches the supersaturation level, where the substrate can be an emulsion or form micelles. This phenomenon, known as interfacial activation (8), is of great importance for the reaction rate. Indeed, the maximal catalytic activity occurs after a characteristic lag period (9) during which the enzyme is stacked at the lipid interface.
To date, two models have been proposed to explain the molecular basis of interfacial activation. According to the “substrate” theory (10), the most important feature is the quality of the interface, whereas the “enzyme” theory proposes that the driving parameter is the conformational change of a protein fragment (the lid) at the adsorption surface (11). Based on crystallographic studies, Derewenda et al. (12) showed the high conformational lability of the lid in the absence of interface, and suggested that the two theories are nonexclusive.
The crystal structure of several lipases has already been solved (12–15). The structure of T. lanuginosa lipase presents the α/β hydrolase fold typical of the family. The TLL structure consists of a central seven-stranded β-sheet surrounded by a large N-terminal α-helix on the distal face and connecting fragments on the proximal face (15). The classical catalytic triad S146, D201, and H258 is buried in a hydrophobic groove. Located on one border of the groove, the lid (residues 85–93) can adopt two major conformations, defined as “open” and “closed” with respect to the active site (16,17). In aqueous conditions, the lid covers the active site and restricts its accessibility to water. In the open conformation, hydrophobic residues that are located at the inner face of the lid are exposed to the solvent. The opening of the lid increases the hydrophobic surface area near the active site, which is then more favorable to lipid binding.
Preliminary works using lipase crystallographic structures have been performed in an attempt to understand the interfacial activation and its associated lid motions. Based on 500 ps molecular dynamics (MD), it has been proposed that the conserved arginine at the lid hinge of Rhizomucor miehei lipase contributes to the stabilization of the open conformation by forming an electrostatic interaction with an aspartate (D62 in T. lanuginosa) (18). Moreover, in a study combining MD approaches with a constrained mechanical protocol, Peters et al. (19) suggested that lipase activation is enhanced in a hydrophobic environment.
Using fast Fourier transform-attenuated total reflection studies, Noinville et al. (20) followed the hydrogen/deuterium exchange reaction for Humicola lanuginosa lipase and proposed a two-step water shuttling mechanism occurring at the interface. They suggested that water molecules can be trapped near the internal cavity of the lipase (the first step) and could be released during the adsorption onto the interface (the second step). Moreover, they proposed that lipases should have a unique structural feature that leads to the release of water molecules during the adsorption/opening process, but did not identify the residues implicated in the process.
A partial answer was proposed by Derewenda et al. (14), who showed that lipases from a fungal family have a buried polar cluster of amino acids. They furthermore revealed the presence of ordered water molecules in protein cavities. No explicit mechanism by which water molecules gain access to the active site has been proposed to date; however, in a recent work on another type of lipase, Linderoth et al. (21) suggested that access of water molecules to the active site is a key step in the hydrolysis mechanism.
In this study, we performed MD simulation runs of TLL complexed to a tributyrylglycerol (TBG) aggregate in water. We modified the water content of the active site in the last two MD runs to focus on the role of water-protein interactions near the active site, and their implications for lipase activity. The different runs correspond to the lipase with an active-site cavity 1), full of water; 2), with only one water molecule in the vicinity of the catalytic triad; and 3), without water. We further analyzed the conformational plasticity of the lid as well as the adsorption and motion of the TBG near the active site.
Materials and methods
Protein model selection
The initial protein structure was an open structure of TLL cocrystallized with lipids (diundecyl phosphatidyl choline (PLC); Protein Data Bank (PDB): 1EIN) (15). This conformation was selected because it contains a phospholipid in the active site and thus could accept ligand within the MD timescale without requiring major modifications to simulate the opening of the lid. Moreover, extensive experimental research has been performed on this enzyme. A major challenge in studying transesterification by MD using actual all-atom force fields is to reproduce experimental conditions with very low water concentrations (<1%) and TGs as solvent molecules. To avoid a fast pressure increase and system instability, we worked on a system consisting of one TLL molecule and a sufficiently large, unstructured TG aggregate in a box of water. The TGs were not constrained or treated as rigid.
TBG model construction
To limit the size of the system while mimicking experimental procedures, we did not use the lipid cocrystallized with the protein; instead we used TBG, which is less complex than PLC and is fluid at 308 K. The structural model (Fig. 1 A) was achieved based on the experimental structure of a truncated tristearylglycerol obtained from the HIC-UP server (22). The TBG GROMACS (23) topology and coordinate files were generated with the PRODRG server (24) using GROMOS96 (G43a1) parameters (25). The atom names, bond and angle definitions, and charge repartition were adapted to fit the atom and bond types defined in the default topology files of the G45a3 version of the force field (26) (see Fig. S1 in the Supporting Material). This version was chosen for its potential for lipid description.
Figure 1.
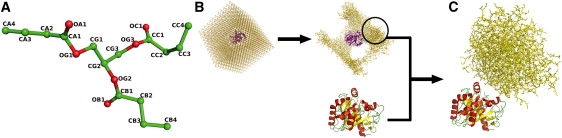
Initial system models. (A) Structural model of a TBG. Oxygen atoms are in red and carbon atoms are in green. (B) Initial system used to obtain the TBG aggregate. (C) Starting system for the first MD run. Protein is in ribbon (yellow, β-sheets; red, α-helices; and green, coils), and the aggregate is in yellow sticks. Water molecules are not represented.
TBG aggregate construction
The TBG aggregate was created using an MD simulation of a complete system (Fig. 1 B). A lipase molecule was put into a 10 nm cubic box and surrounded by 922 TBG molecules. All remaining spaces were filled in with 15,528 simple point charge (SPC) water molecules. The system was minimized by 2000 steps of steepest descent and 1000 steps of conjugate gradient, and then run for a 10 ns simulation with the protein under position restraints in periodic boundary conditions (PBCs) using a 2 fs time step. The temperature was fixed to 343 K to accelerate the TBG aggregation process. Temperature and pressure (1 bar) were maintained using the bath method (27). Electrostatics and van der Waals were treated with a 1.4 nm cutoff. Bonds were maintained with the SHAKE algorithm (28). As expected, individualized lipid molecules clustered rapidly to form an unstructured aggregate. A volume corresponding to a cubic box of 4 nm sides (larger than the entire protein volume) containing only TBG molecules was selected in the vicinity of the protein.
Starting box construction and minimization
A crystallographic lipase structure that had not been subjected to any simulation was placed with the lid helix oriented toward the aggregate (Fig. 1 C). Protonation states of ionizable groups were set according to pH 7. For histidine, the protonation state was determined from the hydrogen (H)-bonding pattern, or if the side chain was solvent-exposed, a proton was put on the Nɛ only (29). The minimal distance between any atom of the protein and the aggregate was set to 4 Å. The protein/TBG complex was then put into a triclinic box at 1 nm from each side of the box filled with 27,981 SPC water molecules, and neutralized using an automatic subroutine replacing solvent molecules by monoatomic ions at the position of the first atoms with the most favorable electrostatic potential. Nine sodium ions were added this way, for a total size of 92,768 atoms. The system was minimized by 2000 steps of steepest descent and 1000 steps of conjugate gradient, and submitted to two consecutive 50 ps heating runs in PBC at 308 K and 1 bar (bath method) with a 2 fs time step. During the first heating run, the protein and all TBG molecules were constrained. The TBG molecules were relaxed in the second run. Electrostatic interactions were treated using the particle mesh Ewald (PME) method, and van der Waals interactions were treated with the shift method. All cutoffs were set to 1.2 nm. Atomic bonds were maintained using the SHAKE algorithm.
Simulation runs
A first run (R1) of 10 ns, with no restraint, was achieved to completely relax the system and leave enough time for the protein and aggregate to move closer together. The last structure was then used as starting point to extend R1 by 20 ns through runs R2–R4. R2 was a continuation of the first 10 ns with no change in the system. The opening of the lid is thought to be a step in interfacial activation and to be enhanced in a hydrophobic environment (19). It is thus likely that the active site contains only a few water molecules after the activation lag time. To test the effect of an active site free of water molecules, we performed two runs. In R3, only one water molecule was left near the active site, for a total system size of 92,726 atoms. During R4, all water molecules between the protein and the aggregate were removed from the active-site groove, leaving the system with 92,723 atoms. These three runs (R2–R4) were carried out with coordinates and velocities generated in the last step of the first trajectory (R1). A 2 fs time step was used in all four simulations. Two repeats of each run were computed using two different sets of velocity for R1. All computations, corresponding to a total time of 210 ns, were performed on four Bi-Xeon Quad core computers.
The trajectories were performed and analyzed with the GROMACS 3.3 tools as well as with in-house-made scripts and softwares, and 3D structures were analyzed with both PYMOL (30) and VMD (31) softwares. All analyses were performed on the triplicated runs independently and yielded similar results. The results are described here on the basis of the first runs only; differences observed in the repeats, if any, are specified in the text.
Results
Evolution of global parameters
To evaluate the stability of the protein structure during the simulations, a root mean-square deviation (RMSD) was calculated on the α-carbons after the superposition of each structure from the trajectory by series of rotations and translations (least-square fit computed on α-carbons) to the crystallographic reference structure using the standard GROMACS subroutine g_rms. During the first simulation (R1), the RSMD reaches a plateau between 1.5 and 2 Å after 2 ns (Fig. 2 A). This plateau is continued through the simulation without water in the active site (R4). During R2 and R3, the RMSD value increases more or less linearly to reach another plateau between 2 and 2.5 Å. It is not surprising that the largest RMS fluctuation (RMSF) values are associated with both the protein loops and the lid (Fig. 2 B). The lowest RMSF values are obtained during R4, indicating a structure that is slightly less labile than in the other simulations. The three residues of the active site (S146, D201, and H258) present an RMSF value of <1 Å in all trajectories.
Figure 2.
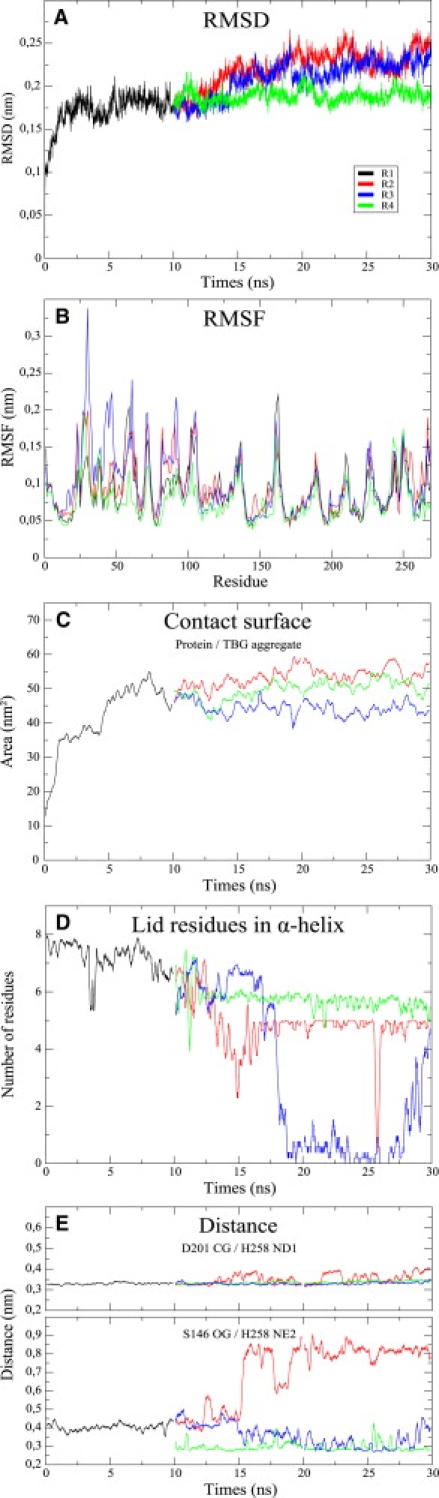
Global parameters. (A) RMSD calculated on α-carbons after a least-square fit on α-carbons of the crystallographic initial structure. (B) RMS fluctuations computed on the α-carbons. (C) Contact surface computed between the protein and the TBG aggregate. (D) Number of lid residues in α-helix computed with DSSP along the trajectories. (E) Active-site residue distances. Upper panel: Distances between CG from D201 and ND1 from H258. Lower panel: Distance between OG from S146 and NE2 from H258. The same color code is adopted in all plots (R1, black; R2, red; R3, blue; and R4, green).
To follow the relative displacement of the protein and the TBG aggregate, we computed the surface of contact shared by these groups averaged over 21 steps. This was computed by subtracting the solvent-accessible surface of the group protein-TBG aggregate from the sum of the solvent-accessible surface of the protein and the TBG aggregate alone. Fig. 2 C shows that the protein and the TBG aggregate get close together in <2 ns and share 45–55 nm2 of their surface after 10 ns. During the other runs, the two groups do not move away from each other and share a surface varying between 40 and 60 nm2.
The helical part of the lid can be defined by residues I86–N92, which correspond to the α3 helix of the protein (15). The number of lid residues (computed between S85 and L93) in helical conformation computed with DSSP software (32) over 21 steps shows that the helix is very stable during R1 (Fig. 2 D). Five to six residues are in helical conformation during R4, whereas the number of helical residues decreases during the first 5 ns of R2. After 5 ns, there are 4–5 residues in helical conformation, except for a short unstructured break around 26 ns. During R3, the helix is stable until 17 ns and then becomes totally unstructured. Its helicity is partially recovered at the end of the simulation (Fig. 2 D). When the lid loses helicity, the unfolding begins from its C-terminal end (Fig. S2).
These results show that the integrity of the protein is conserved during the simulations even in the region close to the aggregate. The lid is known to be very flexible and labile. Even if its structure is not well conserved through time, no movement of large magnitude is observed during the simulation. Repeat runs confirm the global evolution of the parameters. To investigate in detail the reciprocal impact of lipids, water, and protein on each other, we focused on interactions in the active site and at the protein/TBG interface.
Movement of functional residues
The catalytic triad consists of residues S146, D201, and H258. To examine how this triad adapts its conformation to incoming TBG molecules, we analyzed distances between atoms implicated in the processing of the substrate as previously described (33,34), i.e., one D201 side-chain oxygen, two nitrogens from the H258 ring, and the S146 hydroxyl oxygen. Since the two oxygen atoms from the aspartate are indistinguishable, we focused on the D201 CG–H258 ND1 and S146 OG–H258 NE2 distances (Fig. 2 E). In all runs, the distance between D201 CG and H258 ND1 averaged over 21 steps is maintained around 3.2 ± 1 Å. The S146 OG–H258 NE2 distance is subject to more variations. During the first 10 ns (R1), it fluctuates between 3.5 and 4.5 Å. After 10 ns, some differences can be observed between the three runs R2–R4. In R2, the distance increases to 9 Å after 15 ns up to the end of the simulation. In R3, where the starting structure has only one water molecule in the active site, the distance decreases regularly with time down to 3 Å and increases to 4 Å at the end of the simulation. In R4, with no water molecule in the active site, this distance decreases more rapidly than in R3 (in a few picoseconds), and is always lower than 4.0 Å.
These results show that the catalytic triad can adapt to different ligand sizes and to environmental variations.
Run R1 (0–10 ns active site full of water)
During the first 10 ns, the protein and aggregate come close to each other. The average surface of contact between the protein and the TBG aggregate shows that it takes place in <2 ns (Fig. 2 C). Nevertheless, we did not observe any TBG molecule close enough to the active site and in a conformation compatible with the enzymatic reaction. None of the TBG molecules in R1 superimpose exactly on the lipid molecule (PLC) present in the crystallographic structure. However, from time to time, the acyl part of a TBG molecule superimposes partially with part of the PLC molecule (Fig. 3 A).
Figure 3.
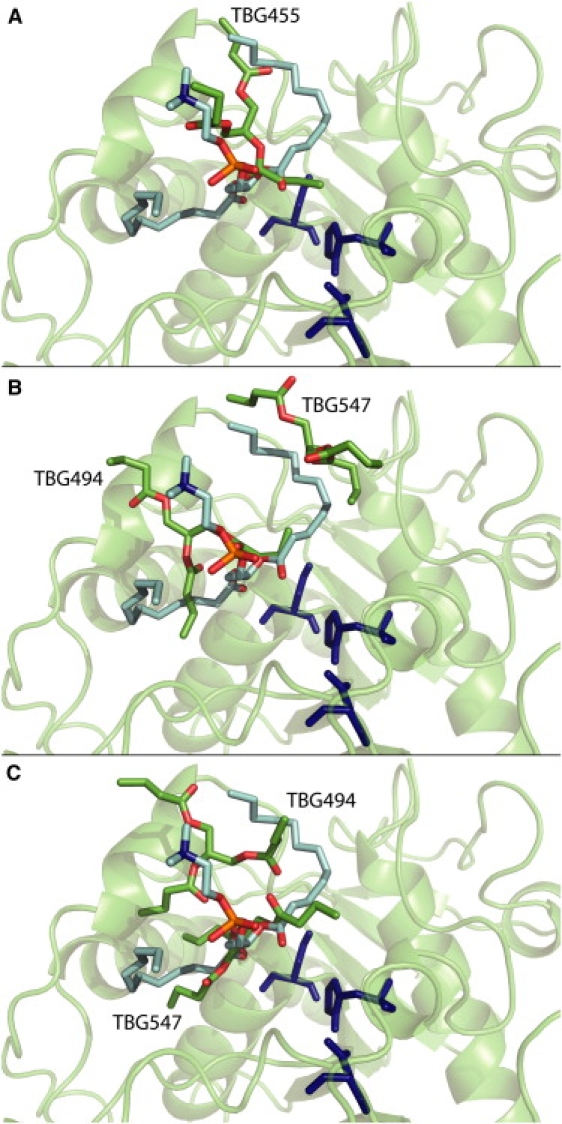
TBG orientation in the active site at (A) 6,610 ps during R1, (B) 13,490 ps during R2, and (C) 17,090 ps during R2. Each structure was superimposed on the reference crystallographic structure (1EIN) shown in green cartoon representation with the active-site residues in dark blue. Carbon atoms of the PLC molecule are colored in light blue, oxygen atoms in red, phosphorus atom in orange, and nitrogen atom in dark blue. Carbon atoms from TBG molecules (labeled as in the simulations) are colored in green, and oxygen atoms in red.
The catalytic triad and substrate can only make indirect interactions through water molecule bridges. This is not surprising given the starting structure, the time of simulation, and the hindrance of the active site by water molecules. Indeed, whereas most water molecules are chased out of the protein/TBG interface, some remain trapped in the active-site groove (Fig. 4 A).
Figure 4.
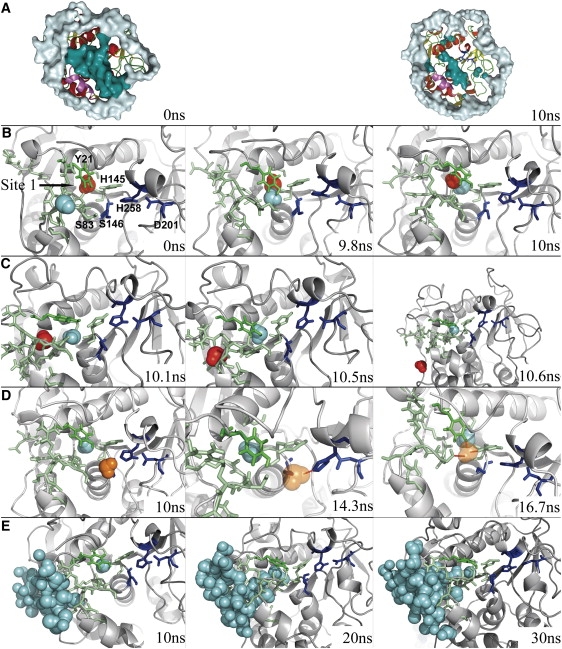
Water molecules at the protein/lipid interface. (A) At 0 and 10 ns (R1) water molecules are represented as a white surface outside and dark cyan surfaces inside the active-site groove. Water molecule motions at the interface near the active site during R1 (B), R2 (C), R3 (D), and R4 (E) are shown. Residues of the channel are shown in green and active-site residues in dark blue. SOL1 is in cyan, SOL2 in red, and SOL3 in orange. H-bonds formed by SOL3 are indicated by dashed lines. Water molecules that fill in the channel in R4 are shown in ball representation.
Our attention was drawn to the motion of a particular molecule of water (SOL1) initially located at the protein/lipid interface. SOL1 is rapidly integrated into a cluster of hydrogen-bonded water molecules in the hydrophobic cavity and is highly mobile from t = 0–9 ns. Meanwhile, the Y21 hydroxyl group makes an H-bond with either the carboxy oxygen or the amide hydrogen of G82, and H145 makes an H-bond with the carboxy oxygen of H258 (Fig. 5 A). After 9 ns, Y21 adopts another conformation and is hydrogen-bonded to the H145 side chain, which is always connected to the H258 backbone (Fig. 5 B). This reorganization lets SOL1 enter a polar channel formed by the side or the main chains of residues S17, Y21, N26, D27, R81–S85, N88, and H145 (Fig. 4 B). When SOL1 enters the channel, it takes the place of a second water molecule, SOL2 (site 1; Fig. 4 B). A comparison of the structures along the trajectory with the crystallographic structure of the lipase shows that despite their low RMSF (Fig. 2 B), three residues (Y21, R81, and R84) located at each extremity can move enough to close or open the channel. At the end of the run, only 14 molecules occupy the active-site groove, and two are in the polar channel (Table 1).
Figure 5.
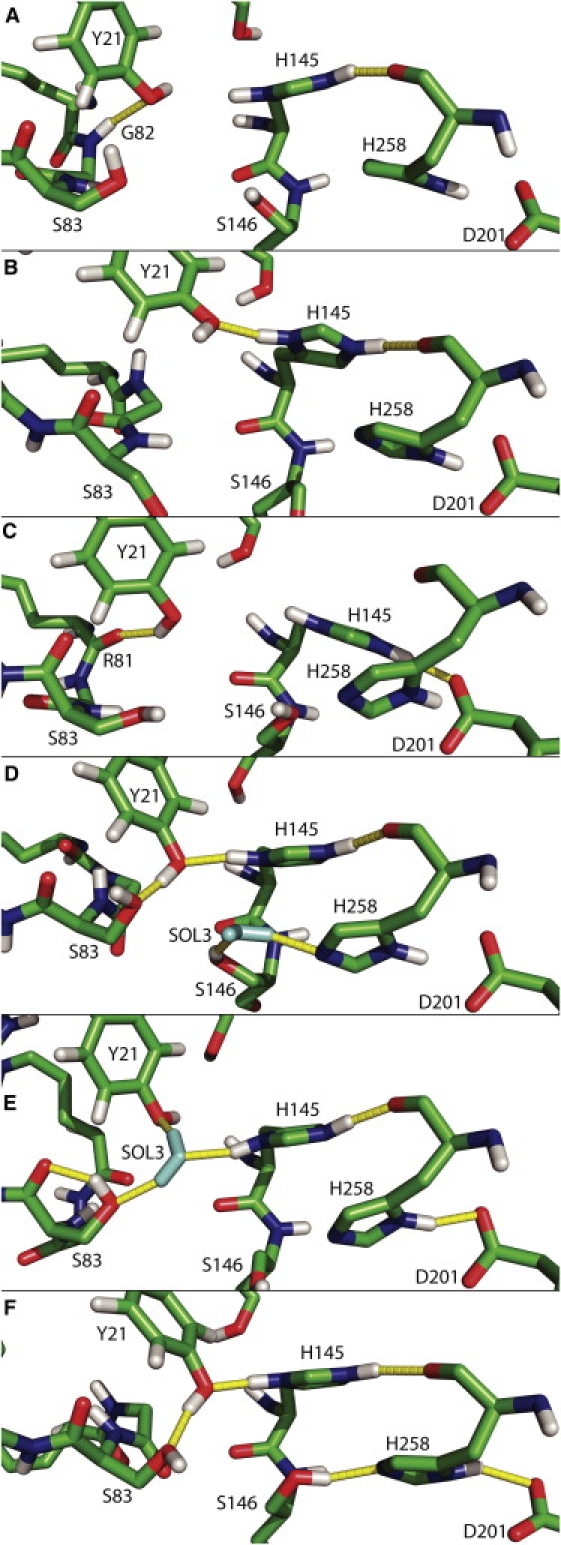
H-bond pattern in the active site at (A) t = 6,070 ps and (B) t = 9,980 ps during R1, (C) t = 12,350 ps during R2, (D) t = 13,220, (E) t = 18,900 ps during R3, and (F) t = 11,810 ps during R4. Carbon atoms are in green, oxygen in red, nitrogen in blue, and hydrogen in white. Water molecules are in cyan and H-bonds are represented as yellow lines.
Table 1.
Active-site water content
| Run |
ti |
tf |
||
|---|---|---|---|---|
| Groove | Channel | Groove | Channel | |
| 1 | Full | 1 | 14 | 2 |
| 2 | 14 | 2 | 11 | 2 |
| 3 | 1 | 1 | 1 | 3 |
| 4 | 0 | 1 | 0 | 7 |
In some lipases, the lid is suspected to play an important role in both the interfacial activation and the adsorption mechanism. It presents two different faces along its helix axis, both of which are accessible to the solvent in the structure used here. Most of the lid residues facing the active site are hydrophobic (I86, W89, I90, N92, L93, and F95). The outer face of the lid, opposed to the active site, is more polar with two asparagines (N88 and N94), S85, E87, and R84, and could interact with solvent molecules at the interface.
Surprisingly, after 5 ns, a TBG molecule (TBG365) interacts with the protein on the outer face. TBG365 is maintained until the end of the run in a pocket defined by residues V60, V63, N88, G91, N92, G112, F113, S116, and L147 (Fig. 6 A). This pocket corresponds to an alternative binding site that was partially described in the crystallographic structures of TLL by Yapoudjian et al. (35), who highlighted interactions between residues N92 and H110 with lipids. In our simulation, the main stabilizing interaction is an H-bond between the OC1 atom from TBG365 (Fig. 1 A) and the amide group from the N88 side chain. Moreover, the residues that border the secondary pocket form a polar ring that could make electrostatic interactions with the remaining oxygen atoms of the TBG molecule. Regarding the orientation of the acyl chain containing OC1, we could assume other possible interactions between this part and the four hydrophobic residues from the bottom of the pocket. During the repeats of this run, the pocket structure is not conserved and no TBG interacts with the lid outer face.
Figure 6.
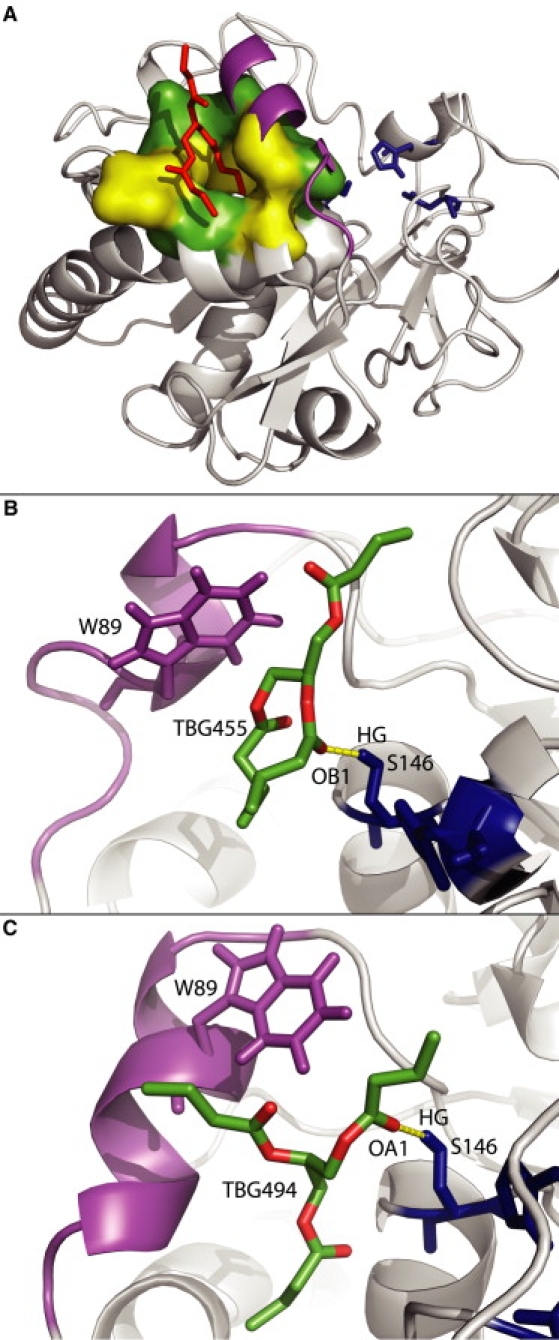
Two TBG-binding sites. (A) Secondary TBG-binding pocket. The more representative snapshot is at 10 ns (R1). The TBG molecule is in red, the lid fragment in magenta, and the catalytic triad in blue. Hydrophobic residues are in yellow and polar residues are in green. A transient H-bond (yellow dashed line) between a TBG molecule (green and red) and S146 from the catalytic triad (blue) is shown at t = 19,570 ps during R3 (B) and t = 11,420 ps during R4 (C).
Run R2 (10–30 ns full of water)
The R2 run was a continuation of R1 under the same conditions (i.e., the same number of atoms, temperature, and pressure; Table 1). The velocities used for the first step of R2 were taken from the last step of R1.
The H-bond between TBG365 and N88 breaks after 12 ns, but the lipid is maintained in the secondary pocket by another H-bond with the D62 backbone nitrogen. The pocket conformation does not vary much during the last few nanoseconds. The V60 side chain, which interacts with an acyl chain of the TBG365, would increase its stabilization in the secondary pocket through additional hydrophobic interactions.
The lid loses its helical conformation from I90 to its C-terminus (Fig. 2 D). This explains the high RMSF value for theses residues during R2 (Fig. 2 B). After 17 ns, there are four to five lid residues in helical conformation, except for a short unstructured break just before 26 ns. During this event, a water molecule is present between the TBG365 and the lid for few picoseconds (Fig. S2).
At the beginning of the run, 14 water molecules are evenly distributed along the active-site groove and two others (SOL1 and SOL2) are present in the polar channel. The water molecule SOL1, which leaves the catalytic groove during R1, replaces SOL2, which is expelled from the protein through the polar channel in few picoseconds (Fig. 4 C). At 23 ns, the water molecule SOL1 is replaced by another molecule from the active-site groove and returns near the catalytic triad.
Thirteen solvent molecules in the active-site groove cluster around the catalytic triad. Only one remains in the hydrophobic part of the site during the whole simulation, stabilized by H-bonds with the carboxyl of V203 and the backbone nitrogen of R175.
Two water molecules of the cluster diffuse at the interface near the C-terminal end of the protein. A third one enters the groove in the same way before the TBG molecules get close enough to the protein to hinder the path. One last water molecule leaves the groove under the lid segment. At the same time, near Y21, a water network is established connecting the cluster to the water molecule in the polar channel (SOL1). The two arginines at the other end of the channel move to open or close the channel during the simulation.
The H-bond between Y21 and H145 side chains breaks after 11 ns and is never observed again. Then, H145 moves slightly and establishes another H-bond with the D201 side chain between 12 and 30 ns (Fig. 5 C).
Finally, at t = 30 ns, one molecule is engaged in the polar channel and a water molecule network is formed between the channel and the active site.
As a result, the water molecules make more contacts with the surrounding polar residues, especially with the serine and the histidine of the catalytic triad, preventing interactions of TBG molecules with the active site. Three major TBG molecules come close enough to the triad to be hydrolyzed, but are not stabilized for more than a few picoseconds. Nevertheless, two TBGs present two of the three acyl chains superimposed on the PLC present in the crystallographic structure (Fig. 3, B and C).
Run R3 (10–30 ns with one water molecule into the active site)
To mimic conditions that could happen during hydrolysis, we conserved only one water molecule (SOL3) in the active site of the last conformation of R1. This molecule was chosen for its location between S146 and H258.
During R3, the lid progressively loses its α-helical conformation from W89 to its C-terminus (Fig. 2 D) and is completely random after 18 ns.
The H-bond between TBG365 and N88 definitely breaks after 11.5 ns and the TBG progressively slides out of the secondary pocket. After 15 ns, the pocket is empty of any TBG molecule and will not be filled again. As the TBG365 leaves the secondary pocket, the N92 side chain rotates to fill in the pocket and makes H-bonds with S116 and N88. The hydrophobic residues of the secondary pocket are then hidden by these polar side chains. Even if no TBG molecule can be superimposed on the PLC present in the crystallographic structure, TBG455 enters the groove near W89 and comes close to the active site. Of interest, it exposes its central ester bond to the catalytic triad and makes transient H-bonds with S146 (Fig. 6 B).
At the beginning of R3, the SOL3 water molecule is located between H258 and S146 (Fig. 4 D). It then moves slightly around this position for the next 5 ns, making transient H-bonds with these two residues. During this time, Y21, H145, and H258 keep the same H-bond network observed at the end of R1 (Fig. 5 D). After 15 ns, SOL3 jumps to another position, still close to S146, where it is maintained by three H-bonds with Y21, S83, and H145 (Figs. 4 D and 5 E). Throughout the 20 ns of R3, no water molecule enters the active site. The polar channel is filled with water from 28 ns to the end of the run. The H-bond between the H145 and H258 backbone is maintained. Y21 adopts the conformation of the crystallographic structure, but the rotation of the hydroxyl hydrogen allows different H-bonds.
Run R4 (10–30 ns with no water molecule into the active site)
To facilitate the interactions between TGs and the protein, all water molecules were removed from the catalytic groove in the last conformation of R1.
As in R3, the H-bond between TBG365 and N88 breaks after 10.3 ns and the TG starts to move out of the secondary pocket after 11.8 ns. However, this pocket is not left empty or filled in by water molecules. TBG303 slowly replaces TBG365 between 13.1 and 19.2 ns. In the meantime, the pocket is obstructed by the two molecules and after 19 ns, the TBG308 molecule enters the pocket, replacing TBG303 and blocking the access of water molecules. At 27 ns, the TBG308 slides out of the pocket, which will not be filled again. Over the 20 ns run of R4, the lid is thus maintained in a hydrophobic environment on both sides and loses one α-helix turn at its C-terminus. During repeats, TBG365 never slides out of the pocket.
All along the trajectory, six TBG molecules interact with the active-site groove (TBG 412, 428, 455, 494, 499, and 525). At the beginning of the simulation, TBG428 is too far away to make contact with the protein. The three other TGs cover the major part of the groove without interacting with the catalytic triad. TBG494 rapidly enters the groove and slides between W89 and the active site, exposing one of its ester bonds to the catalytic triad. A transient H-bond is made between the OA1 atom from the TBG molecule and the S146 side chain (Fig. 6 C).
The active-site cavity remains free of any water molecule all along R4. This is not surprising since the groove entry is blocked by the TBG aggregate on the upper side and by a hydrophobic cap of three aromatic residues (F211, Y213, and F95) in contact with the aggregate on the side opposed to the active site. Although no water molecule enters the active-site groove, a network of H-bonded water molecules enters the protein through the polar channel observed in R1 and R2 (Fig. 4 E). Just before 29 ns, the water molecule of site 1 is replaced by another one from the polar channel and slides out of the protein. Y21 is then maintained by H-bonds with both S83 and H145, the latter being always engaged in another H-bond with the H258 backbone (Fig. 5 F), creating a very polar environment very close to the active site (Fig. 4 E). This conformation is maintained until the end of the run. The two arginines adopt an open conformation, but can adopt a closed one during the repeats.
Discussion
TLL can drive both hydrolysis and transesterification reactions in vitro depending on the water content of the medium. One of the major parameters controlling the efficiency of these reactions is the interfacial activation that occurs between the protein and the lipids. When the enzyme is activated, a pocket composed mainly of hydrophobic residues and lipids is created around the active site. Here we studied the behavior of water molecules as well as TBG molecules and protein structure at the protein/lipid interface by MD in four runs with varying active-site groove water contents. Global evolution parameters, such as RMSD and RMSF, show that the structure is stable throughout the different simulations, except for some protein loops and the lid fragment. Hence, we focused on details near the active site.
Based on in silico studies, it was first proposed that the role of the lid is to protect the active site in highly aqueous situations and to open in a hydrophobic environment during interfacial activation (36). However, later crystallographic studies showed disordered lid conformations in aqueous conditions (12). Based on molecular modeling of the pancreatic lipase and experimental studies, Thomas et al. (37) suggested that the high mobility of the lid fragment could be involved in the extraction of substrate molecules from the lipid aggregate or in the release of the product after hydrolysis.
As expected, it appears that the lid is highly flexible. It can completely lose its helical structure, but can also evolve between an unfolded structure and a partial α-helix in <30 ns (Fig. S2). Moreover, as shown for other lipases (38), the hydrophobic side of this fragment is tightly associated with the aggregate. The TBG repartition around the lid highlights the presence of a secondary binding site partially described in crystallographic structures (35). When TBG molecules enter this pocket and are also located in the active-site groove, the lid fragment is entirely surrounded by lipids. Our simulations show that, in this case, the lid adopts an α-helix conformation containing half of the residues defined as helical in the crystallographic structure. Nevertheless, if only one water molecule moves between the lid and the TBG molecules (Fig. S2), it can lose its structure in a few picoseconds. This effect is more important when the active-site groove is full of water.
Altogether, these results suggest that, at the interface, the lid may move as a hook to maintain interaction with the lipids, rapidly adapting its conformation in response to a variation in environment. We suppose that when the environment does not vary, the lid can evolve toward a stable helix structure as seen in the crystallographic structures, or in experimental work using solid hydrophobic surfaces (20). One possible function of the lid of fungal lipases could be to anchor the protein at the protein/lipid interface, as proposed for colipase in colipase-dependent enzymes (2–4).
The distance between D201 and H258 varies slightly over the four simulations between 3 and 4 Å. The catalytic triad mechanism implies the polarization of the histidine by the aspartate with an H-bond, as described elsewhere (33). Although classical MD simulations cannot solve this type of reaction, it appears that during the runs containing no more than one water molecule in the active site (R3 and R4), the histidine is positioned correctly to activate S146. The distance between these two residues (S146 and H258) is subject to more fluctuations during R2. Since the RMSF computed on α-carbons of the two residues is <0.08 Å (Fig. 2 B), it appears that the sudden increase of the distance to 9 Å is essentially due to side-chain motions; however, no flip of the histidine was observed. This increase is exclusively observed in R2 and its repeats in which no water molecules were manually removed from the active site cavity.
During the 30 ns corresponding to the continuous R1 and R2 runs, the protein and the TBG aggregate come close enough together to form a hydrophobic cap closing the active-site groove. Inevitably, water molecules get trapped in the active site as the system moves, and these molecules could be responsible for the increase in the S146–H258 distance. A cluster of water molecules is indeed formed around the S146 residue, repulsing the side chains of S146 and H258 apart.
Beer et al. (34) demonstrated that an H-bond network including residues corresponding to Y21 and H145 in another fungal species is essential for protein function because it contributes to the rigidity of the catalytic triad and the oxyanion hole. In fact, the bulk of water engages in many interactions with these two residues, disrupting the H-bond network that includes Y21, H145, and the backbone of H258, leading to a greater mobility of the residues of the catalytic triad.
Since this is not observed during runs containing one or no water molecules in the active-site groove, we propose that the first step of the activation process after the adsorption of the protein to the aggregate should be the draining of the active-site groove until an optimal number of water molecules is reached. Therefore, the sequestered water molecules could be responsible for the relatively long lag time observed experimentally, as suggested by Noinville et al. (20).
The study of water molecules around the active site revealed that all along our trajectories, as well as in the repeat runs, one position—site 1—is always occupied by a water molecule. This molecule is located near the Y21 ring at the inner extremity of a polar channel connecting the aqueous phase and the active-site groove (Fig. 4, B–D). Two nonexclusive roles can be proposed for this water molecule. Since it is present in all crystallographic structures, it can be interpreted as structural water that is crucial for the stabilization of this part of the protein. Regarding its location very close to the active site, it could also constitute a preferential position for the water molecule used to hydrolyze the lipid after the formation of the acyl-enzyme.
The elucidation of the structure of a novel Mycobacterium smegmatis enzyme involved in acyl transfer opens new ways to unravel the transesterification mechanism (39). Indeed, based on crystallographic data, Mathews et al. (39) showed that this protein can favor alcoholysis over hydrolysis in water by restricting the access of water to the active site. A hydrophobic channel formed during the oligomerization of the protein controls the access of water and substrate to the active site, regulating the enzyme activity. Such a feature has not heretofore been observed for lipases, but our simulations revealed the presence of a polar channel formed by 11 residues (Fig. 7). Among these, Y21, R81, G82, and H145 are fully conserved through different species; two residues (83 and 85) are always a serine or a threonine, and two residues (84 and 88) are always polar. The three remaining residues (17, 26, and 27) are highly variable. The presence of this channel supports the hypothesis that water molecules in site 1 are preferentially used during the hydrolysis process.
Figure 7.
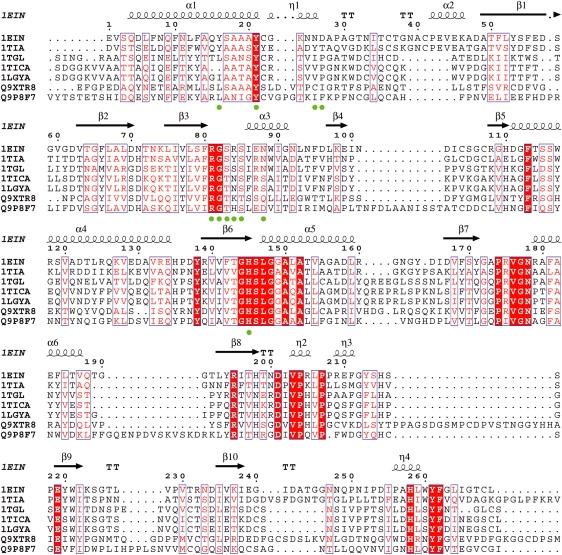
Multiple alignment of lipases from Thermomyces lanuginosa (PDB: 1EIN), Penicillium camembertii (PDB: 1TIA), Rhizomucor miehei (PDB: 1TGL), Rhizopus oryzae (1TIC), Rhizopus niveus (PDB: 1LGY), Caenorhabditis elegans (SwissProt: Q9XTR8), and Yarrowia lipolytica (SwissProt: Q9P8F7) generated with 3D-Coffee (41) and analyzed with ESpript (42). The color code corresponds to the default values of ESpript except for the green dots indicating residues of the polar channel. A red box over a white character highlights a strict identity, a red character indicates a similarity in a group, and a blue frame indicates a similarity across groups (refer to the ESpript manual for more details). The first line shows the secondary structure computed with DSSP (32) on the T. lanuginosa structure (PDB: 1EIN).
In all simulations containing water molecules in the active-site groove (R1, R2, and their repeats), the channel is empty except at site 1. The water content of the channel is more variable during the simulations with less water molecules in the active-site groove. For example, it is entirely filled for more than 10 ns during R4 (Fig. 4 E) but not in the repeats. Conversely, it is empty during the major part of R3 but filled for more than 10 ns during one R3 repeat. Given that a water molecule is only needed in the second part of the hydrolysis mechanism (33), the fact that this channel is not always filled in all R3 and R4 runs is not surprising since the acyl-enzyme formation cannot be reached during the classical MD simulation times. We propose that a balance exists between filled and empty channels until the overall active site conformation becomes suitable for water entry. When the lipid is bound to the protein, the channel could be filled with water molecules; the water molecule present in site 1 could then be “pushed” to the catalytic triad and achieve the hydrolysis of the bond between the serine and the lipid.
Based on short MD runs, the R. miehei residue equivalent to R81 has been proposed to maintain the lid in the open conformation by an electrostatic interaction with D62 (18). This correlates with experimental studies demonstrating the role of arginines in protein activity (40). Our results suggest that the loss of activity could also result from the obstruction of the polar channel since R81 is at the outer channel entry. Moreover, the difference observed experimentally between R. miehei and T. lanuginosa can be explained by the presence of a second arginine (at position 23) at the channel extremity in T. lanuginosa replaced by a glycine in R. miehei. These arginines could directly control the content of water in the polar channel.
At the inner extremity of the channel, opposite from these arginines, a tyrosine (Y21) interacts directly with the site 1 water molecule. On one hand, site-directed mutagenesis in another fungal lipase highlights the role of this residue in the catalytic triad stabilization (34). On the other hand, it appears that during the trajectories Y21 does not limit its contact to H145 or to the site 1 water molecule, and can also interact with other partners such as G82, R81, and S83 (Fig. 5). Our results suggest that the mobility of Y21 is not associated with the destabilization of the catalytic triad when the active site is not full of water. Rather, we propose that it could function as a valve and control the water motion to the active site. When there are too many water molecules in the hydrophobic groove (for example, during the interfacial activation) this valve could open and leave a part of the water molecules to flow out. Since this mechanism is not observed in all of the repeats, it is probable that this is not the preferred way for the water molecules to leave the groove. Although the acyl-enzyme cannot be reached in the simulations, the intrinsic mobility of Y21, as well as the location of site 1, allow us to suppose that this valve, coupled to the polar channel, is used to allow water to enter active site when the acyl-enzyme is formed. This could take place without any contact with TBG and the very hydrophobic environment of the groove. Hence, it should be possible for the protein to control the quantity of water present in the active site depending on its conformation.
This is the first time, to our knowledge, that such a mechanism has been proposed for a lipase. Future investigations, both experimental and in silico, should help to determine the exact implication of the polar channel identified here and the role of Y21 and H145 in balancing the hydrolysis/transesterification process. In fact, transesterification only occurs at very low water concentration and requires heavy experimental conditions to maintain a quasi-anhydrous environment. The control of this balance in aqueous solvent should be of crucial importance for improving lipase efficiency for biotechnological applications as well as for TG modifications at an industrial level.
Supporting Material
Two figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00784-X.
Supporting Material
Acknowledgments
S.S. is supported by a grant from the Gembloux Agricultural University (Belgium), and J.M.C. and B.C. are supported by the Belgian Program on Interuniversity Attraction Poles initiated by the Federal Office for Scientific, Technical and Cultural Affairs (IAP. P6/19 PROFUSA). L.L. is a research associate and R.B. is a research director for the Fonds National pour la Recherche Scientifique (Belgium). A.T. is a research director at the French Institute for Medical Research.
Footnotes
S. Santini and J. M. Crowet contributed equally to this work.
S. Santini's present address is Information Génomique et Structurale, UPR2589, Parc Scientifique de Luminy, Marseille, France.
B. Charloteaux's present address is Center for Cancer Systems Biology (CCSB) and Department of Cancer Biology, Dana-Farber Cancer Institute, and Department of Genetics, Harvard Medical School, Boston, MA.
References
- 1.Ollis D.L., Cheah E., Cygler M., Dijkstra B., Frolow F. The α/β hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 2.Borgstrom B., Erlanson-Albertsson C. Lipases In. In: B. Borgstrom Lipases, Brockman H.L., editors. Elsevier Science Publishers B.V.; Amsterdam: 1984. pp. 152–183. [Google Scholar]
- 3.van Tilbeurgh H., Egloff M.P., Martinez C., Rugani N., Verger R. Interfacial activation of the lipase-procolipase complex by mixed micelles revealed by X-ray crystallography. Nature. 1993;362:814–820. doi: 10.1038/362814a0. [DOI] [PubMed] [Google Scholar]
- 4.van Tilbeurgh H., Sarda L., Verger R., Cambillau C. Structure of the pancreatic lipase-procolipase complex. Nature. 1992;359:159–162. doi: 10.1038/359159a0. [DOI] [PubMed] [Google Scholar]
- 5.Borgstrom B., Erlanson C. Pancreatic lipase and co-lipase. Interactions and effects of bile salts and other detergents. Eur. J. Biochem. 1973;37:60–68. doi: 10.1111/j.1432-1033.1973.tb02957.x. [DOI] [PubMed] [Google Scholar]
- 6.Jaeger K.E., Dijkstra B.W., Reetz M.T. Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu. Rev. Microbiol. 1999;53:315–351. doi: 10.1146/annurev.micro.53.1.315. [DOI] [PubMed] [Google Scholar]
- 7.Villeneuve P. Lipases in lipophilization reactions. Biotechnol. Adv. 2007;25:515–536. doi: 10.1016/j.biotechadv.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Sarda L., Desnuelle P. Action de la lipase pancreatique sur les esters en emulsion. Biochim. Biophys. Acta. 1958;30:513–521. doi: 10.1016/0006-3002(58)90097-0. [DOI] [PubMed] [Google Scholar]
- 9.Patton J.S., Carey M.C. Watching fat digestion. Science. 1979;204:145–148. doi: 10.1126/science.432636. [DOI] [PubMed] [Google Scholar]
- 10.Brockman H.L., Law J.H., Kezdy F.J. Catalysis by adsorbed enzymes. The hydrolysis of tripropionin by pancreatic lipase adsorbed to siliconized glass beads. J. Biol. Chem. 1973;248:4965–4970. [PubMed] [Google Scholar]
- 11.Muderhwa J.M., Brockman H.L. Lateral lipid distribution is a major regulator of lipase activity. Implications for lipid-mediated signal transduction. J. Biol. Chem. 1992;267:24184–24192. [PubMed] [Google Scholar]
- 12.Derewenda U., Swenson L., Wei Y., Green R., Kobos P.M. Conformational lability of lipases observed in the absence of an oil-water interface: crystallographic studies of enzymes from the fungi Humicola lanuginosa and Rhizopus delemar. J. Lipid Res. 1994;35:524–534. [PubMed] [Google Scholar]
- 13.Kohno M., Funatsu J., Mikami B., Kugimiya W., Matsuo T. The crystal structure of lipase II from Rhizopus niveus at 2.2 A resolution. J. Biochem. (Tokyo) 1996;120:505–510. doi: 10.1093/oxfordjournals.jbchem.a021442. [DOI] [PubMed] [Google Scholar]
- 14.Derewenda U., Swenson L., Green R., Wei Y., Dodson G.G. An unusual buried polar cluster in a family of fungal lipases. Nat. Struct. Biol. 1994;1:36–47. doi: 10.1038/nsb0194-36. [DOI] [PubMed] [Google Scholar]
- 15.Brzozowski A.M., Savage H., Verma C.S., Turkenburg J.P., Lawson D.M. Structural origins of the interfacial activation in Thermomyces (Humicola lanuginosa lipase. Biochemistry. 2000;39:15071–15082. doi: 10.1021/bi0013905. [DOI] [PubMed] [Google Scholar]
- 16.Brady L., Brzozowski A.M., Derewenda Z.S., Dodson E., Dodson G. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature. 1990;343:767–770. doi: 10.1038/343767a0. [DOI] [PubMed] [Google Scholar]
- 17.Brzozowski A.M., Derewenda U., Derewenda Z.S., Dodson G.G., Lawson D.M. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature. 1991;351:491–494. doi: 10.1038/351491a0. [DOI] [PubMed] [Google Scholar]
- 18.Peters G.H., van Aalten D.M., Edholm O., Toxvaerd S., Bywater R. Dynamics of proteins in different solvent systems: analysis of essential motion in lipases. Biophys. J. 1996;71:2245–2255. doi: 10.1016/S0006-3495(96)79428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peters G.H., Toxvaerd S., Olsen O.H., Svendsen A. Computational studies of the activation of lipases and the effect of a hydrophobic environment. Protein Eng. 1997;10:137–147. doi: 10.1093/protein/10.2.137. [DOI] [PubMed] [Google Scholar]
- 20.Noinville S., Revault M., Baron M.H., Tiss A., Yapoudjian S. Conformational changes and orientation of Humicola lanuginosa lipase on a solid hydrophobic surface: an in situ interface Fourier transform infrared-attenuated total reflection study. Biophys. J. 2002;82:2709–2719. doi: 10.1016/S0006-3495(02)75612-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linderoth L., Andresen T.L., Jorgensen K., Madsen R., Peters G.H. Molecular basis of phospholipase A2 activity toward phospholipids with sn-1 substitutions. Biophys. J. 2008;94:14–26. doi: 10.1529/biophysj.107.110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleywegt G.J. Crystallographic refinement of ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2007;63:94–100. doi: 10.1107/S0907444906022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 24.Schuttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 25.van Gunsteren W.F., Billeter S.R., Eising A.A., Hünenberger P.H., Krüger P. AG an der ETH Zürich; Zürich, Switzerland: 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Hochschulverlag. [Google Scholar]
- 26.Chandrasekhar I., Kastenholz M., Lins R.D., Oostenbrink C., Schuler L.D. A consistent potential energy parameter set for lipids: dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field. Eur. Biophys. J. 2003;32:67–77. doi: 10.1007/s00249-002-0269-4. [DOI] [PubMed] [Google Scholar]
- 27.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 28.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 29.Lindahl E., Hess B., van Der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 30.deLano W.L. DeLano Scientific; Palo Alto, CA: 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 31.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 33.Verger R. Enzymes lipolytiques et lipolyse. OCL. 1995;2:52–56. [Google Scholar]
- 34.Beer H.D., Wohlfahrt G., McCarthy J.E., Schomburg D., Schmid R.D. Analysis of the catalytic mechanism of a fungal lipase using computer-aided design and structural mutants. Protein Eng. 1996;9:507–517. doi: 10.1093/protein/9.6.507. [DOI] [PubMed] [Google Scholar]
- 35.Yapoudjian S., Ivanova M.G., Brzozowski A.M., Patkar S.A., Vind J. Binding of Thermomyces (Humicola lanuginosa lipase to the mixed micelles of cis-parinaric acid/NaTDC. Eur. J. Biochem. 2002;269:1613–1621. doi: 10.1046/j.1432-1327.2002.02786.x. [DOI] [PubMed] [Google Scholar]
- 36.Norin M., Olsen O., Svendsen A., Edholm O., Hult K. Theoretical studies of Rhizomucor miehei lipase activation. Protein Eng. 1993;6:855–863. doi: 10.1093/protein/6.8.855. [DOI] [PubMed] [Google Scholar]
- 37.Thomas A., Allouche M., Basyn F., Brasseur R., Kerfelec B. Role of the lid hydrophobicity pattern in pancreatic lipase activity. J. Biol. Chem. 2005;280:40074–40083. doi: 10.1074/jbc.M502123200. [DOI] [PubMed] [Google Scholar]
- 38.Peters G.H., Bywater R.P. Influence of a lipid interface on protein dynamics in a fungal lipase. Biophys. J. 2001;81:3052–3065. doi: 10.1016/S0006-3495(01)75944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathews I., Soltis M., Saldajeno M., Ganshaw G., Sala R. Structure of a novel enzyme that catalyzes acyl transfer to alcohols in aqueous conditions. Biochemistry. 2007;46:8969–8979. doi: 10.1021/bi7002444. [DOI] [PubMed] [Google Scholar]
- 40.Holmquist M., Norin M., Hult K. The role of arginines in stabilizing the active open-lid conformation of Rhizomucor miehei lipase. Lipids. 1993;28:721–726. doi: 10.1007/BF02535993. [DOI] [PubMed] [Google Scholar]
- 41.Poirot O., Suhre K., Abergel C., O'Toole E., Notredame C. 3DCoffee@igs: a web server for combining sequences and structures into a multiple sequence alignment. Nucleic Acids Res. 2004;32:W37–W40. doi: 10.1093/nar/gkh382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouet P., Courcelle E., Stuart D.I., Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.