

Abstract
Energy and electron transfer in a Leu M214 to His (LM214H) mutant of the Rhodobacter sphaeroides reaction center (RC) were investigated by applying time-resolved visible pump/midinfrared probe spectroscopy at room temperature. This mutant replacement of the Leu at position M214 resulted in the incorporation of a bacteriochlorophyll (BChl) in place of the native bacteriopheophytin in the L-branch of cofactors (denoted βL). Purified LM214H RCs were excited at 600 nm (unselective excitation), at 800 nm (direct excitation of the monomeric BChl cofactors BL and BM), and at 860 nm (direct excitation of the primary donor (P) BChl pair (PL/PM)). Absorption changes associated with carbonyl (C=O) stretch vibrational modes (9-keto, 10a-ester, and 2a-acetyl) of the cofactors and of the protein were recorded in the region between 1600 cm−1 and 1770 cm−1, and the data were subjected to both a sequential analysis and a simultaneous target analysis. After photoexcitation of the LM214H RC, P∗ decayed on a timescale of ∼6.3 ps to P+BL−. The decay of P+BL− occurred with a lifetime of ∼2 ps, ∼3 times slower than that observed in wild-type and R-26 RCs (∼0.7 ps). Further electron transfer to the βL BChl resulted in formation of the P+βL− state, and its infrared absorbance difference spectrum is reported for the first time, to our knowledge. The fs midinfrared spectra of P+BL− and P+βL− showed clear differences related to the different environments of the two BChls in the mutant RC.
Introduction
The photosynthetic reaction center (RC) of Rhodobacter (Rb.) sphaeroides is the paradigm for understanding photosynthetic charge separation. Important aspects include the role of the special pair of bacteriochlorophylls (BChl) that act as the primary electron donor (P), the origin of the functional asymmetry that produces the so-called “active” branch of cofactors, the mechanism of charge separation between P and the adjacent BChl in the active branch (BL), and the role of the protein medium in facilitating rapid and efficient charge separation. In previous work we have studied the midinfrared (mid-IR) spectral responses of both wild-type (WT) and mutant RCs (1–3) to learn more about the specific environment of each of the chromophores and how the environment changes upon excitation and charge separation. This work describes the IR-signature of the state P+BL−, the charge redistribution within the special pair BChls during charge separation and stabilization, and protein responses to charge separation.
A major issue in the study of the IR-signatures of the states formed during ultrafast charge separation is the assignment of the differential signals to particular cofactors/groups or protein elements. One approach to this issue is alteration of the protein matrix through site-directed mutagenesis, including mutations that alter the cofactor composition of the RC. As an example, replacement of the Leu at position 214 on the M polypeptide by His results in the incorporation of a BChl molecule (denoted as βL) in place of the L-branch bacteriopheophytin (BPhe) in the WT RC (4). The x-ray crystal structure of the Rb. sphaeroides RC shows that a His residue is located adjacent to the Mg2+ of each of the four BChls, and it provides the fifth (axial) ligand to the Mg2+ ion (5). In contrast, the RC BPhes lack a central Mg2+ ion and do not have a metal-coordinating ligand from the protein close to the center of the macrocycle. In the case of the L-branch BPhe (denoted HL), a conserved Leu (residue 214) is located over the center of the BPhe macrocycle (6), and replacement of this Leu by His (denoted LM214H) leads to the incorporation of a BChl (βL) in place of the HL BPhe during assembly of the RC (4). X-ray crystal structures of the LM214H mutant have shown that, with the exception of the Leu to His substitution and with the introduction of a Mg2+ at the center of the adjacent bacteriochlorin macrocycle, the structure of the RC is unaffected by the substitution of the HL BPhe by the βL BChl (7,8).
An extensive investigation of the dynamics of charge separation in LM214H RCs has been reported (4,9–11). The kinetics of electron transfer are significantly perturbed compared to those of WT RCs, although the pigment exchange does not lead to electron transfer to the symmetrical HM BPhe or a completely blocked photoreaction, as evidenced by an appreciable yield of P+QA− formation (∼60%) (4,9–11). An intermediate P+I− state was formed with an ∼100% yield in ∼6 ps (4,9–11), which is a factor of ∼2 longer than the lifetime of primary donor-excited state (P∗) observed in WT RCs (1,12–14). To explain the observed spectral evolution during charge separation in the LM214H RC, it was concluded that the state “I” included the new βL pigment and that P+I− was a quantum mechanical mixture of the states P+BL− and P+βL− (4,9). In addition to describing charge separation in the LM214H RC, this model also confirmed that the energy gap between P∗ and P+BL− is very small in the WT RC (9,11). In a visible-region transient absorption study, spectral features different from those attributable to HL reduction in WT RCs, and consistent with BChl reduction, were observed between 720–840 nm on a timescale of 20 ps (4). From these it was concluded that the P∗ state decays by electron transfer to βL to form P+βL−, with the same quantum yield as that for P+HL− in WT RCs (4,10). The lifetime of the P+βL− state was found to be ∼350 ps. The reduction of QA in ∼600 ps (60% yield) was a factor of ∼3 slower than the reduction in the WT RCs, whereas competing deactivation of P+βL− to the ground state in ∼900 ps (40% yield) was a factor of >10 faster than that of recombination of P+HL− in WT RCs (15–17). As a confirmation of this, the decay of P+βL− had a time constant of ∼850 ps in LM214H RCs, with blocked electron transfer to QA (4).
The free energy of P+βL− with respect to P∗ was determined from delayed fluorescence from P∗ at room temperature, and this energy was found to have a value of ∼75 meV (4). The rate of electron transfer from P∗ to βL (or HL) depends weakly on this free-energy gap, and it changes by a factor of only 2, despite a significant change in ΔG from 160 meV in WT RCs (15,17) to 75 meV in LM214H RCs (4). An additional factor may be the very small pigment-protein reorganization energy (λ) on the timescale of the initial reaction. The activationless nature of the primary charge separation process in LM214H RCs follows from the P∗ lifetime of 2.5 ps at 5 K (4). It could not be decided whether the proposed mixing of the P+BL− and P+βL− states (4,10) is quantum mechanical or a thermal equilibrium, because experiments on LM214H RCs at 5 K and 295 K yielded the same time constant for P+βL− charge recombination of ∼1 ns. The large increase in the charge recombination rate (>10-fold) is compatible with a much smaller change in the rate of initial electron transfer from P∗, because of the different sensitivity of these processes to ΔG°. In addition, slow structural reorganizations after formation of P+βL−, as have been proposed to occur in the case of P+HL− state in WT RCs (15–19), could uncouple the electronic factors for the two processes.
The purpose of this report was to examine charge separation in the LM214H mutant of the Rb. sphaeroides RC using ultrafast visible-pump mid-IR-probe spectroscopy, to identify characteristic absorption features of all the states involved in the primary events in the LM214H RC, including the “new” state P+βL−. The kinetic model used to fit the experimental data was in good agreement with earlier reaction models proposed on the basis of visible region pump-probe and fluorescence data (4,9,11). Signals that reflect responses of the chromophores and protein to charge separation were identified, and possible attributions are proposed. The main results described in this report are: 1) identification of the mid-IR region signature of HL/HL− and βL/βL− by comparing the difference spectra associated with electron transfer to HL in R-26 RCs and to βL in LM214H RCs; 2) observation of the influence of the different environments of the βL and BL BChls by comparing the P+βL− and P+BL− spectra; 3) further confirmation of the mid-IR difference spectrum of PBL/P+BL− reported recently (1); and 4) development of further insights into the protein response to charge separation and charge delocalization within P+.
Materials and Methods
All experiments were performed with a femtosecond visible pump/mid-IR probe laser setup that has been described in previous publications (20,21). Instrument setup and experimental procedures were exactly as those described in detail in two recent reports on visible pump/mid-IR probe spectroscopy of R-26 and mutant RCs (1–3). The LM214H RC was constructed as described by Watson and co-workers (22), and the purification of RCs for spectroscopy was carried out as reported previously (23). For each excitation wavelength experiments were repeated twice using fresh samples, with spectra recorded at 80 different time points. In a single experiment, a spectral probe window of ∼175 cm−1 (between 1598 and 1773 cm−1) was covered. All data were subjected to a global analysis, followed by a target analysis (24,25).
Results
Data acquisition and global analysis
Charge separation in the LM214H RC was investigated using 600 nm, 805 nm and 860 nm excitation, with data collected in the region 1773–1598 cm−1. Two sets of data were recorded employing nonselective excitation of the BL and BM BChls at 805 nm. The data obtained were similar in the two experiments, and there was good correspondence between the line shapes of the evolution associated difference spectra (EADS) and associated lifetimes obtained by global analysis of each data set. The data were compared to data collected previously for the R-26 RC and described in detail in a recent report (1).
Fig. 1 shows two selected time traces at 1684 cm−1 (open squares) and 1715 cm−1 (solid squares) for the LM214H RC and at 1680 cm−1 (open squares) and 1717 cm−1 (solid squares) for the R-26 RC. The presented time traces were taken from experiments involving 600 nm excitation and detection over a ∼175 cm−1 window with a spectral resolution of 6 cm−1. The fit through the data was the result of a global analysis of the whole data set, using a sequential model with increasing lifetimes. The global analysis of the LM214H data yielded five kinetic components with lifetimes of 400 fs, 2 ps, 6.2 ps, 302 ps and 5.2 ns (see Table 1). For R-26 RCs four kinetic components were resolved with lifetimes of 100 fs, 4.4 ps, 222 ps and a nondecaying component (1). As can be seen from Fig. 1, the time traces measured for the LM214H and R-26 RCs were clearly different from each other, reflecting differences in the kinetics of electron transfer in the two RCs. The negative signal before time zero, visible in the traces presented in Fig. 1, originated from perturbed free induction decay (26,27) and was not included in the fit.
Figure 1.
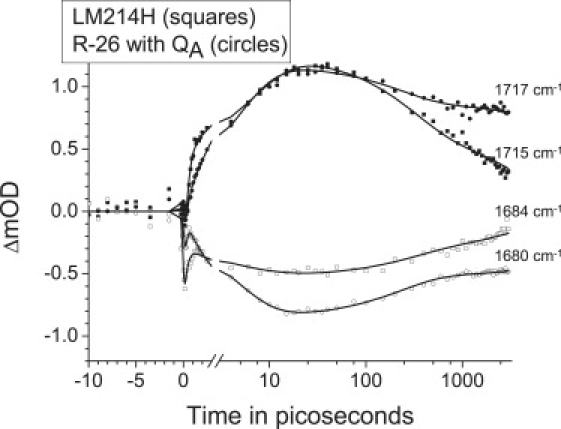
Two representative time traces for LM214H RCs (squares) excited at 600 nm and probed at 1684 cm-1 or 1715 cm−1. For comparison, two time traces for R-26 RCs (circles) excited at 600 nm and probed at ∼1680 cm−1 or ∼1717 cm−1 are shown. The solid lines through the data points are the result of a global fit using a sequential model. The time axis is linear up to 3 ps and logarithmic thereafter. The instrument response function was 150 fs.
Table 1.
Summary of the lifetimes resulting from global analysis of the data obtained in four individual experiments on LM214H RCs
| Excitation wavelength (nm) | IR region /6 cm−1 resolution | Lifetimes resulting from global analysis |
|---|---|---|
| 600 | 1773–1598 | 400 fs, 2 ps, 6.2 ps, 302 ps, 5.2 ns |
| 805 | 1768–1600 | 3 ps, 7 ps, 379 ps, 11 ns |
| 805 | 1768–1600 | 2.6 ps, 6.6 ps, 329 ps, 12.7 ns |
| 860 | 1773–1598 | 700 fs, 1.6 ps, 6.8 ps, 385 ps, 7.5 ns |
Global analysis yielded five (upon excitation at 600 nm and 860 nm) or four (upon excitation at 805 nm) EADS with the associated lifetimes listed in Table 1. An example set of EADS for the LM214H RC excited at 805 nm is shown in Fig. 2. These EADS reflect the spectral evolution of the system in time, but, as discussed in detail elsewhere (24,25), they do not necessarily represent “pure” states. As the resolution of ultrafast energy transfer is hampered in fs mid-IR spectroscopy by a component that follows the instrument response function arising from a coherent interaction of the laser pulses with the sample, the component with a lifetime of a few hundreds of femtoseconds (Table 1) was not shown in any of the EADS or species associated difference spectra (SADS) presented below. Because no reference probe pulse was used, the noise in the measured spectra (not shown) consisted mainly of so-called baseline noise, i.e., a flat, structureless offset in the spectra, which was easily recognized by a singular vector decomposition of the residual matrix. For the time traces in Fig. 1 (and the target analysis described below), the outer product of the first two singular vector pairs of the residual matrix (structureless in the time domain) was subtracted from the data, leading to reduction in the noise by a factor of 2.
Figure 2.
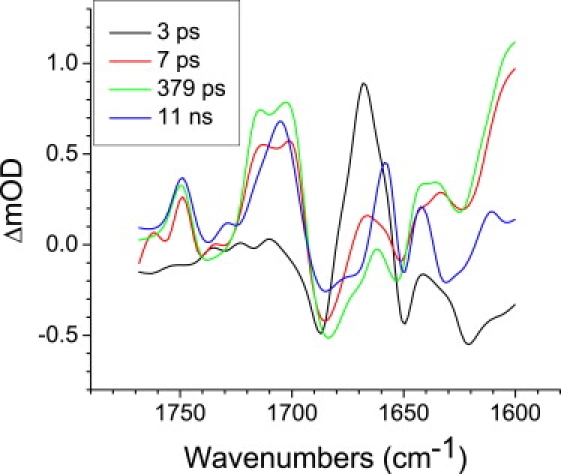
EADS for LM214H RCs excited at 805 nm resulting from global analysis using a sequential model with increasing lifetimes. The measurements were performed over a 1768–1600 cm−1 window with a spectral resolution of 6 cm−1.
Nonselective excitation of BChls BL and BM at 805 nm
Global analysis of each of the two data sets collected in experiments with 805 nm excitation yielded four components with increasing lifetimes of 3 ps, 7 ps, 379 ps, 11 ns in the first experiment and 2.6 ps, 6.6 ps, 329 ps, 12.7 ns in the second experiment (Table 1). The EADS derived from the first data set are shown in Fig. 2. An expected ultrafast component of a few hundred femtoseconds representing a mixture of energy transfer from BM∗ and BL∗ to P was not resolved in this analysis. As with the EADS resulting from 600 nm and 860 nm excitation (see Selective excitation of the P BChls at 860 nm and nonselective excitation at 600 nm), the 3 ps (or 2.6 ps) component represented mainly decay of P∗, and the next 7 ps (or 6.6 ps) component represented a mixture of P∗ and P+BL−. The 379 ps (or 329 ps) component showed features indicative of the P+BL− and P+βL− radical pairs. This spectrum decayed into that of the long-lived charge-separated state P+QA−, the lifetime of which was assumed to be infinite because of the limited time range (11 and 12.7 ns) of the fs mid-IR experiment. The line shapes of the EADS shown in Fig. 2 were similar to those obtained in the experiments with 600 nm and 860 nm excitation (data not shown). In addition, the line shapes of the EADS corresponding to (primarily) the P∗ and P+QA− states were very similar to those described in a previous report on R-26 RCs (1).
In the EADS shown in Fig. 2, negative bands refer to ground-state vibrational modes and positive bands to shifted vibrational modes in excited or charge-separated states. The first EADS that decays in 3 ps represents the excited state of P, plus some amount of charge-separated state (Fig. 2, black). As discussed in previous reports (1,2), the spectrum is dominated by a positive band at 1668 cm−1 and a negative band at 1687 cm−1 assigned to the 9-keto C=O modes of both PL and PM in the excited and ground states, respectively. The 7 ps spectrum (Fig. 2, red) represents a mixture of P∗ and the P+BL− state, and it therefore displays the differential feature attributed to the P/P∗ transition at 1668(+)/1685(−) cm−1, plus positive bands at 1714 cm−1 and 1702 cm−1 attributed to the 9-keto modes of PL+ and PM+, respectively. Further evidence for contribution of the radical pair state was the differential feature at 1740(−)/1749(+) cm−1, which has been attributed to the 10a-ester C=O of PLPM/PL+PM+ (28–31).
The 379 ps EADS (Fig. 2, green) represents a mixture of P+BL− and P+βL− radical pairs. The spectrum was dominated by bands attributable to the P/P+ transition, principally modes at 1685(−)/1714(+) and 1702(+) due to the keto C=O groups, 1738(−)/1749(+) due to the 10a-ester C=O groups, and bands in the 1620–1650 cm−1 region due to the acetyl C=O groups. Modes attributable to the BL/BL− and βL/βL− transitions are considered in the Discussion.
The long-lived spectrum (Fig. 2, blue) represents the P+QA− state. In addition to modes attributable to P/P+ (summarized in Table 2 of Pawlowicz et al. (1,2)) the spectrum contained a number of features attributable to the QA/QA− transition, including signals at 1631(−)/1611(+) cm−1 and 1604(−) cm−1. As discussed in detail in our previous reports (1,2), the signals at 1668(−)/1658(+) cm−1 and 1650(−)/1642(+) cm−1 have been assigned to protein responses to charge separation.
Selective excitation of the P BChls at 860 nm and nonselective excitation at 600 nm
LM214H RCs were also excited at either 860 nm or 600 nm and probed in the region 1773–1598 cm−1. The data collected after 860 nm excitation of the P BChls were analyzed using the global analysis algorithm, which resolved five components with increasing lifetimes of 700 fs, 1.6 ps (P∗), 6.8 ps (P∗ and P+BL−), 385 ps (P+BL− and P+βL−) and a long-lived component (P+QA−) (see Table 1). In the case of 860 nm excitation a large Stokes shift is expected followed by decay of P∗ to the P+BL− radical pair. The further dynamics of the system were expected to remain the same as that in experiments in which 600 nm and 805 nm excitation was used. In agreement with this the line shapes of the EADS (data not shown) were similar to those obtained for the equivalent states in the experiments with 600 nm and 805 nm excitation.
Excitation of LM214H RCs at 600 nm led to nonselective excitation of all four BChls. Global analysis of the data yielded five kinetic components with lifetimes of 400 fs, 2 ps (P∗), 6.2 ps (P∗ and P+BL−), 302 ps (P+BL− and P+βL−), and 5.2 ns (P+QA−) (Table 1). The fastest component that followed the instrument response function described the processes of energy equilibration among all four BChls and formation of P∗.
Target analysis of the four data sets using a single model
The global analysis using a sequential model with increasing lifetimes described in the three preceding paragraphs gave very good fits to the data, but to further disentangle the contributions of various states to the observed spectral evolution, a simultaneous target analysis of all data sets was performed. This analysis included back reactions and a P+BL− intermediate. On the basis of the sequential analysis presented above and results from the literature obtained in visible region pump/probe experiments (see Introduction), rate constants for the forward and backward reactions were estimated and the full kinetic scheme depicted in Fig. 3 was obtained. According to this scheme, decay of the P∗ state proceeds sequentially through the states P+BL−, P+βL−, and P+QA−. This kinetic scheme yielded lifetimes (Fig. 3, right) that agree well with those obtained in published studies employing visible-region pump-probe spectroscopy (4,9–11,32).
Figure 3.
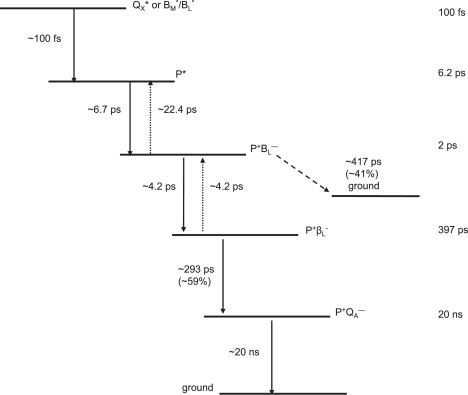
Scheme for energy transfer and electron transfer in LM214H RCs. Solid line arrows (plus associated time constants) refer to forward processes, dotted line arrows to back reactions; ∼40% of the P+BL− state recombined to the ground state with a time constant of ∼417 ps, whereas the remaining ∼60% formed the P+QA− state with a time constant of ∼293 ps. Calculated lifetimes for the various excited or radical pair states are shown on the right.
The kinetic model shown in Fig. 3 was applied simultaneously to all of the data sets described in the previous paragraphs, resulting in lifetimes of 100 fs (Qx∗ or BL∗/BM∗), 6.2 ps (mainly P∗), 2 ps (mainly P+BL−), 397 ps (mainly P+βL−), as well as a nondecaying component (P+QA−). The initial excited states Qx∗ and BL∗/BM∗, observed upon excitation at 600 nm and 805 nm, respectively, are formed during the laser pulse. Energy transfer from BL∗/BM∗ to P, that is expected to take place in ∼100 fs after direct excitation of the accessory BChls, was poorly resolved in our experiments. P∗ decayed with a rate of ∼(6.7 ps)−1 by charge separation to form P+BL−, and this in turn equilibrated with P+βL− in ∼2 ps. P+BL− recombined to the ground state with a rate of ∼(417 ps)−1 (yield of ∼41%), whereas P+βL− formed P+QA− with a rate of ∼(293 ps)−1 (yield of ∼59%). P+QA− was represented by a nondecaying component, because this state decayed on a timescale of ms that is assumed to be infinite, taking into account the time range of our experiment.
SADS arising from the target analysis
A representative set of SADS resulting from the target analysis is shown in Fig. 4 (the SADS corresponding to the ultrafast ∼100 fs component are not shown). These particular SADS were derived from the data obtained with LM214H RCs that had been excited at 805 nm, and very similar SADS were obtained using the remaining data sets.
Figure 4.
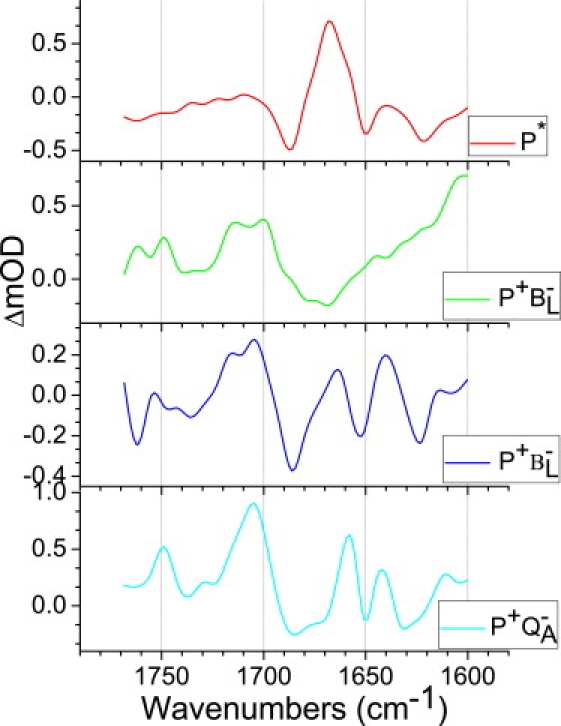
SADS derived from data obtained with LM214H RCs that had been excited at 805 nm. The SADS were estimated from a simultaneous target analysis of all data sets using the kinetic scheme outlined in Fig. 3.
For the P∗, P+BL−, and P+QA− states the line shape of the SADS obtained was similar to that obtained in recent studies on the R-26 RC (1) and a YM210W mutant with slowed charge separation (2). The attribution of bands is discussed in detail in those reports, and is summarized in Table 2 of Pawlowicz et al. (1,2), and only a brief account is given in the remainder of this section. The SADS of the P+βL− state, which is unique to the LM214H RC, is discussed in more depth.
The red spectrum in Fig. 4, which is attributed to P∗, is dominated by a negative band at 1687 cm−1 that is ascribed to the 9-keto modes of PL and PM in the ground state, and this downshifts to 1668 cm−1 in the excited state, giving rise to a positive band. The green spectrum in Fig. 4 is that of the first intermediate in charge separation, P+BL−, and the SADS of this state were generally well reproduced in the simultaneous target analysis of the four data sets. This spectrum contains the ground-state bleaching of the keto C=O groups of PL/PM around 1689 cm−1, plus an additional (main) bleaching in the 1670–1680 cm−1 region that can be ascribed to the keto C=O of BL. A comparison of the SADS of the P+BL− state in R-26 and LM214H RCs is described in more detail in the Discussion. The cyan spectrum in Fig. 4 is that of P+QA−. In addition to modes attributable to the P/P+ transition, this spectrum also contains features that arise from the reduction of QA. The strong band shift signal at 1671(−)/1658(+) cm−1 may be ascribed to a protein amide I transition in response to QA reduction, and it corresponds to a feature at 1666(−)/1656(+) cm−1 in SADS of R-26 RCs (1,33), and at 1668(−)/1657(+) cm−1 in SADS of YM210W RCs (2). A second band shift observed at 1650(−)/1641(+) cm−1 was attributed to a protein backbone C=O H-bonded to QA (28,31,34), and the signals at 1631(−)/1611(+) cm−1 and 1604(−) cm−1 have been assigned to QA/QA− modes (29,31,33,35). The small feature at 1728(+)/1725(−) cm−1 was attributed to a transition of the 10a-ester C=O of the HL cofactor in the electric field of QA− (29,31,36). Because the HL BPhe is replaced by a BChl in the LM214H RC, it seems possible that the 10a-ester C=O of this BChl would respond in a similar way and, if this is the case, then this would underline the conservative nature of the BPhe→BChl substitution indicated by x-ray crystallography (7,8). In Fourier transform infrared spectroscopy experiments (29,31,36) another negative band at 1735 cm−1 was observed in the 10a-ester C=O region. In the fs mid-IR spectrum this band was observed at 1737 cm−1 (Fig. 4, cyan), probably due to an overlap with 10a-ester modes of P, and upshifts to 1749 cm−1 due to P oxidation. The 9-keto mode of P was observed at 1685(−), PM+ at 1705(+) cm−1, whereas the mode of PL+ at 1715(+) cm−1 was reduced in this spectrum.
SADS of the P+βL− radical pair
The blue spectrum in Fig. 4 represents the product of the second step of charge separation, P+βL−. This spectrum, unique to the LM214H RC, shows a number of positive and negative features that can be attributed to the P/P+ transition. The shift of the 9-keto C=O groups of PL/PM manifests by a negative band at 1685 cm−1 (for PL and PM) and positive bands at 1704 cm−1 (for PM+) and 1715 cm−1 (for PL+). The region between 1730 and 1755 cm−1 is expected to contain signals arising from the 10-ester C=O groups of PL/PM and βL, modes of the former shifting up in frequency on formation of PL+/PM+ and of the latter shifting down in frequency on formation of βL−. For the equivalent P+HL− state in R-26 RCs, the assignment of features in this region is further complicated by the presence of two conformational states of the 10-ester C=O of HL, one free from interactions and the other hydrogen bonded to Trp L100 (8,37). Given the degree of structural conservation shown by the LM214H mutant, it is to be expected that the 10-ester C=O of the new βL cofactor will also occupy two equivalent conformational states, complicating the line shape of the SADS of the P+βL− state. In addition, we have shown previously that the line shape of this region of the spectrum is influenced by the resolution of experimental data collected over a narrower window, with 3 cm−1 resolution rather than the 6 cm−1 resolution routinely used, producing a more complex line shape (1). As a result, it is not possible to make a detailed assignment of the bands in the region between 1730 and 1755 cm−1 in the SADS of the P+βL− state, other than the general comment that they constitute overlapping contributions arising from the 10-ester C=O groups of PL/PM and βL. At frequencies below 1680 cm−1 the SADS of P+βL− showed marked differences from those of P+BL− or P+QA−, and these are considered in detail in the next section.
In summary, the SADS obtained for the various states formed during charge separation contained features that correlated well with previous data from fs mid-IR experiments, and most spectral features could be assigned with reasonable confidence. The majority of features arose from frequency shifts associated with oxidation of P or reduction of BL, βL, or QA, with some additional components arising from the response of the protein.
Discussion
Below the key points resulting from the analysis of the experimental data are discussed. The SADS derived from the target analysis are compared to those recently obtained for R-26 RCs (1). Features specific for βL in its ground and anion state observed in fs time-resolved mid-IR spectra are assigned, as are changes in protein conformation due to the electron transfer. Finally, the issue of charge delocalization within the primary electron donor is revisited.
Comparison of LM214H SADS with previous data
SADS of the primary electron donor P∗ and the long-lived radical pair P+QA−
To look at the issue of reproducibility, the SADS for the various states resulting from individual experiments on the LM214H RCs were compared with equivalent spectra obtained in previous work on the R-26 RC (1). Fig. 5 A shows a comparison of the SADS corresponding to P∗ for LM214H (solid line) and R-26 RCs (dashed line), both excited at 860 nm. The spectra are very similar with the main positive band centered at 1668 cm−1, and main negative bands at 1687 and 1650 cm−1. The SADS corresponding to P∗ derived from the data sets measured after 600 nm (not shown) and 805 nm (Fig. 4, red) excitation for the LM214H mutant also had similar line shapes, and we conclude that the spectral signature of this state is very reproducible, apart from the presence of some variation in the underlying baseline.
Figure 5.
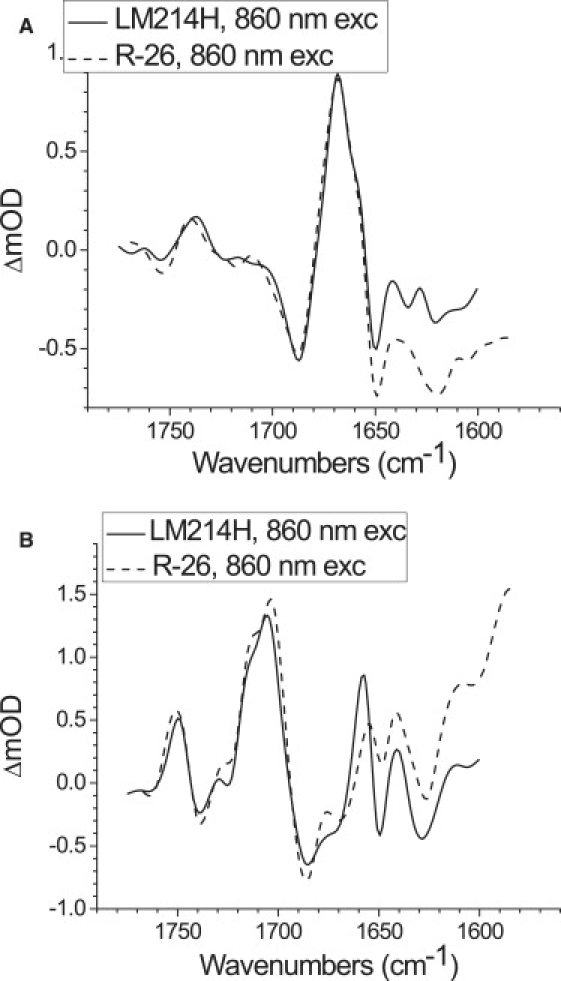
Comparison of the P∗ (A) and P+QA− (B) SADS upon excitation with 860 nm. Each plot presents a comparison of two SADS: for LM214H RCs (solid line) and for R-26 RCs (dashed line).
SADS for the P+QA− state derived from data on LM214H RCs (solid line) and R-26 RCs (dashed line), both excited at 860 nm, are shown in Fig. 5 B. Again the spectra show a good correspondence, with the positions of the multiple positive and negative bands preserved within a few wavenumbers. One point of note is that the amplitude of the 9-keto PL+ band at 1715 cm−1 was somewhat diminished in the LM214H SADS presented in Fig. 5 B compared to the amplitude in the data for the R-26 RC. This 1715 cm−1 feature was almost absent in the SADS for the LM214H RC derived from 805 nm excitation data (Fig. 4, green).
SADS of the short-lived radical pair P+BL−
Fig. 6 compares an average of four SADS for the P+BL− state derived from the four data sets for the LM214H RC recorded in this work (solid line) with an average of six SADS for this state derived from six data sets recorded previously for the R-26 RC (dashed line) (1). The latter utilized data recorded for QA-removed R-26 RCs with excitation at 600, 805 (2 sets), and 860 nm and QA-containing R-26 RCs excited at 600 and 860 nm (see (1) for details).
Figure 6.
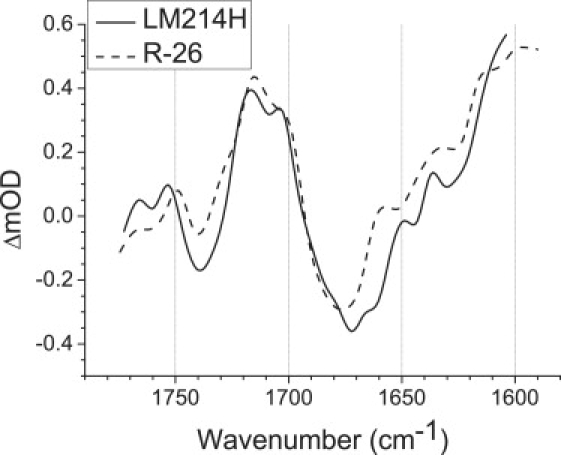
Comparison of averaged SADS for the P+BL− state in R-26 and LM214H RCs. The average of the four SADS obtained for the P+BL− state in LM214H RCs (solid line) is compared with the average of six SADS obtained for the P+BL− state in R-26 RCs (dashed line).
As discussed in previous reports (1–3) the rapid rate of formation of P+BL− and its even more rapid rate of decay indicates that this state does not accumulate at a high level in the WT RC during charge separation. As a result the degree of uncertainty in the SADS of the P+BL− state is much greater than that for the SADS of the P∗, P+HL−, or P+QA− state, each of which builds up to near 100% during the experiment. In the presented case, the level of uncertainty in the P+BL− SADS for the LM214H RC shown in Fig. 6 was somewhat less than that for R-26 RCs, due to the ∼4-fold slower electron transfer from BL− to βL, plus the ∼2-fold slower initial charge separation in the LM214H mutant. Bearing these points in mind, there was a reasonable correspondence between the SADS for the two types of RC (Fig. 6). The pattern of negative and positive bands typical for PL/PM and PL+/PM+, respectively, was reproduced reasonably well, particularly in terms of the transitions of the 9-keto C=O modes of P at 1687(−) cm−1, PL+ at 1715(+) cm−1, and PM+ at 1704(+) cm−1, and the 10a-ester C=O of PL/PM, PL+/PM+ at 1740(−)/1749(+) cm−1, and 2a-acetyl modes of PL/PL+ at 1626(−)/1636(+) cm−1.
The broad negative feature between 1690 and 1660 cm−1 represents the ground-state modes of the 9-keto C=O groups of PLPM and BL. In previous work on the R-26 RC (1), and a mutant YM210W complex (2), we assigned the main negative band between 1670 and 1680 cm−1 to the 9-keto C=O of BL, and the shoulder at ∼1687–1688 cm−1 to the 9-keto C=O groups of PL and PM and these bands are evident in the averaged SADS of P+BL− in the R-26 RC shown in Fig. 6 (dashed line). In the averaged SADS of the LM214H RC, the main negative band was at 1672 cm−1 with a shoulder in the region of 1683 cm−1, and the simplest interpretation of this is that the ground-state mode of both BL and PLPM had both undergone a downshift of several cm−1. The relative amplitude of the PL+ and PM+ bands observed at 1715 and 1700 cm−1 in LM214H SADS was similar to that seen for R-26 RCs, with the 1715 cm−1 band making the major contribution.
SADS of the radical pair P+βL− that is unique to the LM214H RC
Fig. 7 compares the SADS of the state P+βL− that is formed in LM214H RCs (blue) with the SADS of the preceding P+BL− state (green) and the SADS of the equivalent P+HL− state formed in the R-26 RC (cyan), all derived from data with 805 nm excitation (Fig. 7 A). It is clear that in each case the line shape of the P+βL− SADS is significantly different from that of either P+BL− or P+HL−, most notably in the region between 1675 cm−1 and 1630 cm−1. This finding is in agreement with the substitution of the HL BPhe by BChl in the LM214H RC, and it indicates that the spectral differences between the P+BL− or P+HL− SADS in the R-26 RC are partly due to the difference in cofactor at the two positions and partly by differences in the environment of the two cofactors.
Figure 7.
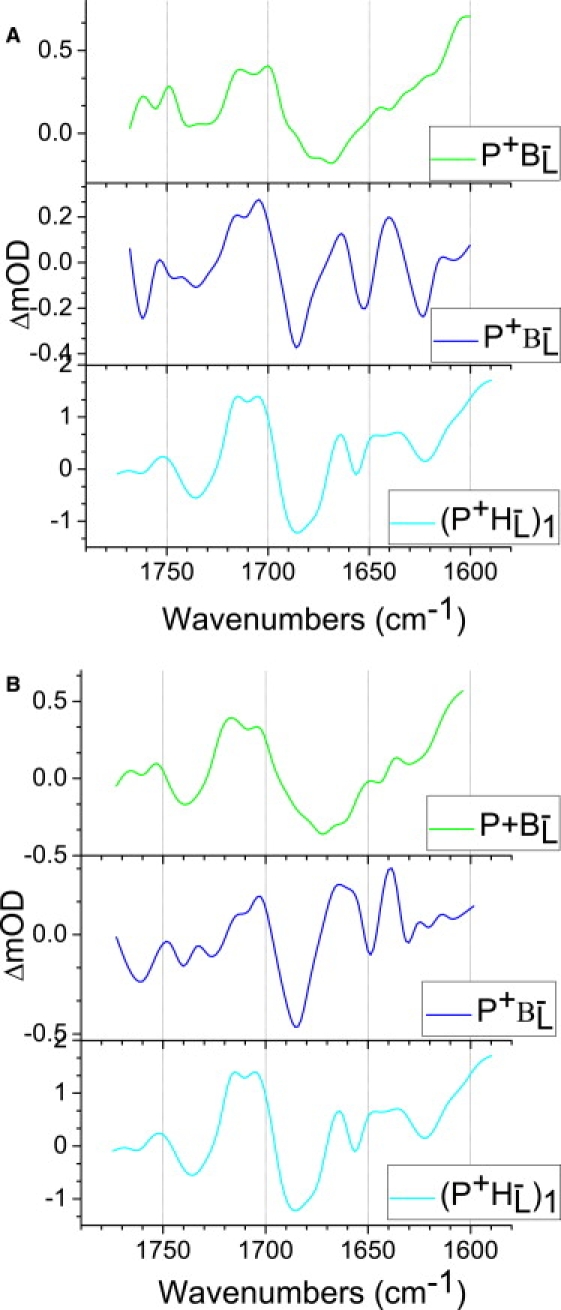
Comparison of the P+BL− and P+βL− SADS resulting from target analysis of the LM214H RCs (green and blue, respectively) and (P+HL−)1 SADS from target analysis of R-26 QA-depleted RCs (cyan): plot A— upon excitation with 805 nm; plot B—average of four P+BL− and P+βL− SADS compared with (P+HL−)1 SADS upon 805 nm excitation.
Because the LM214H mutant lacks a BPhe at the HL position, the negative band at 1675 cm−1 assigned to the 9-keto mode of HL (Fig. 7, cyan spectra) was not observed in the P+βL− SADS (Fig. 7, blue spectra). This negative band, which overlaps with the P ground-state band at 1687 cm−1 in the cyan spectra in Fig. 7, has been assigned to the 9-keto C=O of HL hydrogen bonded to Glu L104 (8). On charge separation this band undergoes a strong downshift to 1591 cm−1 (38). Notably, the negative band at ∼1670 cm−1 in the SADS of the P+BL− state (Fig. 7, green spectra) attributed to the ground-state keto C=O of BL (see above), was also absent from the P+βL− SADS (Fig. 7, blue spectra). Instead, the P+βL− SADS contained a single, particularly intense, negative band at ∼1685 cm−1, which could be a superposition of the ground-state modes of PL/PM and βL. A difference in frequency of 13 cm−1 between the ground-state 9-keto C=O modes of the BChls at the BL and βL positions would be consistent with what is known about the environments of these groups. The keto C=O of BL is proposed to engage in a moderate-strength hydrogen bond interaction with a water molecule located ∼2.9 Å from the keto oxygen, whereas the keto C=O of the HL, and by extension the βL in the LM214H RC, is free from hydrogen bond interactions and therefore might be expected to have a higher frequency C=O stretching mode.
The SADS of P+HL− in the R-26 RC (Fig. 7, cyan spectra) also contain bands at 1665(+)/1656(−) cm−1, which have been ascribed to the response of an amide I C=O to HL reduction (28,38–41). This feature did appear to be present in the SADS of the P+βL− state (Fig. 7, blue spectra) but was slightly shifted to 1664(+)/1652(−) cm−1. However, it has also been proposed that this ground-state mode at 1656 cm−1 is attributable to conformational changes in the protein backbone in response to electron transfer to HL (31) and/or further relaxation of HL− (37), the mode shifting to 1643 cm−1. Examination of the SADS of P+βL− state (Fig. 7, blue spectra) shows the presence of a positive band at ∼1640 cm−1, and therefore the new data on the LM214H mutant does not allow us to distinguish between these assignments. However, the fact that the 1665(+)/1656(−)/1643(+) features were retained despite the replacement of BPhe by BChl in the LM214H mutant does allow us to conclude that they are a response to reduction of the bacteriochlorin at this particular binding site, irrespective of whether it is BPhe or BChl, rather than arising directly from the HL BPhe itself.
In Fourier transform infrared spectroscopy experiments, where the state P+QA− was studied on a slow (ms) timescale, two bands corresponding to the 9-keto mode of P in the ground state were found, at 1692 cm−1 for PL and 1683 cm−1 for PM (29–31). Our experiments show that the observed signal is largely because of the 9-keto C=O of PM, explaining the upshift of the bleaching. As can be seen in the P+BL− (Fig. 7, green spectrum) and P+βL− (Fig. 7, blue spectrum) SADS, the evolution of the relative ratio of the PL+/PM+ bands was now inverted with respect to the P+HL− SADS of R-26 RCs (Fig. 7, cyan spectrum). The PM+ signal at 1705 cm−1 increases in time in the P+BL− and P+βL− SADS. These changes in the exact positions of the PLPM signals, and the charge distribution within P with the final electron hole localization on the PL half of the dimer, must be the result of the different environment of the P upon the BPheo a replacement.
Conclusions and Outlook
Using the fs mid-IR technique, the processes of electron transfer and protein dynamics after excitation in the visible/near-IR region were studied in an Rb. sphaeroides RC in which the HL BPhe was replaced by a BChl. The experimental results were analyzed using a kinetic model based on charge separation dynamics derived from analysis of visible region pump/probe data, and a good agreement was found with time constants obtained from mid-IR probe data. The mid-IR absorption spectrum of P+BL− extracted from a target analysis of data on the LM214H mutant agreed well with equivalent spectrum for the R-26 RC obtained in a previous report (1). The P+I− intermediate state observed in previous experiments (4,9–11) was resolved into two distinct spectral states, P+BL− and P+βL−, and features attributable to transitions specific to BL, βL, and HL present in the fs mid-IR difference spectra were identified, and an influence of the environment of the BL on its vibrational transitions was observed. To further clarify the origin of IR signals that do not arise directly from the electron transfer cofactors and therefore must arise from the protein-solvent environment, a further study of RCs with mutations that change the structure of the protein-cofactor system in the immediate vicinity of the primary reactants is required.
Acknowledgments
This research was supported by the Netherlands Organization of Scientific Research via the Foundation of Earth and Life Sciences, by the European Union (Grant MRTN-CT-2003-505069, Intro2), and by Human Frontiers Scientific Program Research Grant RGP 38/2006. M.R.J. acknowledges financial support from the Biotechnology and Biological Sciences Research Council of the United Kingdom.
References
- 1.Pawlowicz N.P., van Grondelle R., van Stokkum I.H.M., Breton J., Jones M.R. Identification of the first steps in charge separation in bacterial photosynthetic reaction centers of Rhodobacter sphaeroides by ultrafast mid-infrared spectroscopy: electron transfer and protein dynamics. Biophys. J. 2008;95:1268–1284. doi: 10.1529/biophysj.108.130880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlowicz N.P., van Stokkum I.H.M., Breton J., van Grondelle R., Jones M.R. Charge separation in tyrosine M210 to tryptophan mutant of the Rhodobacter sphaeroides reaction center studied with femtosecond mid-infrared spectroscopy. Phys. Chem. Chem. Phys. 2008 doi: 10.1039/b905934b. Submitted. [DOI] [PubMed] [Google Scholar]
- 3.Pawlowicz N.P., van Stokkum I.H.M., Breton J., van Grondelle R., Jones M.R. Multi-phasic radical pair relaxation in bacterial reaction centers that lack the primary quinone studied by femtosecond visible pump/mid-IR probe spectroscopy. Photosynth. Res. 2008 Submitted. [Google Scholar]
- 4.Kirmaier C., Gaul D., DeBey R., Holten D., Schenck C.C. Charge separation in a reaction center incorporating bacteriochlorophyll for photoactive bacteriopheophytin. Science. 1991;251:922–927. doi: 10.1126/science.2000491. [DOI] [PubMed] [Google Scholar]
- 5.Yeates T.O., Komiya H., Chirino A., Rees D.C., Allen J.P. Structure of the reaction center from Rhodobacter sphaeroides R-26 and 2.4.1—protein-cofactor (bacteriochlorophyll, bacteriopheophytin and carotenoid) interactions. Proc. Natl. Acad. Sci. USA. 1988;85:7993–7997. doi: 10.1073/pnas.85.21.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komiya H., Yeates T.O., Rees D.C., Allen J.P., Feher G. Structure of the reaction center from Rhodobacter sphaeroides R-26 and 2.4.1—symmetry relations and sequence comparisons between different species. Proc. Natl. Acad. Sci. USA. 1988;85:9012–9016. doi: 10.1073/pnas.85.23.9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chirino A.J., Lous E.J., Huber M., Allen J.P., Schenck C.C. Crystallographic analyses of site-directed mutants of the photosynthetic reaction center from Rhodobacter sphaeroides. Biochemistry. 1994;33:4584–4593. doi: 10.1021/bi00181a020. [DOI] [PubMed] [Google Scholar]
- 8.Paddock M.L., Chang C., Xu Q., Abresch E.C., Axelrod H.L. Quinone (QB) reduction by B-branch electron transfer in mutant bacterial reaction centers from Rhodobacter sphaeroides: quantum efficiency and X-ray structure. Biochemistry. 2005;44:6920–6928. doi: 10.1021/bi047559m. [DOI] [PubMed] [Google Scholar]
- 9.Kirmaier C., Laporte L., Schenck C.C., Holten D. The nature and dynamics of the charge-separated intermediate in reaction centers in which bacteriochlorophyll replaces the photoactive bacteriopheophytin. 1. Spectral characterization of the transient state. J. Phys. Chem. 1995;99:8903–8909. [Google Scholar]
- 10.Heller B.A., Holten D., Kirmaier C. Control of electron transfer between the L- and M-sides of photosynthetic reaction centers. Science. 1995;269:940–945. doi: 10.1126/science.7638616. [DOI] [PubMed] [Google Scholar]
- 11.Kirmaier C., Laporte L., Schenck C.C., Holten D. The nature and dynamics of the charge-separated intermediate in reaction centers in which bacteriochlorophyll replaces the photoactive bacteriopheophytin. 2. The rates and yields of charge separation and recombination. J. Phys. Chem. B. 1995;99:8910–8917. [Google Scholar]
- 12.Ziolek M., Pawlowicz N., Naskrecki R., Dobek A. Electron transfer in the reaction center of the Rb. sphaeroides R-26 studied by transient absorption. J. Phys. Chem. B. 2005;109:18171–18176. doi: 10.1021/jp050682i. [DOI] [PubMed] [Google Scholar]
- 13.Hamm P., Zinth W. Ultrafast initial reaction in bacterial photosynthesis revealed by femtosecond infrared spectroscopy. J. Phys. Chem. 1995;99:13537–13544. [Google Scholar]
- 14.Zinth W., Knapp E.W., Fischer S.F., Kaiser W., Deisenhofer J. Correlation of structural and spectroscopic properties of a photosynthetic reaction center. Chem. Phys. Lett. 1985;119:1–4. [Google Scholar]
- 15.Woodbury N.W.T., Parson W.W. Nanosecond fluorescence from isolated photosynthetic reaction centers of Rhodopseudomonas sphaeroides. Biochim. Biophys. Acta. 1984;767:345–361. doi: 10.1016/0005-2728(84)90205-6. [DOI] [PubMed] [Google Scholar]
- 16.Woodbury N.W., Allen J.P. The pathway, kinetics and thermodynamics of electron transfer in wild type and mutant bacterial reaction centers of purple nonsulfur bacteria. In: Blankenship R.E., Madigan M.T., Bauer C.E., editors. Anoxygenic Photosynthetic Bacteria. Kluwer Academic; Dordrecht, The Netherlands: 1995. pp. 527–557. [Google Scholar]
- 17.Schenck C.C., Blankenship R.E., Parson W.W. Radical-pair decay kinetics, triplet yields and delayed fluorescence from bacterial reaction centers. Biochim. Biophys. Acta. 1982;680:44–59. [Google Scholar]
- 18.Bixon M., Jortner J., Michel-Beyerle M.E., Ogrodnik A. A superexchange mechanism for the primary charge separation in photosynthetic reaction centers. Biochim. Biophys. Acta. 1989;977:273–286. [Google Scholar]
- 19.Goldstein R.A., Boxer S.G. The effect of very high magnetic fields on the reaction dynamics in bacterial reaction centers—implications for the reaction mechanism. Biochim. Biophys. Acta. 1989;977:78–86. [Google Scholar]
- 20.Groot M.L., van Wilderen L.J.G.W., Larsen D.S., van der Horst M.A., van Stokkum I.H.M. Initial steps of signal generation in photoactive yellow protein revealed with femtosecond mid-infrared spectroscopy. Biochemistry. 2003;42:10054–10059. doi: 10.1021/bi034878p. [DOI] [PubMed] [Google Scholar]
- 21.Groot M.L., Breton J., van Wilderen L.J.G.W., Dekker J.P., van Grondelle R. Femtosecond visible/visible and visible/mid-IR pump-probe study of the Photosystem II core antenna complex CP47. J. Phys. Chem. B. 2004;108:8001–8006. [Google Scholar]
- 22.Watson A.J., Fyfe P.K., Frolov D., Wakeham M.C., Nabedryk E. Replacement or exclusion of the B-branch bacteriopheophytin in the purple bacterial reaction centre: the HB cofactor is not required for assembly or core function of the Rhodobacter sphaeroides complex. Biochim. Biophys. Acta Bioenergs. 2005;1710:34–46. doi: 10.1016/j.bbabio.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 23.McAuley-Hecht K.E., Fyfe P.K., Ridge J.P., Prince S.M., Hunter C.N. Structural studies of wild type and mutant reaction centers from an antenna-deficient strain of Rhodobacter sphaeroides: monitoring the optical properties of the complex from bacterial cell to crystal. Biochemistry. 1998;37:4740–4750. doi: 10.1021/bi971717a. [DOI] [PubMed] [Google Scholar]
- 24.van Stokkum I.H.M., Beekman L.M.P., Jones M.R., van Brederode M.E., van Grondelle R. Primary electron transfer kinetics in membrane-bound Rhodobacter sphaeroides reaction centers: a global and target analysis. Biochemistry. 1997;36:11360–11368. doi: 10.1021/bi9707943. [DOI] [PubMed] [Google Scholar]
- 25.van Stokkum I.H.M., Larsen D.S., van Grondelle R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta Bioenerg. 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Lozovoy V.V., Pastirk I., Comstock M.G., Dantus M. Cascaded free-induction decay four-wave mixing. Chem. Phys. 2001;266:205–212. [Google Scholar]
- 27.Mitsumori Y., Fukushima H., Ogura M., Kuroda T., Koshihara S. Excitonic free induction decay studied with femtosecond pulse pairs. J. Lumin. 1996;66&67:81–83. [Google Scholar]
- 28.Nabedryk E., Bagley K.A., Thibodeau D.L., Bauscher M., Mantele W. A protein conformational change associated with the photoreduction of the primary and secondary quinones in the bacterial reaction center. FEBS Lett. 1990;266:59–62. doi: 10.1016/0014-5793(90)81506-j. [DOI] [PubMed] [Google Scholar]
- 29.Breton J., Thibodeau D.L., Berthomieu C., Mantele W., Vermeglio A. Probing the primary quinone environment in photosynthetic bacterial reaction centers by light-induced FTIR difference spectroscopy. FEBS Lett. 1991;278:257–260. doi: 10.1016/0014-5793(91)80129-q. [DOI] [PubMed] [Google Scholar]
- 30.Bagley K.A., Abresch E., Okamura M.Y., Feher G., Bauscher M. FTIR studies of the D+QA− and D+QB− states in reaction centers from Rb. sphaeroides. In: Baltscheffsky M., editor. Current Research in Photosynthesis. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1990. pp. 77–80. [Google Scholar]
- 31.Nabedryk E. Light-induced Fourier transform infrared difference spectroscopy of the primary electron donor in photosynthetic reaction centers. In: Mantsch H.H., Chapman D., editors. Infrared Spectroscopy of Biomolecules. Wiley-Liss; New York: 1996. pp. 39–81. [Google Scholar]
- 32.Shkuropatov A.Y., Shuvalov V.A. Electron transfer in pheophytin a-modified reaction centers from Rhodobacter sphaeroides (R-26) FEBS Lett. 1993;322:168–172. doi: 10.1016/0014-5793(93)81561-d. [DOI] [PubMed] [Google Scholar]
- 33.Breton J., Burie J.R., Berthomieu C., Berger G., Nabedryk E. The binding-sites of quinones in photosynthetic bacterial reaction centers investigated by light-induced FTIR difference spectroscopy— assignment of the QA vibrations in Rhodobacter sphaeroides using O18-labeled or C13-labeled ubiquinone and vitamin K1. Biochemistry. 1994;33:4953–4965. doi: 10.1021/bi00182a026. [DOI] [PubMed] [Google Scholar]
- 34.Breton J., Nabedryk E. Protein-quinone interactions in the bacterial photosynthetic reaction center: light-induced FTIR difference spectroscopy of the quinone vibrations. Biochim. Biophys. Acta Bioenerg. 1996;1275:84–90. [Google Scholar]
- 35.Breton J., Bauscher M., Berthomieu C., Thibodeau D.L., Andiranambinintsoa S., Dejonghe D., Mantele W., Nabedryk E. FTIR difference spectroscopy of mena-quinone photo-reduction in bacterial reaction centers. In: Hester R.E., Girling R.B., editors. Spectroscopy of Biological Molecules. The Royal Society of Chemistry; Cambridge, UK: 1991. pp. 43–46. [Google Scholar]
- 36.Breton J., Nabedryk E., Allen J.P., Williams J.C. Electrostatic influence of QA reduction on the IR vibrational mode of the 10a-ester C=O of HA demonstrated by mutations at residues Glu L104 and Trp L100 in reaction centers from Rhodobacter sphaeroides. Biochemistry. 1997;36:4515–4525. doi: 10.1021/bi962871k. [DOI] [PubMed] [Google Scholar]
- 37.Mantele W.G., Wollenweber A.M., Nabedryk E., Breton J. Infrared spectroelectrochemistry of bacteriochlorophylls and bacteriopheophytins—implications for the binding of the pigments in the reaction center from photosynthetic bacteria. Proc. Natl. Acad. Sci. USA. 1988;85:8468–8472. doi: 10.1073/pnas.85.22.8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nabedryk E., Andrianambinintsoa S., Dejonghe D., Breton J. FTIR Spectroscopy of the photoreduction of the bacteriopheophytin electron acceptor in reaction centers of Rhodobacter sphaeroides and Rhodopseudomonas viridis. Chem. Phys. 1995;194:371–378. [Google Scholar]
- 39.Maiti S., Cowen B.R., Diller R., Iannone M., Moser C.C. Picosecond infrared studies of the dynamics of the photosynthetic reaction center. Proc. Natl. Acad. Sci. USA. 1993;90:5247–5251. doi: 10.1073/pnas.90.11.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maiti S., Walker G.C., Cowen B.R., Pippenger R., Moser C.C. Femtosecond coherent transient infrared spectroscopy of reaction centers from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 1994;91:10360–10364. doi: 10.1073/pnas.91.22.10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thibodeau D.L., Breton J., Berthomieu C., Mantele W., Nabedryk E. FTIR studies on quinone containing photosynthetic reaction centers. In: MichelBeyerle M.-E., editor. Reaction Centres of Photosynthetic Bacteria. Springer; Berlin, Germany: 1990. pp. 87–98. [Google Scholar]