

Abstract
The widely used hydrophobic cannabinoid ligand CP-55,940 partitions with high efficiency into biomembranes. We studied the location, orientation, and dynamics of CP-55,940 in POPC bilayers by solid-state NMR. Chemical-shift perturbation of POPC protons from the aromatic ring-current effect, as well as 1H NMR cross-relaxation rates, locate the hydroxyphenyl ring of the ligand near the lipid glycerol, carbonyls, and upper acyl-chain methylenes. Order parameters of the hydroxyphenyl ring determined by the 1H-13C DIPSHIFT experiment indicate that the bond between the hydroxyphenyl and hydroxycyclohexyl rings is oriented perpendicular to the bilayer normal. 2H NMR order parameters of the nonyl tail are very low, indicating that the hydrophobic chain maintains a high level of conformational flexibility in the membrane. Lateral diffusion rates of CP-55,940 and POPC were measured by 1H magic-angle spinning NMR with pulsed magnetic field gradients. The rate of CP-55,940 diffusion is comparable to the rate of lipid diffusion. The magnitude of cross-relaxation and diffusion rates suggests that associations between CP-55,940 and lipids are with lifetimes of a fraction of a microsecond. With its flexible hydrophobic tail, CP-55,940 may efficiently approach the binding site of the cannabinoid receptor from the lipid-water interface by lateral diffusion.
Introduction
The ligand CP-55,940 (Fig. 1 a) is a widely used synthetic agonist of the human central cannabinoid receptor CB1 (1) and the peripheral cannabinoid receptor CB2 (2,3), which belong to the class of heptahelical G-protein-coupled receptors (GPCRs). Cannabinoid ligands, including the endocannabinoids 2-AG (4,5) and anandamide (6), are known to be highly lipophilic (the octanol-water partition coefficient of CP-55,940 is 1.35 × 106) (7). It is assumed that incorporation of lipophilic ligands into the plane of membranes containing the receptors facilitates the approach to the binding site of the receptor through lateral diffusion (8,9). A similar diffusion-controlled mechanism of receptor approach was also proposed for binding between amphipathic neuropeptides and GPCRs (10). These peptides possess significant affinity for membranes, and molecular details of the peptide-lipid interactions are being elucidated (10–13). The binding crevice for ligands of cannabinoid receptors is expected to be located among transmembrane helices III, V, VI, and VII near the extracellular face of the receptor (14), as was recently found for the crystal structures of the β2-adrenergic receptor (15) and the human A2A adenosine receptor (16). Most likely, the ligand needs to approach the receptor at a particular depth in the lipid matrix and in particular orientation(s) to be able to reach the binding pocket efficiently. The conformational flexibility of the ligand could be critical for reducing steric hindrance during entry from the lipid matrix and allowing the ligand to adjust to the geometry of the binding site. Understanding interactions between CP-55,940 and lipids is thus an important subject for biophysical research that contributes to our understanding of ligand-receptor binding. The conformation of CP-55,940 in organic solvent used as a membrane mimetic environment was reported previously and a model of bilayer incorporation was suggested (17). However, the location, orientation, and dynamics of the ligand in actual membranes have not yet been investigated.
Figure 1.
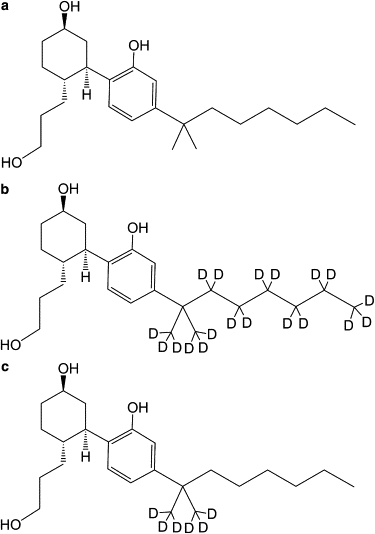
Chemical structure of (a) synthetic cannabinoid ligand CP-55,940 and its deuteration at the nonyl tail (b) CP-55,940-d19 and (c) CP-55,940-d6.
In this work, the interaction of CP-55,940 with bilayers of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was studied. Using solid-state NMR methods, we determined 1), the location of the hydroxyphenyl ring of CP-55,940 in bilayers; 2), the orientation of the hydroxyphenyl ring with respect to the bilayer normal; 3), the conformation and dynamics of the nonyl tail; 4), the lateral diffusion coefficients of the ligand and lipids; and 5), the perturbation of the acyl-chain order of lipids from CP-55,940 incorporation.
The location of the hydroxyphenyl ring of CP-55,940 in bilayers is critical for understanding CP-55,940-lipid interactions. The locations of simple aromatic rings such as benzene (18), phenol (19), indole (19–21), and pyrene (22) in bilayers were investigated previously. Although benzene locates preferentially in the hydrophobic core (18), the addition of a directly bonded hydroxyl group (as in phenol) results in a location near the lipid-water interface (19). It is also known that the hydroxyphenyl ring of tyrosine in proteins and peptides is found at high probability near the lipid-water interface of membranes (23). The aromaticity of the hydroxyphenyl ring of CP-55,940 is not only important for its interaction with lipids, it also provides a convenient way to specify the location of the ligand in bilayers by NMR (21,24). In this work, we determined the location of the hydroxyphenyl ring of CP-55,940 at atomic resolution via its ring-current effect on the lipid 1H chemical shifts (21), and by measuring cross-relaxation rates between the ring and lipid protons (24). High resolution of the NMR signals was achieved by magic-angle spinning (MAS) using rotor inserts to create a small spherical volume in the center of the rotor.
The orientation of cannabinoid ligands in bilayers was previously investigated for the cannabis ingredients Δ9-THC (25,26), Δ8-THC (26–28), and a functionally inactive analog, O-methyl-Δ8-THC (Me-Δ8-THC) (26,28,29). Small-angle x-ray diffraction at ∼6 Å resolution and 2H NMR quadrupolar splittings of the isotope-labeled tricyclic rings showed that Δ9-THC and Δ8-THC are located near the water-membrane interface and oriented with the bond connecting the hydroxyphenyl and cyclohexenyl rings perpendicular to the bilayer normal (25–28). With this orientation, the hydroxyl group at the phenyl ring is exposed to the water phase. In contrast, Me-Δ8-THC, a methylated analog at the hydroxyl group, has an orientation of the same bond parallel to the bilayer normal (26,28,29). It was suggested that the orientation of the functionally active Δ9-THC and Δ8-THC may be critical for ensuring efficient access to the receptor-binding pocket from the lipid matrix (26). Here we determined the orientation of the hydroxyphenyl ring of CP-55,940 in POPC bilayers by measuring the 1H-13C dipolar interactions with the dipolar and chemical-shift (DIPSHIFT) correlation experiment (30). The magnitude of the 1H-13C dipolar interactions in the ring is a function of the orientation of the ring, i.e., the angle between the 1H-13C bond and the bilayer normal. Since the ligand may approach the receptor from the lipid matrix, we measured the lateral diffusion rates of CP-55,940 and POPC by 1H MAS NMR with application of pulsed magnetic field gradients (PFGs).
Another important structural feature of CP-55,940 is the conformational state of the nonyl tail. Because of its hydrophobicity, the nonyl tail is expected to have a tendency to point toward the bilayer core like an anchor. The efficiency of anchoring should be linked to the location and orientation of the hydroxyphenyl ring to which it is bonded. The nonyl tail was selectively deuterated (Fig. 1, b and c), and 2H NMR quadrupolar splittings were measured and analyzed for orientation and chain isomerization. The 2H NMR method was also applied to the lipids that had a perdeuterated palmitoyl chain (POPC-d31) to sensitively monitor perturbations of lipid packing from CP-55,940 incorporation.
Materials and Methods
Materials
CP-55,940 ((1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol); Fig. 1 a), was purchased from Tocris Bioscience (Ellisville, MO). CP-55,940 with a deuterated alkyl tail (CP-55,940-d19 and -d6; Fig. 1, b and c) was synthesized as described elsewhere (31). POPC and POPC with a perdeuterated palmitoyl chain (POPC-d31) were obtained from Avanti Polar Lipids (Alabaster, AL). Deuterium-depleted H2O (2–3 ppm D), D2O (99.9%D), and methanol-d4 (99.8% D with 0.05% v/v tetramethylsilane) were obtained from Cambridge Isotope Laboratories (Andover, MA).
Multilamellar vesicles (MLVs) containing various concentrations of the ligand (0, 15, and 30 mol %) were prepared as follows: First, 7 mg of lipid and a proper amount of the ligand were dissolved in methanol, and the solvent was removed in a stream of nitrogen gas to form a dry film. MLVs were formed by addition of an equal volume of water, and transferred to a 4-mm outer-diameter zirconia MAS rotor with an insert made of Kel-F (Bruker Biospin, Billerica, MA) that kept the sample centered within the rotor. The DIPSHIFT experiment was conducted on a sample of ∼25 mg of POPC at a POPC/CP-55,940 molar ratio of 7:3, using a MAS rotor with an insert for a 50-μL sample volume. For 2H NMR experiments on deuterated ligand or lipid, samples were prepared with deuterium-depleted water.
1H MAS NMR
Solid-state 1H MAS NMR spectra were recorded on a Bruker AV800 spectrometer operating at a resonance frequency of 800.18 MHz. Sample spinning at 10 kHz was accomplished with a Bruker 4-mm-diameter double gas bearing MAS probehead. The temperature inside the spinning rotor was 17°C, which was calibrated by recording the proton spectra of 1-stearoyl-2-oleoyl-sn-glyero-3-phosphocholine (SOPC) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) as a function of temperature to detect the gel-to-liquid crystalline phase transition at 6.6°C (32) and 23.5°C (33), respectively. The temperature was controlled to ±0.1°C. The chemical-shift scale was calibrated by setting the resonance of the terminal methyl group of the lipid acyl chains to 0.885 ppm (24).
1H MAS nuclear Overhauser enhancement spectroscopy NMR
1H NMR cross-relaxation rates reflecting ligand-lipid interactions were measured on lipid bilayers containing 30 mol % ligand. Two-dimensional phase-sensitive 1H MAS nuclear Overhauser enhancement spectroscopy (NOESY) NMR experiments were recorded at 17°C and a MAS frequency of 10 kHz on a Bruker AV800 spectrometer. The pulse sequence (90° − t1 − 90° − τm − 90° − acquire [t2])n was used with a 2.7-μs 90° pulse, 512 t1 increments, 4096 t2 data points, and 16 scans per t1 increment. Spectra for the mixing times, τm, of 5, 50, 100, 150, 200, 250, 300, and 350 ms were recorded. The intensities of the diagonal- and cross-peaks were determined by volume integration and cross-relaxation rates calculated by a matrix algorithm as reported previously (24). To aid signal assignment, a high-resolution spectrum of CP-55,940 in methanol was recorded on the same spectrometer.
2H NMR
2H NMR powder spectra of POPC-d31 bilayers containing 0, 15, or 30 mol % CP-55,940, and POPC bilayers containing 30 mol % CP-55,940-d19 or -d6 were acquired on the Bruker AV800 spectrometer operating at a resonance frequency of 122.83 MHz. The MAS probehead was used without sample spinning. Sample temperature was varied from 5°C to 45°C in increments of 5°C. A quadrupolar echo sequence (34) was used with two 5.2-μs 90° pulses and an interpulse delay of 25 μs. Typically 10,240 scans at a spectral width of 500 kHz and a delay time of 0.2 s were acquired.
Analysis of the 2H NMR spectra was conducted as follows: Spectra were recorded and the signals phase-corrected to minimize intensity in the imaginary channel. The echo maximum in the real channel was determined with a resolution of 1/10 of a dwell-time unit. A time-base-corrected free induction decay (FID) was calculated by spline interpolation between data points to begin exactly at the echo maximum. An exponential line-broadening of 100 Hz was applied to the FID before the Fourier transformation. 2H NMR powder pattern spectra were dePaked (35,36) to obtain spectra that corresponded to the 0° orientation of the bilayer normal with respect to the external magnetic field. The order parameter S(n) of the methylene or methyl groups at the carbon number n of the palmitoyl chain were then determined from the quadrupolar splitting ΔνQ(n) as
| (1) |
where e2qQ/h is the quadrupolar coupling constant (167 kHz for deuterons in the C-2H bond). Smoothed order parameter profiles for the carbon sites from C2 to C11 giving unresolved signals were calculated according to Lafleur et al. (37). All data processing starting from the manipulation of the FID was done with a program written for Mathcad (MathSoft, Cambridge, MA).
DIPSHIFT
To determine the orientation of the hydroxyphenyl ring of CP-55,940 with respect to the bilayer normal, 1H-13C dipolar interactions of the ring were measured on a liposome sample containing 30 mol % CP-55,940. Measurements were performed with the DIPSHIFT experiment (30) on the Bruker AV800 spectrometer with a Bruker 4-mm-diameter MAS probehead operated at the 13C resonance frequency of 201.21 MHz. The sample temperature was 25.0°C as verified by the 1H chemical shift of water immediately before and after the DIPSHIFT experiment. The MAS frequency was set to 3,994 Hz to fit seven cycles of the MREV-8 pulse train into one rotor period. After the first 2.8-μs 90° pulse on 1H, cross polarization from 1H to 13C was applied at 42 kHz for 1 ms, ramped from 100% to 75%. The evolution time t1 of the spin system under 1H-13C dipolar interaction was altered by varying the number of 37.1-μs MREV-8 cycles from zero to seven within one rotor period. The decoupling power on the 1H channel for the homonuclear decoupling with MREV-8 and for continuous wave decoupling after MREV-8 was 105 kHz. The duration of continuous wave decoupling was 40 ms. The π-pulse on the 13C channel had a length of 10 μs. For each of the t1 increments, the natural-abundance 13C spectrum was acquired with 3000 scans with a delay time of 4 s. The magnitude of the 1H-13C dipolar interactions was determined by comparing the dipolar dephasing curves with theoretical curves that were calculated as reported previously (30). The chemical-shift scale was calibrated by setting the resonance of the terminal methyl group of the lipid acyl chain to 14.1 ppm (38).
1H MAS NMR PFG diffusion experiments
1H MAS NMR PFG diffusion experiments on bilayers containing 15 mol % CP-55,940 were performed on a Bruker DMX500 at a proton frequency of 500.17 MHz with a Bruker 4-mm PFG-MAS probe with a magic-angle gradient. Sample spinning was set to 5 kHz. The temperature inside the spinning rotor was 22°C. Spectra were acquired with a stimulated-echo sequence (39) using sine-shaped bipolar gradient pulses with a length of 5 ms and a diffusion time of 50 ms. The spectra were acquired at 16 different values of gradient strength varying from to 0.007 to 0.368 T/m. A longitudinal eddy-current delay of 5 ms was used. Diffusion constants were determined by fitting measured intensities to the following equation (40):
| (2) |
where D is the diffusion constant and k is a factor that depends on the pulse sequence and instrumental settings. For the stimulated-echo sequence with sine pulses, k = 4γ2g2δ2(Δ − (T/2) − (δ/8)), where γ is the gyromagnetic ratio of protons, g is the gradient strength, δ is the gradient pulse length, Δ is the diffusion time, and T is the time between the gradient pulses sandwiching the 180° pulses.
Results and Discussion
Lipid order after CP-55,940 insertion
Perturbation of lipid packing in the bilayers from CP-55,940 incorporation was monitored by measuring the 2H NMR quadrupolar splittings, ΔνQ, of the perdeuterated palmitoyl chain of POPC-d31. The shape of the order parameter profile and the magnitude of ΔνQ are a reflection of the phase state of lipids (41), whereas the perturbation of ΔνQ along the acyl chain after ligand incorporation reflects the location of the ligand.
The spectra acquired for the POPC-d31 bilayers at CP-55,940 concentrations of 0, 15, or 30 mol % at 25°C (see Fig. S1 in the Supporting Material) demonstrate the integrity and homogeneity of the fluid bilayers. We have also observed that the functional integrity of the recombinant pheripheral cannabinoid receptor CB2, reconstituted in lipid bilayers, is fully maintained by addition of CP-55,940 in this concentration range (data not shown). The quadrupolar splittings, ΔνQ, decrease with increasing ligand content, indicating reduced order parameters. Order parameter profiles S(n) calculated as described in the Materials and Methods section are plotted in the top panel of Fig. 2. The reduction of order takes place along the entire chain. The same trend was observed regardless of the temperature in the investigated range of 5–45°C (see Fig. S2). The overall reduction of order indicates that CP-55,940 incorporation has a tendency to increase the lateral area and reduce the thickness of the bilayers.
Figure 2.
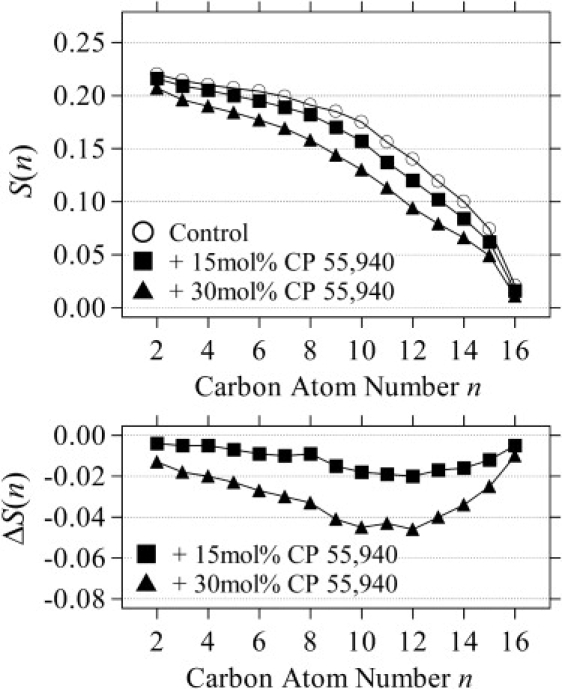
Order parameters S(n) of the POPC-d31 palmitoyl chain at 0, 15, and 30 mol % of CP-55,940 at 25°C (top panel). Ligand-induced changes of order parameters, ΔS(n), are shown in the bottom panel.
Changes of the order parameters, ΔS(n), due to CP-55,940 incorporation are plotted in the bottom panel of Fig. 2. The order reduction is larger from the middle of the chain toward the terminal methyl group. The reduction close to the chain terminus is smaller because of generally low chain order in this region. The observation is in contrast to a previously reported CP-55,940-induced order (ΔS(n) > 0) near the surface of DMPC-DHPC bicelles (42), whereas the overall trend of larger disordering toward the bilayer center is similar. The degree of disordering can be quantified by comparing averages of order parameter changes, 〈ΔS(n)〉, for the upper half (C2-8) and lower half (C9-16) of the chain (Table S1). The larger disordering in the lower half of the chains supports a location of CP-55,940 at the lipid-water interface, as would intuitively be expected from its amphipathic structure. The ligand incorporation at the interface introduces a void volume in the bilayer center, causing progressive disordering toward the chain termini. With increasing temperature, hydrocarbon chains become more disordered by enhanced thermal motions, which diminish the effect from ligand incorporation (compare also Fig. 2 and Fig. S2).
Location of the hydroxyphenyl ring
The location of the CP-55,940 hydroxyphenyl ring in POPC bilayers was determined by analyzing the ring-current-induced chemical-shift perturbation (43) of POPC protons measured by 1H MAS NMR (21), as well as by measuring ligand-to-lipid cross-relaxation rates with the two-dimensional 1H MAS NOESY experiment (24). Here we approximate that the ring-current effect of CP-55,940 is dominant in perturbing lipid 1H chemical shifts. The validity of the approximation was confirmed by the congruence with results from the cross-relaxation rates.
Ring-current-induced chemical-shift changes
The ring-current-induced chemical-shift changes, Δδ, depend solely on the mutual orientation of the ring and protons of lipids in proximity. The ring current shifts the resonances downfield upon interaction with the ring edge, and upfield upon interaction with the ring center (43). The 1H spectra of POPC-d31 bilayers in the absence and presence of 30 mol % CP-55,940 are presented in Fig. 3, a and b, respectively. Assignment of the lipid signals is shown at the chemical structure. For comparison, a high-resolution 1H NMR spectrum of CP-55,940 in methanol-d4 solution is shown in Fig. 3 c. The hydroxyphenyl ring of CP-55,940 gave signals at 6.67 and 6.86 ppm (see Fig. 3 b). Lipid signals do not have overlap with CP-55,940 signals, except for the alkyl chain resonances at 0.8–2.0 ppm.
Figure 3.
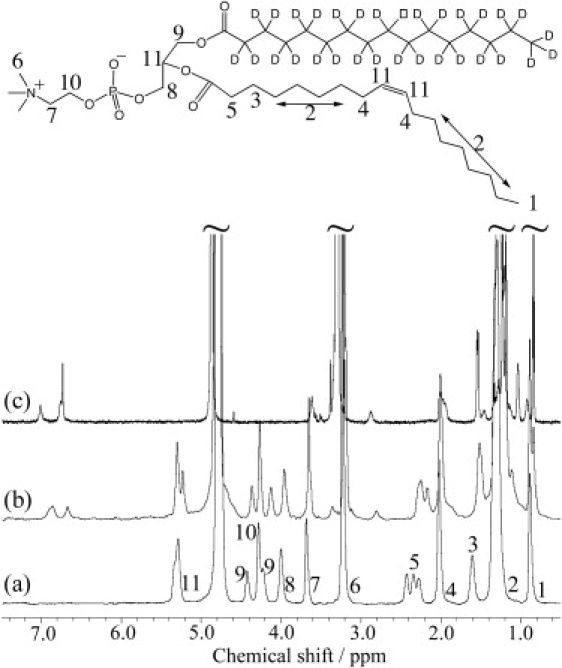
Solid-state 1H MAS NMR spectra of (a) POPC-d31 MLVs and (b) POPC-d31 MLVs with 30 mol % CP-55,940. (c) High-resolution solution-state 1H NMR spectrum of 1 mM CP-55,940 in methanol-d4.
The proton-site dependence of Δδ at concentrations of 15 and 30 mol % CP-55,940 is illustrated as a bar graph in Fig. 4. Upon binding of CP-55,940 to the bilayers, an upfield shift is observed for all POPC resonances near the lipid-water interface. The induced shifts Δδ are largest for protons of the upper acyl-chain C2, C3, and of the glycerol carbon, g1. Hence it is concluded that the preferred location of the hydroxyphenyl ring is centered near the lipid ester groups between the glycerol and upper acyl chain regions. The location is not dependent on the ligand concentration. The upfield direction of the induced shifts indicates that the shielding ring-current effect is dominant; that is, the ring plane orients parallel to the bilayer normal. It is known that the probability of locating water molecules in a phosphatidylcholine membrane decays to nearly zero at C2, whereas there is a significant probability at the ester carbonyl groups (44). Therefore, the amphipathic hydroxyphenyl ring of CP-55,940 is located in a region where the degree of hydration is notable.
Figure 4.
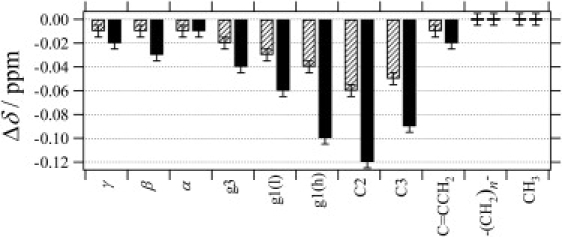
1H chemical-shift changes (Δδ) of POPC-d31 induced upon incorporation of 15 mol % (slashed bars) and 30 mol % (solid bars) of CP-55,940.
1H NMR cross-relaxation rates
1H NMR cross-relaxation rates depend primarily on the distance between the interacting protons, but also on the correlation time of motions of the vector that links those protons (45). A typical 1H MAS NOESY spectrum observed for POPC-d31 bilayers at the CP-55,940 concentration of 30 mol % is presented in Fig. S3. The spectral region that shows the diagonal and cross peaks for the hydroxyphenyl ring of the ligand is magnified on the left. The signal threshold was adjusted to a lower contour level for clarity. The diagonal and cross peaks were integrated, and normalized cross-relaxation rates between the protons of the hydroxyphenyl ring and the lipid were determined by matrix analysis (24). The mixing-time dependences of the cross- and diagonal-peak volumes used in the matrix equation are shown in Fig. S4, e.g., for protons of the hydroxyphenyl ring (l) at 6.86 ppm and the lipid C2 and g1(h), where the largest ring-current effect on the chemical shift was observed. Fig. 5 provides a summary of the normalized cross-relaxation rates (the lipid C3, -(CH2)n-, and ω-CH3 were excluded from analysis because of signal overlap with other protons of the ligand molecule). The use of CP-55,940-d19 (Fig. 1 b) allowed us to eliminate the overlap at ω-CH3 and confirm that cross-relaxation at this site is negligible. The hydroxyphenyl ring resonance at higher field (6.67 ppm; open bars in Fig. 5) has the highest cross-relaxation rate toward the C2 resonance, and lower rates toward the end of the headgroup. For the other hydroxyphenyl ring resonance at lower field (6.86 ppm, solid bars in Fig. 5), the cross-relaxation rate is highest at the g1 resonance. The preferred location of the ring is thus centered at the lipid carbonyl groups between the glycerol and the upper acyl chain regions. This conclusion is in excellent agreement with the result from the ring-current effect.
Figure 5.
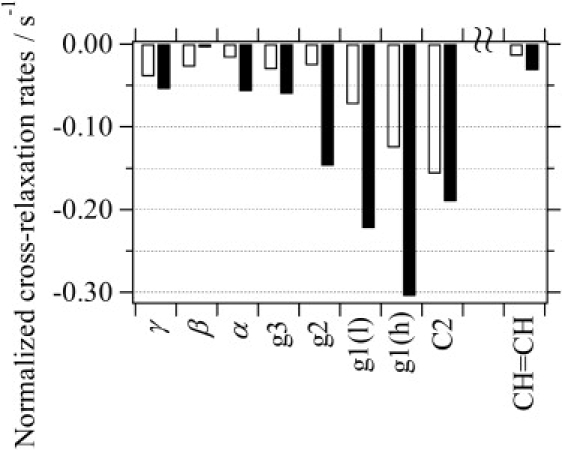
Normalized 1H NMR cross-relaxation rates (per proton) between the hydroxyphenyl ring of CP-55,940 and POPC. Open bars are for the ring signal (h) at 6.67 ppm, and solid bars are for the ring signal (l) at 6.86 ppm. Values for C3, -(CH2)n-, and ω-CH3 were not obtained, due to superposition with ligand protons. The use of CP-55,940-d19 allowed the overlap for ω-CH3 to be circumvented, and cross relaxation at this site was confirmed to be negligible.
The magnitude of the 1H cross-relaxation rates between the CP-55,940 hydroxyphenyl ring and the lipid is comparable to the magnitude of cross-relaxation rates between lipids. We reported previously that the magnitude of intermolecular cross-relaxation rates of lipids is likely to be primarily determined by the lateral diffusion rates (46). The comparable magnitude of cross-relaxation rates for CP,55-940-lipid and lipid-lipid interactions, as well as the similar lateral diffusion rates of CP-55,940 and lipids as shown in the later section (Lateral diffusion rate), suggests that the lifetime of associations between CP-55,940 and lipid is on the order of a fraction of a microsecond (47).
Orientation of the hydroxyphenyl ring
The orientation of the hydroxyphenyl ring of CP-55,940 in POPC bilayers was determined by measuring the 1H-13C dipolar interactions at three different sites of the ring (sites 1–3; Fig. 6) with the DIPSHIFT experiment. The 13C MAS NMR spectra of the hydroxlphenyl ring recorded (a) without the MREV-8 pulse train, and (b) with four MREV-8 cycles are shown in Fig. 6. The peak intensities differ somewhat between the aromatic ring carbons because of differences in spin-spin relaxation rates. Attenuation of the signals observed by MREV-8 is a result of dephasing of the 13C magnetization due to 1H-13C dipolar interactions. The signal intensities were plotted as a function of the number of MREV-8 cycles, and corresponding dipolar curves are given in Fig. 7. The strength of 1H-13C dipolar interactions is ∼3.5 kHz for all three protonated carbon atoms of the hydroxyphenyl ring. The experimental uncertainty is on the order of ±0.5 kHz.
Figure 6.
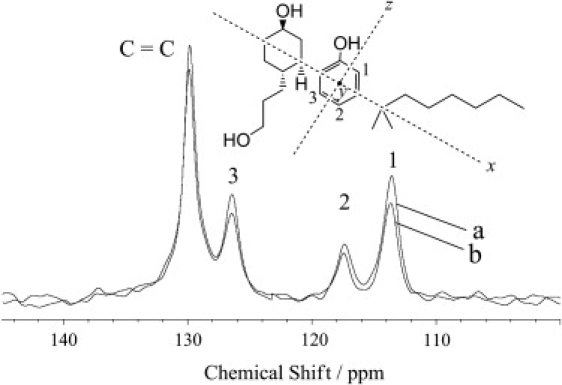
Natural-abundance 13C MAS NMR spectra of the hydroxyphenyl ring of CP-55,940 measured in the DIPSHIFT experiment at the MAS frequency of 3,994 Hz: (a) without MREV-8 cycles (t1 = 0, cross-polarization only), and (b) with four MREV-8 cycles (t1 = 148.6 μs). The chemical structure above the spectra shows the orientations of the hydroxyphenyl ring with respect to the bilayer normal (x, y, or z axis) that could possibly yield identical 1H-13C dipolar interactions for sites 1–3 (see text).
Figure 7.
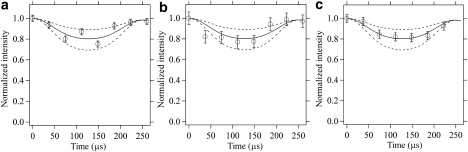
1H-13C dipolar dephasing curves (solid lines) for the carbon atoms at sites (a) 1, (b) 2, and (c) 3 of the hydroxyphenyl ring of CP-55,940 obtained with the DIPSHIFT experiment; see Fig. 6 for the carbon assignments. Intensities of the 13C NMR signals were plotted as a function of the evolution period t1 of the 13C magnetization under 1H-13C dipolar interaction; the length of t1 corresponds to the number of MREV-8 cycles (from 0 to 7) applied within one rotor period τr = 250 μs. Signal intensities were normalized to the intensity at t1 = 0. The broken lines above and below the solid line show the effect of a ±1 kHz change of the dipolar interaction from the best fit (see text). The error bars represent the spectral noise level.
The rigid-limit coupling (order parameter S = 1) of a 1H-13C bond in the ring with a length of 1.1 Å is 22.7 kHz (48). Under application of the semi-windowless MREV-8 sequence, this dipolar interaction is attenuated by a factor of 0.536 (48). The attenuation factor under our experimental conditions was 0.567, as determined on a sample of 13C-labeled cholesterol, yielding an effective rigid-limit coupling of 12.9 kHz. Therefore, an order parameter S = 0.27 ± 0.04 is obtained for the observed dipolar coupling of 3.5 ± 0.5 kHz.
Ring orientations that could possibly yield nearly identical order parameters at the three sites are shown in Fig. 6 as corresponding axes of the bilayer normal: 1), orientation z (bilayer normal perpendicular to the bond between the hydroxyphenyl- and hydroxycyclohexyl rings); 2), orientation x (bilayer normal parallel to the bond between the hydroxyphenyl and hydroxycyclohexyl rings); 3), orientation y (bilayer normal perpendicular to the plane of the hydroxylphenyl ring); and 4), orientations in between z and y (obtained by tilting the ring plane). Here rapid rotational diffusion of the ligand about the bilayer normal is assumed. Orientation y can be excluded immediately because it does not agree with the observation from the ring-current-induced chemical-shift perturbation; the ring plane orients parallel to the bilayer normal. Orientations close to y in between orientations z and y can also be excluded for the same reason. Order parameters for orientations z and x are easily calculated. Orientation z yields an angle θ between the 1H-13C bonds and the bilayer normal axis of 30°, corresponding to the order parameter S(θ) = (3 cos2θ − 1)/2 = 0.625. Orientation x yields θ = 60° and |S(θ)| = 0.125, which is much lower than the experimentally determined value of 0.27; note that the calculated value should be higher than the experimentally observed value because the calculation does not account for molecular wobble. Therefore, we can conclude that orientation z, or orientations close to z with some tilt of the ring plane agree with the observation. In orientation z, the hydroxyphenyl ring is oriented such that the bond to the hydroxycyclohexyl ring is perpendicular to the bilayer normal. With this orientation, it is likely that the hydroxyl group attached to the phenyl ring is directed toward the water phase. The wobble motion of the hydroxyphenyl ring reduces the order parameter from 0.625 to 0.27. This reduction of order by a factor of 0.43 is in accordance with a location of the hydroxyphenyl ring near the lipid-water interface, for which similar molecular order parameters have been reported.
A structural model of the peripheral cannabinoid receptor CB2 based on homology modeling to the crystal structure of the GPCR bovine rhodopsin was previously proposed (14). The putative solvent-accessible ligand-binding site is located on the extracellular side between helices III, V, VI, and VII. The cavity possesses a clear amphipathic character with a hydrophilic center framed by polar residues near the edges of the transmembrane helices and a hydrophobic cleft surrounded by aromatic residues. The location and orientation of CP-55,940 at the water-membrane interface determined in this work suggest that ligand access to the receptor-binding pocket from the lipid matrix is possible. The entry to the binding pocket may take place between helices VI and VII, considering the absence of a network of hydrogen bonds between these helices in the CB2 model (14) and the prominent movement of helix VI toward V upon receptor activation, as previously found for bovine rhodopsin (49).
Conformation of the nonyl tail
CP-55,940 has a nonyl chain linked to the hydroxyphenyl ring. The hydrophobic chain is expected to contribute to membrane binding by anchoring itself in the membrane core. Here we examine the structural features of this anchoring through order parameter analysis. The order parameters of the nonyl chain were determined by measuring the 2H NMR quadrupolar splittings, ΔνQ, using CP-55,940 specifically deuterated at the chain.
The 2H NMR powder spectrum of 30 mol % CP-55,940-d19 (Fig. 1 b) in POPC bilayers recorded at 25°C is shown in Fig. S5 a. To assign signals of the branched methyl groups in the nonyl chain, CP-55,940 deuterated only at those methyls was also synthesized (CP-55,940-d6; Fig. 1 c). The spectrum for CP-55,940-d6 is shown in Fig. S5 b. CP-55,940-d6 gives a distinct quadrupolar splitting of ΔνQ = 5.6 kHz, corresponding to the order parameter S = 0.045 (the weak signals near the center of the spectrum are from residual deuterated water and minor sample inhomogeneity). A distinct splitting of a similar value, ΔνQ = 5.0 kHz, is observed in the spectrum of CP-55,940-d19. All of the quadrupolar splittings in the CP-55,940-d19 spectrum are ∼8 kHz or less. Therefore, the order parameters of the nonyl chain are S = 0.064 or smaller. For comparison, the quadrupolar splittings of the upper half of the POPC palmitoyl-chain segments where the nonyl chain locates are in the range of ∼20–25 kHz, corresponding to an order parameter S = ∼0.16–0.20. It is concluded that the nonyl chain has a high level of conformational flexibility.
If the nonyl chain points toward the bilayer center with the orientation of the hydroxylphenyl ring determined as above, then the order parameter of the branched methyl groups is predicted to be Smol = 0.056. It should be noted that, with this ring orientation, free rotation about the C-C bond linking the nonyl chain to the hydroxyphenyl ring also yields an order parameter Smol = 0.056 for the branched methyl groups. The value Smol = 0.056 is higher than the measured value Smeas = 0.045, indicating that order is further reduced by nonyl chain dynamics. It is likely that a rotation about the C-C bond linking the nonyl chain to the hydroxyphenyl ring significantly contributes to the overall low order of the chain. The high conformational flexibility of the hydrophobic tail of CP-55,940 in membranes could be critical for reaching the binding site of the cannabinoid receptor.
Lateral diffusion rate
To investigate the possibility that the ligand can efficiently approach its receptor from the bilayer by lateral diffusion, we measured the diffusion coefficients of CP-55,940 and POPC by 1H MAS NMR with application of PFGs. The experiments were conducted at 22°C on 15 mol % CP-55,940 in POPC bilayers. The diffusion coefficient of CP-55,940 was 0.74(±0.05) × 10−11 m2/s (Fig. S6 a), compared with 0.56(±0.05) × 10−11 m2/s for POPC (Fig. S6 b). The diffusion rate of POPC in the presence of CP-55,940 agrees within limits of experimental error with the rate observed in the absence of the ligand (0.64(±0.05) × 10−11 m2/s). At the recorded rate, the CP-55,940 molecules travel 5 μm/s on average in the bilayers, which suggests that a diffusion-controlled approach could be an efficient mechanism.
Conclusions
Our experiments were driven by the hypothesis that a widely used synthetic cannabinoid ligand, CP-55,940, may approach the binding site on the receptor from the lipid matrix because of the marked lipophilic nature of the ligand. The location, structure, and dynamics of CP-55,940 in a membrane were studied by solid-state NMR and the results are summarized in Fig. 8.
Figure 8.

Schematic illustration of the structural features of CP-55,940 in PC bilayers determined by solid-state NMR. The location of the hydroxyphenyl ring with respect to the bilayer normal axis is highlighted with a gray band. The fast lateral diffusion of the ligand takes place along this band. The orientation of the hydroxylphenyl ring is shown by an arrow with the degree of director fluctuation. The nonyl tail has high conformational flexibility.
CP-55,940 incorporates homogeneously into the lipid matrix at concentrations as high as 30 mol %. The aromatic ring-current-induced shifts of lipid resonances and the cross-relaxation rates locate the hydroxyphenyl ring of the ligand in a region near the lipid ester groups between glycerol and upper segments of the hydrocarbon chains shown as a gray band in Fig. 8. Within this band the ligand performs rapid lateral movements with rates similar to or slightly higher than those of the matrix lipid. A structural model of the peripheral cannabinoid receptor CB2 based on homology modeling to the crystal structure of the GPCR bovine rhodopsin was previously proposed, and the putative binding site of the ligand was shown to be located on the extracellular side among helices III, V, VI, and VII (14). The location of CP-55,940 at the water-membrane interface determined in this work is favorable for the ligand access to the receptor-binding pocket from the lipid matrix. The 2H NMR measurements on POPC-d31 showed that incorporation of CP-55,940 into bilayers has a tendency to reduce the order of lipid acyl chains. The reduced lipid order in the vicinity of the ligand may also relate to the efficient configurational search for the ligand entry into the receptor, and to induction of the activated receptor conformation.
Both the location and the orientation of the ligand with respect to the bilayer normal determine the efficiency of the approach of the binding site on the receptor. The order parameters of the hydroxyphenyl ring indicate that the ligand has a distinct orientation in the bilayer such that the bonds linking the hydroxyphenyl ring to the hydroxycyclohexyl ring and to the nonyl chain are oriented perpendicular to the bilayer normal. In this orientation, the hydrophobic nonyl chain tends to be located near the membrane interface. The observed low order parameters of the nonyl chain indicate that the chain has high conformational flexibility. This is likely to be advantageous for enabling the ligand to gain access to the binding pocket among the transmembrane helices, and to adjust to the geometry of the binding site.
Supporting Material
Six figures and a table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00776-0.
Supporting Material
Acknowledgments
We thank Walter Teague for assistance with the spectrometer hardware, and Olivier Soubias for aiding implementation of the DIPSHIFT experiment.
This work was supported by the Intramural Research Program of the National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health.
References
- 1.Matsuda L.A., Lolait S.J., Brownstein M.J., Young A.C., Bonner T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Yeliseev A.A., Wong K.K., Soubias O., Gawrisch K. Expression of human peripheral cannabinoid receptor for structural studies. Protein Sci. 2005;14:2638–2653. doi: 10.1110/ps.051550305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugiura T., Kondo S., Sukagawa A., Nakane S., Shinoda A. 2-Arachidonoylgylcerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 5.Mechoulam R., Ben-Shabat S., Hanus L., Ligumsky M., Kaminski N.E. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 6.Devane W.A., Hanus L., Breuer A., Pertwee R.G., Stevenson L.A. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 7.Thomas B.F., Compton D.R., Martin B.R. Characterization of the lipophilicity of natural and synthetic analogs of δ-9-tetrahydrocannabinol and its relationship to pharmacological potency. J. Pharmacol. Exp. Ther. 1990;255:624–630. [PubMed] [Google Scholar]
- 8.Makriyannis A., Tian X., Guo J. How lipophilic cannabinergic ligands reach their receptor sites. Prostaglandins Other Lipid Mediat. 2005;77:210–218. doi: 10.1016/j.prostaglandins.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Mason R.P., Rhodes D.G., Herbette L.G. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J. Med. Chem. 1991;34:869–877. doi: 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- 10.Sargent D.F., Schwyzer R. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc. Natl. Acad. Sci. USA. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura T. Human opioid peptide Met-enkephalin binds to anionic phosphatidylserine in high preference to zwitterionic phosphatidylcholine: natural-abundance 13C NMR study on the binding state in large unilamellar vesicles. Biochemistry. 2006;45:15601–15609. doi: 10.1021/bi061641v. [DOI] [PubMed] [Google Scholar]
- 12.Kimura T., Okamura E., Matubayasi N., Asami K., Nakahara M. NMR study on the binding of neuropeptide achatin-I to phospholipid bilayer: the equilibrium, location, and peptide conformation. Biophys. J. 2004;87:375–385. doi: 10.1529/biophysj.103.038950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura T., Ninomiya K., Futaki S. NMR investigation of the electrostatic effect in binding of a neuropeptide, achatin-I, to phosphatidylcholine bilayers. J. Phys. Chem. B. 2007;111:3831–3838. doi: 10.1021/jp067100x. [DOI] [PubMed] [Google Scholar]
- 14.Xie X.-Q., Chen J.-Z., Billings E.M. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 15.Cherezov V., Rosenbaum D.M., Hanson M.A., Rasmussen S.G.F., Thian F.S. High-resolution crystal structure of an engineered human β(2)-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaakola V.P., Griffith M.T., Hanson M.A., Cherezov V., Chien E.Y.T. The 2.6 Angstrom crystal structure of a human A(2A) adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie X.-Q., Melvin L.S., Makriyannis A. The conformational properties of the highly selective cannabinoid receptor ligand CP-55,940. J. Biol. Chem. 1996;271:10640–10647. doi: 10.1074/jbc.271.18.10640. [DOI] [PubMed] [Google Scholar]
- 18.Bassolino-Klimas D., Alper H.E., Stouch T.R. Mechanism of solute diffusion through lipid bilayer-membranes by molecular-dynamics simulation. J. Am. Chem. Soc. 1995;117:4118–4129. [Google Scholar]
- 19.Kachel K., Asuncion-Punzalan E., London E. Anchoring of tryptophan and tyrosine analogs at the hydrocarbon-polar boundary in model membrane vesicles: parallax analysis of fluorescence quenching induced by nitroxide-labeled phospholipids. Biochemistry. 1995;34:15475–15479. doi: 10.1021/bi00047a012. [DOI] [PubMed] [Google Scholar]
- 20.Gaede H.C., Yau W.-M., Gawrisch K. Electrostatic contributions to indole-lipid interactions. J. Phys. Chem. B. 2005;109:13014–13023. doi: 10.1021/jp0511000. [DOI] [PubMed] [Google Scholar]
- 21.Yau W.-M., Wimley W.C., Gawrisch K., White S.H. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 22.Hoff B., Strandberg E., Ulrich A.S., Tieleman D.P., Posten C. 2H-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys. J. 2005;88:1818–1827. doi: 10.1529/biophysj.104.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wimley W.C., White S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 24.Huster D., Arnold K., Gawrisch K. Investigation of lipid organization in biological membranes by two-dimensional nuclear Overhauser enhancement spectroscopy. J. Phys. Chem. B. 1999;103:243–251. [Google Scholar]
- 25.Makriyannis A., Banijamali A., Jarrell H.C., Yang D.-P. The orientation of (-)-δ-9-tetrahydrocannabinol in DPPC bilayers as determined from solid-state 2H-NMR. Biochim. Biophys. Acta. 1989;986:141–145. doi: 10.1016/0005-2736(89)90283-6. [DOI] [PubMed] [Google Scholar]
- 26.Yang D.-P., Banijamali A., Charalambous A., Marciniak G., Makriyannis A. Solid state 2H-NMR as a method for determining the orientation of cannabinoid analogs in membranes. Pharmacol. Biochem. Behav. 1991;40:553–557. doi: 10.1016/0091-3057(91)90362-6. [DOI] [PubMed] [Google Scholar]
- 27.Mavromoustakos T., Yang D.-P., Charalambous A., Herbette L.G., Makriyannis A. Study of the topography of cannabinoids in model membranes using X-ray diffraction. Biochim. Biophys. Acta. 1990;1024:336–344. doi: 10.1016/0005-2736(90)90363-s. [DOI] [PubMed] [Google Scholar]
- 28.Yang D.-P., Mavromoustakos T., Beshah K., Makriyannis A. Amphipathic interactions of cannabinoids with membranes. A comparison between δ-8-THC and its O-methyl analog using differential scanning calorimetry, X-ray diffraction and solid-state 2H-NMR. Biochim. Biophys. Acta. 1992;1103:25–36. doi: 10.1016/0005-2736(92)90053-o. [DOI] [PubMed] [Google Scholar]
- 29.Mavromoustakos T., Yang D.P., Makriyannis A. Small-angle X-ray-diffraction and differential scanning calorimetric studies on O-methyl-(-)-δ (8)-tetrahydrocannabinol and its 5′-iodinated derivative in membrane bilayers. Biochim. Biophys. Acta. 1995;1237:183–188. doi: 10.1016/0005-2736(95)00101-8. [DOI] [PubMed] [Google Scholar]
- 30.Kolbert A.C., de Groot H.J.M., Levitt M.H., Munovitz M.G., Roberts J.E. Two-dimensional dipolar-chemical shift NMR in rotating solids. In: Granger P., Harris R.K., editors. Multinuclear Magnetic Resonance in Liquids and Solids—Chemical Applications. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1990. pp. 339–354. [Google Scholar]
- 31.Itagaki N., Sugahara T., Iwabuchi Y. Expedient synthesis of potent cannabinoid receptor agonist (-)-CP55,940. Org. Lett. 2005;7:4181–4183. doi: 10.1021/ol051570c. [DOI] [PubMed] [Google Scholar]
- 32.Boggs J.M., Tummler B. Interdigitated gel phase bilayers formed by unsaturated synthetic and bacterial glycerolipids in the presence of polymyxin-B and glycerol. Biochim. Biophys. Acta. 1993;1145:42–50. doi: 10.1016/0005-2736(93)90379-e. [DOI] [PubMed] [Google Scholar]
- 33.Caffrey M., Hogan J. LIPIDAT: a database of lipid phase-transition temperatures and enthalpy changes. DMPC data subset analysis. Chem. Phys. Lipids. 1992;61:1–109. doi: 10.1016/0009-3084(92)90002-7. [DOI] [PubMed] [Google Scholar]
- 34.Davis J.H., Jeffrey K.R., Bloom M., Valic M.I., Higgs T.P. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 1976;42:390–394. [Google Scholar]
- 35.Sternin E., Bloom M., Mackay A.L. De-Pake-ing of NMR spectra. J. Magn. Reson. 1983;55:274–282. [Google Scholar]
- 36.McCabe M.A., Wassall S.R. Fast-Fourier-transform dePaking. J. Magn. Reson. B. 1995;106:80–82. [Google Scholar]
- 37.Lafleur M., Fine B., Sternin E., Cullis P.R., Bloom M. Smoothed orientational order profile of lipid bilayers by 2H-nuclear magnetic resonance. Biophys. J. 1989;56:1037–1041. doi: 10.1016/S0006-3495(89)82749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamilton J.A., Small D.M. Solubilization and localization of triolein in phosphatidylcholine bilayers: a 13C NMR study. Proc. Natl. Acad. Sci. USA. 1981;78:6878–6882. doi: 10.1073/pnas.78.11.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotts R.M., Hoch M.J.R., Sun T., Markert J.T. Pulsed field gradient stimulated echo methods for improved NMR diffusion measurements in heterogeneous systems. J. Magn. Reson. 1989;83:252–266. [Google Scholar]
- 40.Gaede H.C., Gawrisch K. Lateral diffusion rates of lipid, water, and a hydrophobic drug in a multilamellar liposome. Biophys. J. 2003;85:1734–1740. doi: 10.1016/S0006-3495(03)74603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis J.H. Deuterium magnetic resonance study of the gel and liquid crystalline phases of dipalmitoyl phosphatidylcholine. Biophys. J. 1979;27:339–358. doi: 10.1016/S0006-3495(79)85222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiburu E.K., Bass C.E., Struppe J.O., Lorigan G.A., Avraham S. Structural divergence among cannabinoids influences membrane dynamics: a 2H solid-state NMR analysis. Biochim. Biophys. Acta. 2007;1768:2049–2059. doi: 10.1016/j.bbamem.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 43.Johnson C.E., Bovey F.A. Calculation of nuclear magnetic resonance spectra of aromatic hydrocarbons. J. Chem. Phys. 1958;29:1012–1014. [Google Scholar]
- 44.Wiener M.C., White S.H. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of X-ray and neutron diffraction data. III. Complete structure. Biophys. J. 1992;61:434–447. doi: 10.1016/S0006-3495(92)81849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gawrisch K. The dynamics of membrane lipids. In: Yeagle P.L., editor. The Structure of Biological Membranes. 2nd ed. CRC Press; Boca Raton, FL: 2004. pp. 147–171. [Google Scholar]
- 46.Yau W.-M., Gawrisch K. Lateral lipid diffusion dominates NOESY cross-relaxation in membranes. J. Am. Chem. Soc. 2000;122:3971–3972. [Google Scholar]
- 47.Feller S.E., Huster D., Gawrisch K. Interpretation of NOESY cross-relaxation rates from molecular dynamics simulation of a lipid bilayer. J. Am. Chem. Soc. 1999;121:8963–8964. [Google Scholar]
- 48.Hong M., Gross J.D., Griffin R.G. Site-resolved determination of peptide torsion angle phi from the relative orientations of backbone N-H and C-H bonds by solid-state NMR. J. Phys. Chem. B. 1997;101:5869–5874. [Google Scholar]
- 49.Knierim B., Hofmann K.P., Ernst O.P., Hubbell W.L. Sequence of late molecular events in the activation of rhodopsin. Proc. Natl. Acad. Sci. USA. 2007;104:20290–20295. doi: 10.1073/pnas.0710393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.