

Abstract
Cell types from many tissues respond to changes in substrate stiffness by actively remodeling their cytoskeletons to alter spread area or adhesion strength, and in some cases changing their own stiffness to match that of their substrate. These cell responses to substrate stiffness are linked to substrate-induced changes in the state, localization, and amount of numerous proteins, but detailed evidence for the requirement of specific proteins in these distinct forms of mechanical response are scarce. Here we use microfluidics techniques to produce gels with a gradient of stiffness to show the essential function of filamin A in cell responses to mechanical stimuli and dissociate cell spreading and stiffening by contrasting responses of a pair of human melanoma-derived cell lines that differ in expression of this actin cross-linking protein. M2 melanoma cells null for filamin A do not alter their adherent area in response to increased substrate stiffness when they link to the substrate only through collagen receptors, but change adherent area normally when bound through fibronectin receptors. In contrast, filamin A-replete A7 cells change adherent area on both substrates and respond more strongly to collagen I-coated gels than to fibronectin-coated gels. Strikingly, A7 cells alter their stiffness, as measured by atomic force microscopy, to match the elastic modulus of the substrate immediately adjacent to them on the gradient. M2 cells, in contrast, maintain a constant stiffness on all substrates that is as low as that of A7 cells on the softest gels examined (1000 Pa). Comparison of cell spreading and cell stiffening on the same gradient substrates shows that cell spreading is uncoupled from stiffening. At saturating collagen and fibronectin concentrations, adhesion of M2 cells is reduced compared to that of A7 cells to an extent approximately equal to the difference in adherent area. Filamin A appears to be essential for cell stiffening on collagen, but not for cell spreading on fibronectin. These results have implications for different models of cell protrusion and adhesion and identify a key role for filamin A in altering cellular stiffness that cannot be compensated for by other actin cross-linkers in vivo.
Introduction
The mechanical properties of a cell's microenvironment can have as great an impact on cell structure and function as soluble stimuli and cell-cell contacts (1). Many cell types alter their morphology when grown on substrates of different stiffness (2–8). Cells grown on stiff substrates assemble actin stress fibers (5), exhibit a more spread phenotype (9), activate signaling pathways characteristic of contractility (2,10), and upregulate expression of cytoskeletal proteins (11,12) and integrins (13). Not all cell types appear to be sensitive to substrate stiffness, and not all mechanosensitive cell types respond similarly to changes in stiffness. However most cell types studied thus far spread more and adhere better to harder matrices. Some cell types cannot grow on very soft (<50 Pa) surfaces (3,5,6,8,9,14), and other cell types such as mesenchymal stem cells survive in a quiescent state when the elastic modulus of their substrates is sufficiently low (15).
Neither the physical nor molecular mechanisms by which cells sense the stiffness of their environments or by which they respond to externally imposed forces are thoroughly understood. A current hypothesis to explain increased spreading on stiffer adhesive surfaces is that by pulling on the matrix at focal adhesions, anchorage points to the underlying substrate, the cell creates tension within its membrane and underlying cortical actin mesh (16); the tension depends on the inherent material properties of the matrix, a relatively stiff matrix will resist cellular force more than a soft one. In cell types that grow preferentially on hard matrices, the tension will stimulate such a cell to extend its periphery increasing its spread area (17). A correlation between cell spreading and stiffening has been reported in fibroblasts (12,18) and can be accounted for by a theoretical model (19), but whether cells necessarily stiffen as they spread is not clear.
From a physical perspective, cells might sense stiffness by mechanisms analogous to the two design principles of rheometers. In one mechanism, cells could apply a measured stress (e.g., using acto-myosin motors) and detect the amount of deformation (strain) imposed on their surroundings. In the other, they could impose a constant strain (e.g., by moving a fixed lever arm through a given angle) and detect the amount of work required. Both mechanisms require an elastic material within the cell that deforms to an extent dependent on the opposing stiffness of the extracellular matrix or on an external force applied to the cell. Several candidate molecules and signaling pathways have been proposed to constitute the hypothetical force generators and deformation sensors. These include protein phosphatases (20); small GTPases; Ca2+ channels; and several cytoskeletal proteins including talin, filamin, and dystrophin (summarized in a recent review (21)).
Filamin A is the most efficient actin cross-linking protein in vitro (22–26), producing elastic networks that cannot be formed by other cross-linkers such as alpha actinin (25,27) or temporary branching proteins such as the Arp2/3 complex (28). Previous work has shown that loss of filamin A in a cell line derived from a primary human melanoma decreased the stiffness of these cells by a factor of ∼2 (29) and that loss of Dictyostelium filamin decreases the elastic modulus of these cells (30). Loss of filamin in glomerular podocytes derived from a mouse model of HIV-associated nephropathy (HIVAN) is associated with a decrease in the elastic modulus of these cells by a factor of 4 compared to wild-type podocytes (31). In contrast, studies using magnetic twisting rheometry (32) report insignificant mechanical differences in cells devoid of filamin A compared to their wild-type controls, raising issues as to whether filamin A or any other actin cross-linker can determine the stiffness of the cytoskeleton or overall cell stiffness (33).
In addition to its actin filament cross-linking function, filamin A binds numerous other proteins including some integrins to mediate the link between cytoskeleton and cell membrane (34). Filamin is specifically recruited to sites where stress is applied to integrins by magnetic beads coated with collagen, and melanoma cells lacking filamin cannot mount this mechanoprotective response (35). Several protein complexes involved in signaling cytoskeletal reorganization in response to force bind to filamin A including ras/Erk (36), Rho (37), and ion channels (38–42). These data indicate that filamin A is a good candidate for mediating cellular responses to substrate stiffness, especially responses involving beta 1 integrins that bind filamin A at their cytoplasmic tails (43,44). In this report, we systematically test the role of filamin A in the response of cells to substrates of different stiffnesses and in the ability of cells to alter their own stiffness to match that of their substrate.
Experimental methods
Fabrication of microfluidic channels
Gradient generators made from polydimethyl siloxane (PDMS) microfluidic channels were fabricated using standard photolithography techniques (45,46). Photomasks of the gradient generator (Fig. 1) were designed in AutoCAD and printed (Advance Reproductions, North Andover, MA). SU-8 2100 negative photoresist (Microchem, Newton, MA) was spin coated onto a silicon wafer, baked for 15 min at 65°C, and then baked again at 95°C for 60 min. The photoresist was exposed to 365 nm ultraviolet (UV) light through the photomask to cross-link the exposed parts of the photoresist. A postbake process was then performed at 95°C for 25 min before the coated wafer was immersed in SU-8 developer for 8 min to remove the uncross-linked parts and leave on the wafer a pattern of the designed microfluidic channels called the master. PDMS solution (Sylgard 184, Dow Corning, Midland, MI) was poured over the master and baked at 70°C for 2 h to produce a negative replica of the channels. PDMS was removed from the master and bound to glass coverslips. The gradient-mixer region was bonded to a plain coverslip using a mild oxygen plasma treatment (45 W for 40 s), and the outlet region was bonded using a high-power (75 W) plasma treatment for 50 s to a coverslip activated with (3-aminopropyl)trimethoxysilane (Sigma (St. Louis, MO) 281778) and glutaraldehyde (Sigma G7651) (9,46). Activating the coverslip promotes adhesion of polyacrylamide gels, and the reversible bonding enables separation of the PDMS from the activated coverslip.
Figure 1.
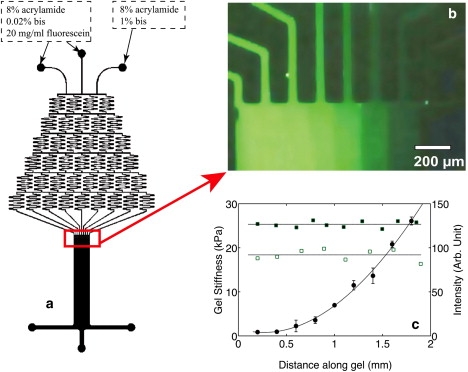
(a) Gradient generator, which has three inlets supplying the relative amounts of bisacrylamide or fluorescent dye shown and one wide outlet. (b)The chemical gradient produced at the outflow from the mixing apparatus of the microfluidic system visualized by doping the bisacrylamide solution with a fluorescent dye. Higher fluorescence intensities represent lower bisacrylamide concentration, and hence lower gel stiffness. (c) Local elastic modulus (solid circles) measured by AFM across a 2 mm wide gradient gel. Fluorescence intensity profile across a gradient gel coated with rhodamine-labeled fibronectin (solid squares) and a gradient gel coated with nonfluorescent fibronectin (open squares) to correct for gel autofluorescence.
Polyacrylamide gel with stiffness gradients
The strategy for making stiffness gradients was developed from the design of Zaari et al (46). The stiffness of polyacrylamide gels was tuned by varying the concentration of bisacrylamide at a fixed acrylamide concentration (13). Three solutions with the same acrylamide (Bio-Rad, Hercules, CA) concentration but different N,N′-methylene-bisacrylamide (Bio-Rad) concentrations were injected into the gradient generator. Each solution had an acrylamide concentration of 8% and a 2,2-diethoxyacetophenone (Sigma 227102) photoinitiator concentration of 0.5%. The bisacrylamide concentrations of the three inlets were: 0.02%, 0.02%, and 1%. Fluorescein (Sigma F6377) was added to the 0.02% bis-acrylamide solution to evaluate the gradient of bis-acrylamide concentration upon polymerization (Fig. 1 b). The solutions were then driven through the microfluidic channels by syringe pumps (Harvard Apparatus, Holliston, MA) at the same flow rate of 3 μL. Once the flow in the outlet channel reached a steady state, a UV light was shined on the outlet region for 8 min. The syringe pumps were stopped after the outlet region was exposed in the UV light for 10 s. Peeling off the PDMS leaves the gel stuck on the activated coverslip. The resulting gel, 1.8 mm in width and 2 cm in length, was immediately immersed in PBS buffer for 12 h to remove the unreacted photoinitiators.
Once the gradient PA gels were fabricated, stiffness across the gel was characterized using atomic force microscopy (AFM) (Fig. 1 c). The gradual transition in bis-acrylamide concentration, which correlates with gel stiffness, was also evaluated by fluorescence (Fig. 1 b). The gel surface was then activated with sulfo-SANPAH, and adhesion proteins were covalently ligated through free amino groups by succinimide chemistry (47). Previous studies using fluorescently labeled fibronectin and quantifying adhesive ligand density by fluorescence microscopy (48) show that incubation of 0.1 mg/ml protein solutions for 30 min on the gel is sufficient to produce a saturating level of adhesion protein on gels independent of their stiffness. Gradient gels were coated with 0.1 mg/ml salmon fibronectin (Sea Run Holdings, Freeport, ME) 0.1 mg/ml rat-tail collagen (BD Bioscience, San Diego, CA) or a combination of both proteins, each at 0.1 mg/ml.
Ligand density across the coated gradient gel was evaluated using fluorescence microscopy. Fluorescence images of a gradient gel coated by 0.1 mg/ml rhodamine labeled fibronectin were acquired using a Coolsnap Hq charge-coupled device camera (Roper Scientific, Trenton, NJ) mounted on an Axio Observer Z1 microscope (Carl Zeiss, Jena, Germany). A total of seven images across the gel were taken and stitched together to reconstruct an image of the whole gel. Each image was acquired at a different location of the gel using the same acquisition parameters so that the overlap regions of the images have the same intensity level. A fluorescence intensity profile across the gel was evaluated by plotting the mean intensity of a selected square region, 100 μm2 in area, against the distance of its centroid from the soft edge of the gel. A constant fluorescence intensity level across the gel (Fig. 1 c) indicates uniform distribution of ligand density.
Cell culture
The M2 cell line was originally identified in a study of cells cultured from primary and metastatic human melanomas as a cell type with impaired motility (49). The abnormal motility and morphology of M2 cells was later shown to be associated with their lack of expression of ABP280 (29), the actin cross-linker now called filamin A (50). Whereas motile melanoma cell lines cultured from human tumors had filamin/actin ratios ranging from 1:80 to 1:140, M2 cells had strongly reduced mRNA for filamin A and no protein detectable by an anti-filamin A antibody. A7 cells, derived from M2 cells by stable reexpression of filamin A, have actin levels comparable to motile melanoma cells and a filamin/actin ratio of 1:160, close to the range found in untransfected filamin A+ melanoma cells (29).
M2 (filamin A deficient) and A7 (filamin A expressing) melanoma cells were cultured in DMEM (BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) at 37°C with 5% CO2. Acrylamide gels, either single stiffness or stiffness gradients, were prepared for cell culture by coating with 0.5 mg/ml sulfo-SANPAH (Pierce Biotechnology, Rockford, IL) and activated using UV for 10 min. Cells were then added to gels coated with adhesion proteins as described above and allowed to attach and spread for 24 h before experiments were performed. Immediately before experimentation, cells were placed in CO2 independent buffer (PBS +Ca, +Mg) to prevent changes in pH of the cellular environment.
AFM and fluorescent microscopy
AFM was done with a DAFM-2X Bioscope (Veeco, Woodbury, NY) mounted on an Axiovert 100 microscope (Zeiss, Thornwood, NY) using silicon nitride cantilevers (196 μm long, 23 μm wide, 0.6 μm thick) with a bead tip (1 μm diameter) for indentation. The spring constant of the cantilever, calibrated by resonance measurements, was typically 0.06 N/m (Novascan, Ames, IA). A grid with 300 μm divisions was placed length-wise beneath the gel, and ∼3–4 cell/gel stiffness measurements are made within each division. Correlation between cell and gel stiffness was determined by indenting the cells at three distinct points and the gel at three points proximal to the attached cell.
To quantify stiffness (elastic modulus), the first 500 nm of tip deflection was fit with the Hertz model for a sphere
where fbead is the force on the bead, k is the spring constant of the cantilever, dcantilever is the deflection of the cantilever measured by the AFM, E is the Young's modulus, v is the Poisson ratio, R is the radius of the bead, and δ is the vertical indentation of the material. δ is determined by subtracting dcantilever from the distance traveled by the cantilever during the indentation process. Previous studies have shown that this simple approximation, although not exact for our conditions, is correct within a small fraction (<15%) of values determined by conventional rheometry of identical gels (51). Phase contrast images of cells, including those measured by AFM, were analyzed using Image J to determine adherent area.
For evaluation of actin organization of M2 and A7 cells on varying gel stiffness, samples were fixed with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) for 30 min at 37° and stained with 1:40 FITC-labeled phalloidin (Invitrogen, Carlsbad, CA) in PBS for 30 min.
Measurement of cell adhesion
Equal numbers of M2 and A7 cells (two 104 cells/well) were plated in triplicate wells of 96-well plates. Groups of wells contained serial dilutions of collagen or fibronectin ranging from 0–10 μg/ml. The cells were allowed to adhere to the wells for 30 min in a tissue culture incubator. After 30 min, the plates were washed, and the cells that remained were stained with crystal violet. The plates were dried and the stain solubilized with methanol-acetic acid, and the plates read in a microplate reader (52).
Results
The specific function of filamin A in the ability of cells to sense or respond to substrate stiffness is illustrated by comparison of the two human melanoma-derived cell lines M2 and A7 (29). M2 cells are derived from a primary human melanoma, and although transformed and robust in culture have highly impaired motility because they lack filamin A expression (Fig. 2 e). A7 cells are derived from M2 cells by stably expressing approximately wild-type levels of filamin A. A typical response of A7 cells to the changes in stiffness within a gradient gel coated with fibronectin and collagen is shown in Fig. 2 a. On the softest region of the gel, which has a Young's modulus of ∼1 kPa, measured by small amplitude indentation at a frequency of 1 HZ, A7 cells are round and lack actin bundles or stress fibers that are visible by fluorescence microscopy after staining with rhodamine-phalloidin. As stiffness increases to levels of ∼30 kPa on the right side of the gel shown, the A7 cells develop a spread, fan-shaped morphology and assemble actin bundles (Fig. 2, a and f). These morphological changes are very similar to those seen in fibroblasts and other cell types. M2 cells form few if any stress fibers on any gel at any level of stiffness under these conditions (Fig. 2, b and g).
Figure 2.
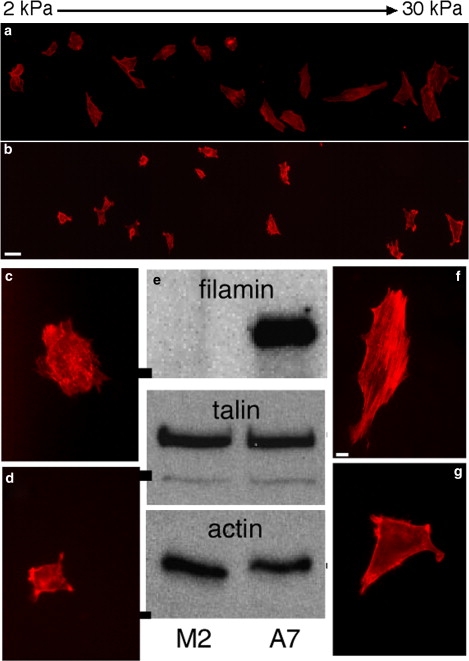
Morphological change of melanoma cells on stiffness gradient gels. Shapes of A7 (a) and M2 (b) cells bound to a PA gradient gel coated with collagen I and fibronectin, taken 24 h after plating. Gel stiffness increases from left to right. Scale bar is 40 μm. The full width of the gel is 1.8 mm, and its stiffness ranges from 1 to 30 kPa. Higher magnification images show examples of A7 (c and f) and M2 (d and g) cells on soft (c and d, 0.5 kPa) and stiff (f and g, 15 kPa) gels. Scale bar for these images is 10 μm Western blot (e) shows equal levels of talin and actin expression in M2 and A7 cells, but no filamin A expression in M2 cells. Molecular mass bars on left of image are 200 kDa for filamin and talin, and 40 kDa for actin.
Function of filamin in response to substrate stiffness
The potential of filamin A to mediate the integrin-specific stiffness sensing-response function of cells is demonstrated by the data in Fig. 3. When A7 cells are cultured on gels coated with collagen I, fibronectin, or a combination of both proteins, they increase both their own cortical stiffness (Fig. 3 a) and their adherent area (Fig. 3 c) but to very different extents, depending on the nature of the adhesive ligand. Cell stiffness is greater and increases more strongly with substrate stiffness when A7 cells are grown on collagen I, and like fibroblasts (51), these cell types modulate their cytoskeletal stiffness to match that of their underlying substrate. A7 cell stiffening on fibronectin is much more gradual, and when plated on both ligands, the cellular response is intermediate between those of either ligand alone. This result suggests that activation of fibronectin receptors, presumably β3 integrins, interferes with the response to collagen receptors, presumably β1 integrins.
Figure 3.
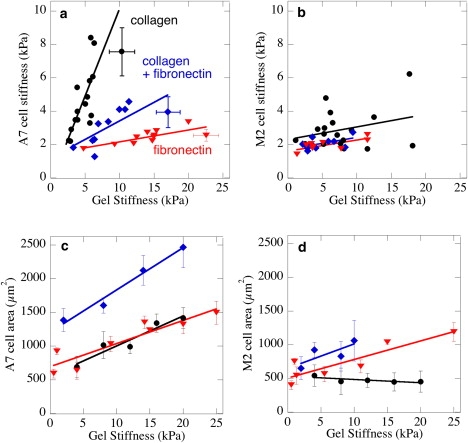
Cellular stiffness measured by AFM (a and b) and adherent area (c and d) of A7 (a, c) and M2 (b and d) melanoma cells cultured for 24 h on polyacrylamide gels laminated with collagen I (circles), fibronectin (triangles), or mixture of collagen I and fibronectin (diamonds). Both proteins were added at saturating concentrations to the gels using methods described in (47). Error bars representing standard errors (n = 3) for stiffness measurements of individual cells and the adjacent gel shown in panel a are representative for data in panels a and b. Error bars in panels c and d represent larger populations of cells (n = 30–50) within gels of the average stiffness shown on the abscissa.
Direct comparison of spread area and cell stiffness for the same population of cells shows that spreading and stiffening are not linked, and in the case of A7 cells show opposite responses to changes in adhesive ligand. Whereas A7 cells were stiffest on collagen, their spread area was the same on collgen-coated and fibronectin-coated gels (Fig. 3 c) and gels with both collagen and fibronectin lead to spread areas that were approximately the sum of the area on either ligand alone. The coating density of both collagen and fibronectin was sufficiently high to be saturating for both collagen and fibronectin, as determined by previous studies and confirmed by the data of Fig. 4.
Figure 4.
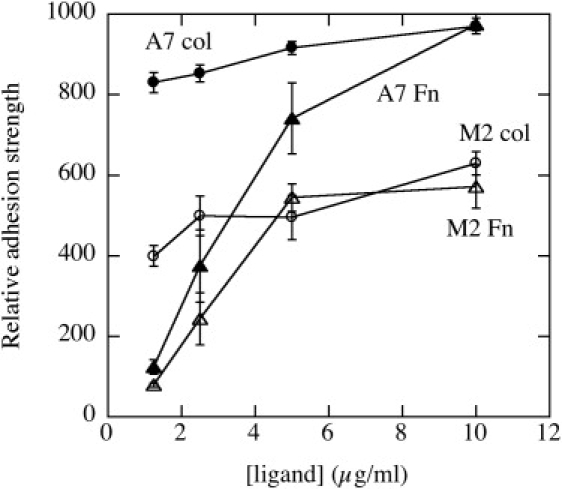
Adhesion strength of M2 and A7 cells to surfaces coated with different densities of fibronectin and collagen. Relative cell adhesion strength is calculated as the intensity of cell staining on surfaces after nonadherent cells are removed by washing with medium.
Filamin-A null M2 cells responded very differently to changes in substrate stiffness that again depended on the nature of the adhesive ligand. Fig. 3 b shows that M2 cells maintain a constant low value of cytoskeletal stiffness independent of the stiffness of their substrate. There appears to be a slight trend toward stiffening on collagen, but not on fibronectin. However, like A7 cells, M2 cells increased their spread area as the stiffness increased on gels that contained fibronectin, consistent with a previous report (48), but not on collagen alone. These findings are consistent with reports that filamin might be the primary cytoskeletal linker to integrin subtypes that promote tight adhesion and inhibit motility, whereas filamin A might be replaced by talin as the cytoskeletal linker when the cell binds fibronectin and initiates a motile phenotype (43). Similar to fibroblasts (51), A7 cells alter their stiffness to match that of their substrate when bound by collagen receptors, but have a much weaker stiffening response when bound to fibronectin-coated gels (Fig. 3 a). M2 cells maintain their low elastic modulus on all substrates (Fig. 3 b). The stiffness values reported here were determined by AFM indentation of gradient gels, with the AFM probing both the cell and the adjacent substrate.
Absence of filamin weakens but does not prevent adhesion to collagen- or fibronectin-coated surfaces
Changes in spread area and potentially cell stiffness might result from differences in the ability of cells to adhere to their substrate. To test for differences in adhesion strength independent of stiffness differences, M2 and A7 cells were plated on plastic surfaces coated with serial dilutions of collagen I or fibronectin. Adherent cells were subjected to uniform fluid shear stress and their abilities to maintain adhesion on surfaces coated with different densities of fibronectin or collagen I are shown in Fig. 4.
At the densities of ligand used for the studies shown in Fig. 3 (>10 μg/ml) M2 cells' adhesion strength is ∼60% that of A7 cells on both types of ligands. Previous studies show that total adhesion strength scales with the number of ligated integrins (53), and therefore at saturating adhesion protein densities, approximately with the adherent area. The M2 cells have a clear adhesion defect on collagen compared to the A7 cells. At the maximum fibronectin concentration, the adhesion of A7 is similar to that of A7 cells on collagen, and adhesion of the M2 cells is similar to that of the M2 cells on collagen. Even though M2 cells spread better on fibronectin than collagen (Fig. 3 d) to the point that their spreading behavior approaches that of A7 cells, their adhesive properties remain defective compared to A7 cells. The difference in adhesion strength between M2 and A7 cells is consistent with their differences in adherent area on the stiffest substrates shown in Fig. 3, suggesting that both cell types are able to engage their substrate effectively, but the lack of spreading leads to overall less adhesion. The lack of filamin A, presumably needed for coupling the membrane receptors to the cytoskeleton, leads to differences in stiffening and spreading as substrate stiffness is altered.
Discussion
The stiffness of tissues is tightly controlled, and the elastic modulus of most normal tissues generally ranges from ∼100 Pa for the softest tissues, such as fat and bone marrow, to tens of thousands of Pa for muscle, and an order of magnitude greater for cartilage and tissues such as bone that are composed more of extracellular matrix than of cells (reviewed in (54)). The stiffening of tissues resulting from fibrosis or other diseases is commonly thought to be merely an inevitable late-stage consequence of the pathologic process, but recent work supports the hypothesis that changes in tissue stiffness have a more active, causative role in disease, as they can exacerbate or even initiate cellular changes coincident with chemical signaling (2,55,56). The molecular determinants of cellular and extracellular matrix elasticity are extensively studied, but a universally accepted model to explain how changes in protein assembly and organization translate into macroscopic or microscopic elasticity has been elusive. Intracellular elasticity is generally thought to rely on the formation of cytoskeletal networks, especially the cortical actin meshwork, a dynamic system of polymerizing and depolymerizing filaments cross-linked to each other and to the plasma membrane by a host of actin binding proteins. The relative efficiency of different purified actin cross-linkers has been documented in vitro, and filamin A is consistently found to be the most efficient cross-linker, not only in terms of forming actin filament networks, but also of inducing stiffening of these networks as they deform (25,28). Whether or not filamin A is essential for stiffness sensing appears to depend on the context, with different studies reporting that filamin A expression is either essential (35,57) or unnecessary (48,58) for cellular response to changes in stiffness or response to external forces.
Cellular responses to matrix stiffness depend not only on the cell type and its expression of cytoskeletal protein but also on the types of extracellular matrix and therefore the type of adhesion receptors expressed, usually integrins, by which the cell binds its substrate. Previous studies have shown that fibroblasts, endothelial cells, and several other cell types all increase in spread area as a function of substrate stiffness, but to different extents, depending on whether they bind through collagen, fibronectin, or laminin receptors (1). Similarly, the data in Figs. 2 and 3 show that melanoma cells, whether or not they express filamin A, respond differently to fibronectin- and collagen-coated surfaces, but that spreading, stiffening, and adhesion (Fig. 4) on collagen-coated surfaces shows an absolute requirement for filamin A.
The results in Fig. 3 are consistent with and help explain the apparently discrepant results of two studies of mechano-sensing in filamin-deficient melanoma cells. Glogauer et al. (35) showed that filamin-expressing A7 cells increased actin assembly and activated myosin contractility at sites where magnetic collagen-coated beads applied force to the surface of the cell, whereas M2 cells, devoid of filamin, did not. In contrast, Giannone et al. (58) reported that M2 cells responded nearly normally to force application by fibronectin-coated beads, whereas deletion of talin abolished the response to force in this system. Because talin, but not filamin, activates β3 integrins (59), the receptors for fibronectin in most cell types (60) the mechanosensing required for shape changes (spreading) in response to substrate stiffness is intact in M2 cells on an Fn matrix. However, when the only adhesive ligand is collagen, which engages largely β1 integrin that binds primarily to filamin A, the mechanosensing in filamin A-deficient cells is abolished, and they do not spread, stiffen, or adhere normally. It is notable that although talin appears to be able to compete with filamin for β1 integrins (44), it alone is not capable of transmitting or transducing the collagen and integrin β1-dependent signal required for shape change (spreading) in response to stiffness differences.
The data in Fig. 3 also show that cell spreading and stiffening can be independently controlled. This finding has implications for differentiating among different models of how cells adhere to and move on surfaces. Several studies suggest that application of internal tension (prestress) is required for a cell to spread, and that a consequence of prestress is stiffening of the cytoskeleton. This mechanism is inconsistent with the data in Fig. 3 a and c, which show that A7 cells stiffen more on collagen than on fibronectin, but spread equally well on fibronectin or collagen. Alternative models posit that cortical tension, and therefore a stiffer cell, counters cell spreading and that softening of the cytoskeleton alters the balance between cytoskeletal elasticity and membrane adhesion energy, allowing the cell to spread more. The data in Figs. 3 and 4 also indicate that adhesion strength and spreading can be dissociated, because despite increased spreading of M2 cells on fibronectin compared to collagen (Fig. 3 d), their adhesive properties on collagen and fibronectin are similar. The different responses to fibronectin- and collagen-coated gels are consistent with a model by which activation of the integrins specific for these ligands initiates signals that support different cellular functions with different requirements for plasticity and solidity. The finding that the combination of collagen and fibronectin produces a greater spread area than either ligand alone (Fig. 3 c), but a stiffness intermediate between that of each ligand separately (Fig. 3 a) suggests that ligation of the Fn receptor might promote cell softening, whereas ligation to collagen promotes stiffening, without a change in adherent area. The differential behavior of cells on matrices with different compositions (collagen versus fibronectin) may also have implications for the behavior of cells in wounds and in response to inflammation.
Acknowledgments
We thank Jerry P. Gollub and Douglas J. Durian for microfluidics advice.
This work was supported by National Science Foundation grant MRSEC/DMR05-20020, including the microfluidics seed to Drs. Golub and Durian, and by National Institutes of Health grant GM083272.
Footnotes
Fitzroy J. Byfield and Qi Wen contributed equally to this work.
References
- 1.Discher D.E., Janmey P., Wang Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 2.Paszek M.J., Zahir N., Johnson K.R., Lakins J.N., Rozenberg G.I. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Engler A.J., Griffin M.A., Sen S., Bonnemann C.G., Sweeney H.L. Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004;166:877–887. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flanagan L.A., Ju Y.E., Marg B., Osterfield M., Janmey P.A. Neurite branching on deformable substrates. Neuroreport. 2002;13:2411–2415. doi: 10.1097/01.wnr.0000048003.96487.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Georges P.C., Janmey P.A. Cell type-specific response to growth on soft materials. J. Appl. Physiol. 2005;98:1547–1553. doi: 10.1152/japplphysiol.01121.2004. [DOI] [PubMed] [Google Scholar]
- 6.Reinhart-King C., Dembo M., Hammer D.A. Endothelial cell traction forces on RGD-derivatized polyacrylamide substrata. Langmuir. 2003;19:1573–1579. [Google Scholar]
- 7.Saez A., Buguin A., Silberzan P., Ladoux B. Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J. 2005;89:L52–L54. doi: 10.1529/biophysj.105.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H.B., Dembo M., Wang Y.L. Substrate flexibility regulates growth and apoptosis of normal but not transformed cells. Am. J. Physiol. Cell Physiol. 2000;279:C1345–C1350. doi: 10.1152/ajpcell.2000.279.5.C1345. [DOI] [PubMed] [Google Scholar]
- 9.Pelham R.J., Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pourati J., Maniotis A., Spiegel D., Schaffer J.L., Butler J.P. Is cytoskeletal tension a major determinant of cell deformability in adherent endothelial cells? Am. J. Physiol. 1998;274:C1283–C1289. doi: 10.1152/ajpcell.1998.274.5.C1283. [DOI] [PubMed] [Google Scholar]
- 11.Hinz B., Celetta G., Tomasek J.J., Gabbiani G., Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang N., Stamenovic D. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. 2000;279:C188–C194. doi: 10.1152/ajpcell.2000.279.1.C188. [DOI] [PubMed] [Google Scholar]
- 13.Yeung T., Georges P.C., Flanagan L.A., Marg B., Ortiz M. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 14.Lo C.M., Wang H.B., Dembo M., Wang Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winer J.P., Janmey P.A., McCormick M.E., Funaki M. Bone marrow-derived human mesenchymal stem cells become quiescent on soft substrates but remain responsive to chemical or mechanical stimuli. Tissue Eng. Part A. 2008;15:147–154. doi: 10.1089/ten.tea.2007.0388. [DOI] [PubMed] [Google Scholar]
- 16.Beningo K.A., Lo C.M., Wang Y.L. Flexible polyacrylamide substrata for the analysis of mechanical interactions at cell-substratum adhesions. Methods Cell Biol. 2002;69:325–339. doi: 10.1016/s0091-679x(02)69021-1. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz U.S., Safran S.A. Elastic interactions of cells. Phys. Rev. Lett. 2002;88:048102. doi: 10.1103/PhysRevLett.88.048102. [DOI] [PubMed] [Google Scholar]
- 18.Thoumine O., Cardoso O., Meister J.J. Changes in the mechanical properties of fibroblasts during spreading: a micromanipulation study. Eur. Biophys. J. 1999;28:222–234. doi: 10.1007/s002490050203. [DOI] [PubMed] [Google Scholar]
- 19.McGarry J.G., Prendergast P.J. A three-dimensional finite element model of an adherent eukaryotic cell. Eur. Cell. Mater. 2004;7:27–33. doi: 10.22203/ecm.v007a03. [DOI] [PubMed] [Google Scholar]
- 20.von Wichert G., Haimovich B., Feng G.S., Sheetz M.P. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 2003;22:5023–5035. doi: 10.1093/emboj/cdg492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janmey P., McCulloch C. Cell mechanics: integrating cell responses to mechanical stimuli. Annu. Rev. Biomed. Eng. 2007;9:1–34. doi: 10.1146/annurev.bioeng.9.060906.151927. [DOI] [PubMed] [Google Scholar]
- 22.Hartwig J.H., Stossel T.P. Isolation and properties of actin, myosin, and a new actinbinding protein in rabbit alveolar macrophages. J. Biol. Chem. 1975;250:5696–5705. [PubMed] [Google Scholar]
- 23.Janmey P.A., Hvidt S., Lamb J., Stossel T.P. Resemblance of actin-binding protein/actin gels to covalently crosslinked networks. Nature. 1990;345:89–92. doi: 10.1038/345089a0. [DOI] [PubMed] [Google Scholar]
- 24.Gardel M.L., Nakamura F., Hartwig J., Crocker J.C., Stossel T.P. Stress-dependent elasticity of composite actin networks as a model for cell behavior. Phys. Rev. Lett. 2006;96:088102. doi: 10.1103/PhysRevLett.96.088102. [DOI] [PubMed] [Google Scholar]
- 25.Gardel M.L., Nakamura F., Hartwig J.H., Crocker J.C., Stossel T.P. Prestressed F-actin networks cross-linked by hinged filamins replicate mechanical properties of cells. Proc. Natl. Acad. Sci. USA. 2006;103:1762–1767. doi: 10.1073/pnas.0504777103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura F., Osborn T.M., Hartemink C.A., Hartwig J.H., Stossel T.P. Structural basis of filamin A functions. J. Cell Biol. 2007;179:1011–1025. doi: 10.1083/jcb.200707073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett J.P., Zaner K.S., Stossel T.P. Isolation and some properties of macrophage alpha-actinin: evidence that it is not an actin gelling protein. Biochemistry. 1984;23:5081–5086. doi: 10.1021/bi00316a039. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura F., Osborn E., Janmey P.A., Stossel T.P. Comparison of filamin A-induced cross-linking and Arp2/3 complex- mediated branching on the mechanics of actin filaments. J. Biol. Chem. 2002;277:9148–9154. doi: 10.1074/jbc.M111297200. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham C.C., Gorlin J.B., Kwiatkowski D.J., Hartwig J.H., Janmey P.A. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- 30.Eichinger L., Koppel B., Noegel A.A., Schleicher M., Schliwa M. Mechanical perturbation elicits a phenotypic difference between Dictyostelium wild-type cells and cytoskeletal mutants. Biophys. J. 1996;70:1054–1060. doi: 10.1016/S0006-3495(96)79651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tandon R., Levental I., Huang C., Byfield F.J., Ziembicki J. HIV infection changes glomerular podocyte cytoskeletal composition and results in distinct cellular mechanical properties. Am. J. Physiol. Renal Physiol. 2007;292:F701–F710. doi: 10.1152/ajprenal.00246.2006. [DOI] [PubMed] [Google Scholar]
- 32.Coughlin M.F., Puig-de-Morales M., Bursac P., Mellema M., Millet E. Filamin-A and rheological properties of cultured melanoma cells. Biophys. J. 2006;90:2199–2205. doi: 10.1529/biophysj.105.061267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lenormand G., Fredberg J.J. Deformability, dynamics, and remodeling of cytoskeleton of the adherent living cell. Biorheology. 2006;43:1–30. [PubMed] [Google Scholar]
- 34.Kim H., Sengupta A., Glogauer M., McCulloch C.A. Filamin A regulates cell spreading and survival via beta1 integrins. Exp. Cell Res. 2008;314:834–846. doi: 10.1016/j.yexcr.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 35.Glogauer M., Arora P., Chou D., Janmey P.A., Downey G.P. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J. Biol. Chem. 1998;273:1689–1698. doi: 10.1074/jbc.273.3.1689. [DOI] [PubMed] [Google Scholar]
- 36.Zhu T.N., He H.J., Kole S., D'Souza T., Agarwal R. Filamin a-mediated downregulation of the exchange factor Ras-GRF1 correlates with decreased matrix metalloproteinase-9 expression in human melanoma cells. J. Biol. Chem. 2007;282:14816–14826. doi: 10.1074/jbc.M611430200. [DOI] [PubMed] [Google Scholar]
- 37.Ohta Y., Hartwig J.H., Stossel T.P. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat. Cell Biol. 2006;8:803–814. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- 38.Gravante B., Barbuti A., Milanesi R., Zappi I., Viscomi C. Interaction of the pacemaker channel HCN1 with filamin A. J. Biol. Chem. 2004;279:43847–43853. doi: 10.1074/jbc.M401598200. [DOI] [PubMed] [Google Scholar]
- 39.Petrecca K., Miller D.M., Shrier A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J. Neurosci. 2000;20:8736–8744. doi: 10.1523/JNEUROSCI.20-23-08736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampson L.J., Leyland M.L., Dart C. Direct interaction between the actin-binding protein filamin-A and the inwardly rectifying potassium channel, Kir2.1. J. Biol. Chem. 2003;278:41988–41997. doi: 10.1074/jbc.M307479200. [DOI] [PubMed] [Google Scholar]
- 41.Huang C., Wu Z., Hujer K.M., Miller R.T. Silencing of filamin A gene expression inhibits Ca2+ -sensing receptor signaling. FEBS Lett. 2006;580:1795–1800. doi: 10.1016/j.febslet.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 42.Kim E.Y., Ridgway L.D., Dryer S.E. Interactions with filamin A stimulate surface expression of large-conductance Ca2+-activated K+ channels in the absence of direct actin binding. Mol. Pharmacol. 2007;72:622–630. doi: 10.1124/mol.107.038026. [DOI] [PubMed] [Google Scholar]
- 43.Calderwood D.A., Huttenlocher A., Kiosses W.B., Rose D.M., Woodside D.G. Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat. Cell Biol. 2001;3:1060–1068. doi: 10.1038/ncb1201-1060. [DOI] [PubMed] [Google Scholar]
- 44.Kiema T., Lad Y., Jiang P., Oxley C.L., Baldassarre M. The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell. 2006;21:337–347. doi: 10.1016/j.molcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Dertinger S.K.W., Chiu D.T., Jeon N.L., Whitesides G.M. Generation of gradients having complex shapes using microfluidic networks. Anal. Chem. 2001;73:1240–1246. [Google Scholar]
- 46.Zaari N., Rajagopalan P., Kim S.K., Engler A.J., Wong J.Y. Photopolymerization in microfluidic gradient generators: microscale control of substrate compliance to manipulate cell response. Adv. Mater. 2004;16:2133–2137. [Google Scholar]
- 47.Kandow C.E., Georges P.C., Janmey P.A., Beningo K.A. Polyacrylamide hydrogels for cell mechanics: steps towards optimization and alternative uses. Methods Cell Biol. 2007;83:29–46. doi: 10.1016/S0091-679X(07)83002-0. [DOI] [PubMed] [Google Scholar]
- 48.Georges P.C., Rojas W.D.J., Levental Ilya, Solon J., Byfield J., Janmey P.A. Effect of substrate stiffness on the structure and function of cells. Biophys. Rev. and Lett. 2006;1:401–410. [Google Scholar]
- 49.Byers H.R., Etoh T., Doherty J.R., Sober A.J., Mihm M.C. Cell-migration and actin organization in cultured human primary, recurrent cutaneous and metastatic melanoma - time-lapse and image-analysis. Am. J. Pathol. 1991;139:423–435. [PMC free article] [PubMed] [Google Scholar]
- 50.Stossel T.P., Condeelis J., Cooley L., Hartwig J.H., Noegel A. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 51.Solon J., Levental I., Sengupta K., Georges P.C., Janmey P.A. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miao H., Strebhardt K., Pasquale E.B., Shen T.L., Guan J.L. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. Role of Rho family small GTPases. J. Biol. Chem. 2005;280:923–932. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]
- 53.Garcia A.J., Huber F., Boettiger D. Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J. Biol. Chem. 1998;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- 54.Levental I., Georges P.C., Janmey P.A. Soft biological materials and their impact on cell function. Soft Matter. 2007;1:299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 55.Engler A.J., Sen S., Sweeney H.L., Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 56.Georges P.C., Hui J.J., Gombos Z., McCormick M.E., Wang A.Y. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;6:G1147–G1154. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 57.Kainulainen T., Pender A., D'Addario M., Feng Y., Lekic P. Cell death and mechanoprotection by filamin a in connective tissues after challenge by applied tensile forces. J. Biol. Chem. 2002;277:21998–22009. doi: 10.1074/jbc.M200715200. [DOI] [PubMed] [Google Scholar]
- 58.Giannone G., Jiang G., Sutton D.H., Critchley D.R., Sheetz M.P. Talin1 is critical for force-dependent reinforcement of initial integrin-cytoskeleton bonds but not tyrosine kinase activation. J. Cell Biol. 2003;163:409–419. doi: 10.1083/jcb.200302001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petrich B.G., Fogelstrand P., Partridge A.W., Yousefi N., Ablooglu A.J. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J. Clin. Invest. 2007;117:2250–2259. doi: 10.1172/JCI31024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boettiger D., Lynch L., Blystone S., Huber F. Distinct ligand-binding modes for integrin alpha(v)beta(3)-mediated adhesion to fibronectin versus vitronectin. J. Biol. Chem. 2001;276:31684–31690. doi: 10.1074/jbc.M103997200. [DOI] [PubMed] [Google Scholar]