

Abstract
Studies of peptide-based nanostructures provide general insights into biomolecular self-assembly and can lead material engineering toward technological applications. The diphenylalanine peptide (FF) self-assembles into discrete, hollow, well ordered nanotubes, and its derivatives form nanoassemblies of various morphologies. Here we demonstrate for the first time, to our knowledge, the formation of planar nanostructures with β-sheet content by the triphenylalanine peptide (FFF). We characterize these structures using various microscopy and spectroscopy techniques. We also obtain insights into the interactions and structural properties of the FF and FFF nanostructures by 0.4-μs, implicit-solvent, replica-exchange, molecular-dynamics simulations of aqueous FF and FFF solutions. In the simulations the peptides form aggregates, which often contain open or ring-like peptide networks, as well as elementary and network-containing structures with β-sheet characteristics. The networks are stabilized by polar and nonpolar interactions, and by the surrounding aggregate. In particular, the charged termini of neighbor peptides are involved in hydrogen-bonding interactions and their aromatic side chains form “T-shaped” contacts, as in three-dimensional FF crystals. These interactions may assist the FF and FFF self-assembly at the early stage, and may also stabilize the mature nanostructures. The FFF peptides have higher network propensities and increased aggregate stabilities with respect to FF, which can be interpreted energetically.
Introduction
The self-assembly of oligopeptides into crystals (1,2), amyloids (3–7), nanotubes (3,4,8,9), and systems responsive to external stimuli (e.g., pH, temperature, and concentration of specific solutes) (10) can provide insights into the formation of nanostructures such as amyloid fibers, and has potential applications in biomaterial synthesis, nanodevice fabrication, and tissue engineering. For these reasons, it has been the focus of extensive experimental (3,4,6–8,10–15) and computational studies (8,11,16–25) in recent years.
One of the most studied short building blocks is the diphenylalanine peptide (L-Phe-L-Phe; FF), the core recognition motif of the Alzheimer's β-amyloid peptide. A series of studies have shown that the peptide and its derivatives can self-assemble into highly ordered structures and other forms with nanoscale order, such as hydrogels (3,9,27–33). A unique property of the FF building block is its ability to form hollow tubular assemblies, most likely by the closure of two-dimensional surfaces (29,34). These assemblies can serve as casts for the fabrication of silver nanowires (9) and have remarkable stiffness (35). The vibrational spectra and birefringence of nanotubes suggest that they may share some structural properties with amyloid fibrils (9). On the other hand, work by Görbitz (36) has provided evidence that nanotubes may be structurally similar to three-dimensional FF crystals, which contain parallel helical channels with six peptides per helical turn and a diameter of 9.2 Å (1,36,37).
In recent studies (38,39), aromatic dipeptide nanotubes (ADNTs) were used to improve an enzyme-based biosensor. The electrode surface of the biosensor was modified with the use of ADNTs, resulting in extremely sensitive glucose and ethanol detection. Furthermore, several methods for aligning and patterning ADNTs have been developed. Axial unidirectional growth of a dense array of the ADNTs, and horizontal alignment of the tubes by coating them with a ferrofluid and applying an external magnetic field were demonstrated (40). Patterning of the ADNTs was achieved by the use of a simple inkjet technology. The ADNTs, which readily self-assemble in solution, were used as “ink” and patterned on transparency foil and ITO plastic (27).
Currently, our understanding of the factors that are responsible for the formation and stabilization of the FF nanotubes is still not complete. For example, the peptide Ac-Phe-Phe-NH2 self-assembles into highly ordered tubular structures despite the lack of charge in its termini (41). Other chemical modifications of the termini promote the formation of macroscopic hydrogels (3), amyloid-like fibers (41), or closed-cage nanostructures (27); thus, the nanostructures seem to depend on the chemical nature and interactions of both the termini and side chains.
In the work presented here, we used experimental and computational methods to study the formation of ordered structures in aqueous solutions of FF or the related peptide triphenylalanine (FFF). Using electron and laser microscopy methods, we demonstrate for the first time (to our knowledge) that triphenylalanine also self-assembles into nanostructures. These nanostructures are rather planar and will be referred to as “nanoplates” here. A structural analysis of the FFF assemblies by Fourier-transformed infrared (FTIR) spectroscopy showed that they are rich in β-sheet, as was previously concluded for FF nanotubes (9).
To gain a better understanding of the structural and energetic factors that control the formation of nanostructures by phenylalanine oligopeptides, we investigated model FF or FFF aqueous solutions by molecular-dynamics simulations using a replica-exchange method (42–44) and a high-accuracy implicit-solvent model (45,46).
The FF simulations reproduced several intermolecular interactions and structural properties seen in the FF crystals (1,36,37). In particular, the individual peptides were frequently and spontaneously arranged into open or closed (ring-like) linear networks, with head-to-tail interactions linking consecutive molecules and aromatic side chains of different peptides interacting extensively in approximately “T-shaped” orientations. We also observed the formation of more-complex structures in which individual peptides or networks were arranged into β-sheets, a feature that is consistent with spectroscopic results in this work and a previous study (9). Analogous structures were observed in the FFF simulations. Notably, the FFF aggregates were characterized by higher stability and peptide-network propensity. The origin of these differences and the connection to our experimental results from FF and FFF nanostructures are discussed further below.
Materials and Methods
Experimental methods
The FF and FFF systems were studied by transmission electron microscopy (TEM), scanning electron microscopy (SEM), environmental SEM (ESEM), confocal laser microscopy, and FTIR spectroscopy. A detailed presentation of the experimental methods is given in the Supporting Material.
Computational methods
Systems
We simulated two aqueous solutions with 12 FF or eight FFF peptides by the replica-exchange method. The peptides were placed in cubic boxes with a size of 57 Å, replicated by periodic boundary conditions. The corresponding peptide concentrations were 34 mg/mL (FF) and 33 mg/mL (FFF). The FF and FFF molecules had charged N-terminal (NH3+) and C-terminal (COO−) ends, as in the experiments. Peptide interactions were modeled by the CHARMM22 all-atom force field (47). Solvent effects were taken into account by the GBSW implicit-solvent model (45) with the optimized parameters and backbone torsional (CMAP) corrections of Chen et al. (46). All simulations were performed with the CHARMM program, v. c33b1 (48). Details of the simulation methods are given in the Supporting Material.
Results
Experimental formation of ordered assemblies by FF and FFF
We experimentally studied and compared the formation of ordered nanostructures by two short aromatic peptides: FF and FFF. Both peptides were dissolved in fluorinated alcohol and then diluted in water. FF (9) self-assembled into ordered discrete peptide nanotubes (Fig. 1 A), whereas FFF, under similar conditions, self-assembled to form plate-like nanostructures. Transmission electron microscopy (TEM) analysis with negative staining indicated that FFF formed well-ordered elongated assemblies (Fig. 1 B). The persistence length of the assemblies appears to be on the order of micrometers, as evidenced by microscopic observation. The formation of the assemblies was very efficient, and almost no amorphous aggregates were observed. We used SEM to further study the FFF assemblies. The SEM of areas filled with individual assemblies further demonstrated that the assemblies, in similarity to the FF nanotubes, are relatively homogeneous entities with a persistence length on the order of micrometers (Fig. 1 C). ESEM demonstrated the assembly morphology in aqueous solution (Fig. 1 D).
Figure 1.

Self-assembly of phenylalanine oligopeptides into ordered nanostructures. (A) ESEM of elongated, discrete FF nanotubes. (B) TEM of FFF plate-like assemblies as individual entities. (C) SEM exhibits the characteristic planar nanostructures of the FFF assemblies. (D) ESEM image of the FFF assemblies.
Experimental secondary structure studies
Previous studies, demonstrated that FF nanotubes are characterized by β-sheet secondary structure (9); ordered structures of a single nanotube were demonstrated with the use of electron diffraction analysis (40). In this work, we are interested in the secondary structure of the self-associated FFF assemblies. A common method for quantitative assessment of amyloid fibrils (β-sheet-rich protein) is the Thioflavin T (ThT) fluorescence assay, which reflects the change in fluorescence of the dye upon its interaction with the ordered assemblies. Here we used the ThT characteristic to visualize the FFF assemblies, which presented an excitation shift typical of amyloid fibril binding. Fluorescence confocal microscopy analysis of the fibrils dyed with ThT showed the presence of elongated ordered structures and implied that they have β-sheet content (Fig. 2, A and B).
Figure 2.
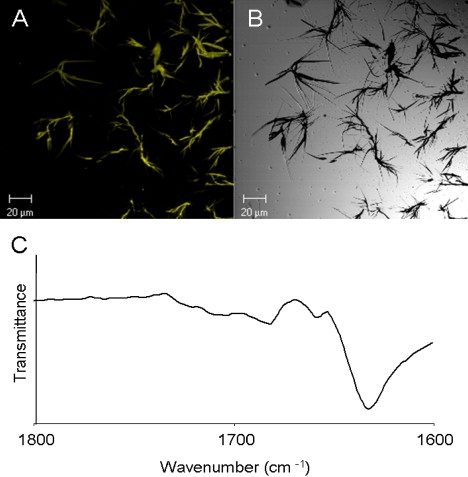
Secondary-structure studies of FFF assemblies. (A) Fluorescent image of fibrils dyed with ThT. (B) Microscopic image of fibrils. (C) FTIR spectral analysis of the amide I region of the formed structures. The amide I peaks corresponds to the vibration of the peptide bonds. The peak of IR absorbance is consistent with an extended β-sheet structure of the peptide chain.
Furthermore, to gain insight into the molecular configuration of the assembled structures, we used FTIR spectroscopy. Spectral analysis of the FFF assemblies showed a sharp 1630 cm−1 peak at the amide I region (Fig. 2 C), which was similar to the FF amide I region peak (9). This peak is consistent with a sheet-like conformation of the amide bond, as previously suggested for other peptide nanotubes consisting of larger building blocks (49,50).
Simulations of the FF and FFF aqueous solutions
Starting from random initial positions and orientations, the peptides coalesced rapidly (within ∼150 ps) into aggregates during the equilibration stage.
Description of the aggregates
The radius-of-gyration of the FF and FFF systems is plotted against temperature in the left panel of Fig. 3. The rise in the average and standard deviation (SD, error bars) reflects the gradual increase in aggregate fluctuations with temperature. The FFF values are smaller in the entire temperature range, suggesting that the FFF aggregate was somewhat more stable. The aggregates were still maintained at the highest simulated temperatures (the SD is 3–4 Å at 405–416 K, <15% of the box half-size); nevertheless, the peptides diffused and reorganized extensively. The right panel of Fig. 3 shows the “time” dependence of the radius-of-gyration at 300 K and at the highest simulated temperatures. The large fluctuations at 405 K (FF) and 416 K (FFF) reflect the frequent detachment and reattachment of monomers; at least one peptide was separated from the aggregates in 84.3% (FF) and 61.5% (FFF) of the frames (not shown).
Figure 3.
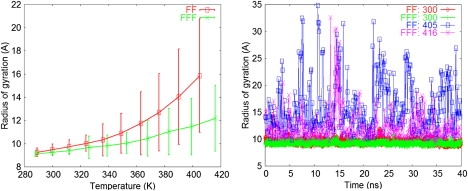
(Left) Temperature dependence of the average radius of gyration for the FF and FFF systems. The corresponding SD values are shown as error bars. (Right) “Time” dependence of the instantaneous FF and FFF radius of gyration, at the physiological temperature (T = 300 K) and at the highest simulated temperatures (405 K and 416 K). At each of the three temperatures, the corresponding simulation segments from all replicas were concatenated.
The FF and FFF aggregates had similar densities (1.779 ± 0.007 g/mL and 1.782 ± 0.006 g/mL, respectively, at 300 K) and approximately ellipsoidal shapes, which became somewhat more prolate at higher temperatures. The polar solvent-accessible surface area (PSA) of the FF and FFF aggregates was 29.0% and 24.4% of the total solvent-accessible surface area (SA) at 300 K. In separate simulations of the isolated FF and FFF monomers with the GBSW implicit model, the monomer PSA was 42.2% and 34.6%, respectively, of the SA at the same temperature. Thus, the polar groups were less accessible to solvent in the aggregates because they formed networks with extensive intra- and intermolecular interactions (see below).
Peptide organization in the aggregates
Crystallographic studies have shown that the FF peptides can form three-dimensional crystal structures (37). The crystals contain rings of hexagonal symmetry in which six peptides are linked by head (NH3+) to tail (−OOC) hydrogen bonds. Adjacent rings are juxtaposed to form channels with a hydrophilic 9.2 Å diameter pore containing mainly disordered water. The peptide side chains are positioned at the exterior of the channels and form an intricate three-dimensional stacking arrangement, which presumably stabilizes the parallel arrangement of adjacent channels in the crystals.
Some of these features were observed in the simulations, as we analyze below and discuss further in the Discussion section. The peptides were frequently arranged into linear networks in which consecutive peptides were linked by intermolecular head (NH3+) to tail (−OOC) hydrogen-bonding interactions. Network definitions and examples are given in the Supporting Material.
The backbone moieties of several adjacent peptides (2–6) often formed open (O) or closed (ring-like (R)) linear networks. Fig. 4 shows representative ring networks of six peptides from the 300 K simulations. The FF network (left panel) is also superposed against the hexagonal peptide ring of the FF crystals (37). Other examples of ring networks are shown in Fig. S2 of the Supporting Material.
Figure 4.
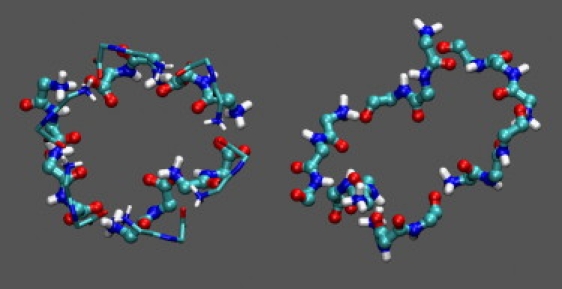
Representative ring-like networks of six peptides observed in the FF (left) and FFF (right) 300 K simulations. Only the main-chain atoms of the networks are shown in licorice/CPK representation; other network atoms and the surrounding aggregate are omitted. For comparison, the hexagonal ring of the FF crystals (1,36,37) (backbone only, in thin licorice) is also superposed against the simulation ring.
Table 1 lists the average number of networks per simulation snapshot at 300 K. Open networks are more frequent compared to ring networks with the same number of peptides. This is due to entropic reasons; as we show below, ring networks have lower association energies than open networks of the same size. The ratios of network populations (corrected to account for the different number of monomers) are included in the last column of Table 2. The FFF networks have a higher relative propensity, which increases with the number of constituent peptides. The overall head-to-tail propensity (HT row in Table 1) is also higher in FFF. An energetic analysis (below) shows that the FFF networks have a lower formation energy, which could partly explain their larger relative propensity.
Table 1.
Statistics of peptide networks observed in the 300 K simulations
| Network | N(FF) | N(FFF) | N(FFF):N(FF)‡ |
|---|---|---|---|
| O2∗ | 2.7(1.4) † | 2.0 (1.2) † | 2 |
| R2 | 1.2(1.0) | 0.39 (0.57) | 1 |
| O3 | 0.88(0.81) | 0.56 (0.66) | 3 |
| R3 | 0.11(0.32) | 0.055 (0.229) | 2 |
| O4 | 0.41(0.56) | 0.24 (0.43) | 4 |
| R4 | 0.04(0.20) | 0.010 (0.101) | 2 |
| O5 | 0.19(0.40) | 0.11(0.31) | 8 |
| R5 | 0.011(0.105) | 0.0068 (0.0819) | 9 |
| O6 | 0.13(0.33) | 0.050 (0.218) | 13 |
| R6 | 0.0014(0.0374) | 0.0023 (0.0474) | 53 |
| HT§ | 9.94(2.58) | 5.53(1.68) | 1.3 |
OX and RX denote open and closed (ring-like) networks, respectively, of X peptides.
Average number of networks per conformation. The SDs are included in parentheses. The numbers are computed from 20,000 snapshots of the 300 K simulations, spaced at 2-ps intervals.
Relative (FFF/FF) network propensity. The ratios N(FFF)/N(FF) (from columns 2 and 3) of networks with X peptides have been multiplied by a combinatorial factor to account for the larger total number of peptides in the FF solution (12) compared to the FFF solution (8).
H and T denote “head” (NH3+) and “tail” (COO−) groups, respectively.
Table 2.
Types and statistics of two- and three-stranded β-structures in the 300 K simulations
|
β-strand type† |
|||||||
|---|---|---|---|---|---|---|---|
| System | Conformation | No. of events∗ | O1/O1 | O1/N | N/N | O1/O1/O1 | O1/N/N |
| FF | Two-stranded β-sheet | 4030(4022) | 893 | 1352 | 1785 | — | — |
| Three-stranded β-sheet | 545 | — | — | — | 471 | 74 | |
| FFF | Two-stranded β-sheet | 6108(6108) | 2181 | 3027 | 900 | — | — |
| Three-stranded β-sheet | 24(14)‡ | — | — | — | 23 | — | |
Total number of occurrences of the listed β-sheet. In parentheses are included the occurrences of antiparallel sheets. The analysis was performed on 20,000 snapshots, spaced at 2-ps intervals.
Number of β-sheet occurrences, where one of β-strands is either a peptide network (N) or a single monomer (O1).
In nine events the strands are arranged in mixed orientation.
The network propensity decreased with temperature in both FF and FFF aggregates, but the higher relative propensity of FFF with respect to FF was maintained at all temperatures (Fig. S3).
Intermolecular β-sheet conformations
The peptide networks possess elementary structural patterns that may reflect early formations in the aggregation process, or may be maintained in FF nanotubes (9) and/or FFF nanoplates. In fact, peptide chains with head-tail interactions are fundamental elements of dipeptide crystals (see Görbitz (1) and references therein). At the same time, the spectroscopic results presented here and in a previous study (9) suggest that FFF nanoplates and FF nanotubes are rich in β-sheet. Analysis of the secondary-structure content of the FF and FFF simulation aggregates with the STRIDE (51) module of VMD (52) and in-house routines showed the existence of conformations with β-sheet content, by pairs of individual peptides or peptide networks. The STRIDE β-sheet formation criteria are defined in Fig. 4 of Frishman and Argos (51).
The properties of the β-sheet conformations in the 300 K simulations are summarized in Table 2. The observed sheets consist of two or (less often) three strands; more-extended sheets are seldom observed, presumably due to the small number of peptides in the simulation systems and the restricted duration of the simulations. In a large fraction of the sheets, one of the strands is a network (N) of at least two peptides (columns 4–8).
The vast majority of two-stranded sheets are antiparallel (99% and 100%, respectively, in FF and FFF). In the case of FFF, a fraction of the three-stranded sheets have mixed character (Table 2). The antiparallel orientation is not necessarily the thermodynamically preferred state in larger-scale and/or higher-symmetry structures (e.g., in FF crystals (37), adjacent peptide rings along a channel have parallel orientations).
Table 3 lists the families of the two-stranded sheets, classified according to the interstrand hydrogen-bonding patterns, and Fig. 5 shows representative conformations. In the majority of FF sheets (families 1 and 3–5 of Table 3), the two strands are unaligned, enabling the formation of interactions between the charged ends and the internal peptide groups. Specifically, in family 1 (Fig. 5 A) the tail of each strand interacts with the head and the internal carbonyl of the opposite strand; in families 3 and 5 (Fig. 5, B and C) the strand heads interact with one and two internal carbonyls, respectively. Family 2 (Fig. 5 D) consists of aligned strands.
Table 3.
Classes of two-stranded β-sheets in the 300 K simulations
| Average distances (SDs) of hydrogen-bonded atoms |
|||||
|---|---|---|---|---|---|
| System | No. of family: hydrogen-bonded atom-pairs: occurrence probability (%) | First bond | Second bond | Third bond | Fourth bond |
| FF | 1: HA-TB and N2A-TB and TA-N2B and TA-HB: 68% | 2.8 (0.1) | 2.9 (0.2) | 2.9 (0.2) | 2.8 (0.1) |
| 2: HA-TB and O1A-N2B: 19%∗ | 2.9 (0.3) | 3.2 (0.4) | — | — | |
| 3: HA-O1B and TA-HB: 14%∗¶ | 3.2 (0.4) | 2.9 (0.3) | — | — | |
| 4: only (HA-TB and TA-N2B): 6% | 2.9 (0.3) | 3.3 (0.4) | — | ||
| 5: HA-O1B and O1A-HB: 3%¶ | 3.4 (0.4) | 3.4 (0.4) | |||
| FFF | 1: O1A-N3B and N3A-O1B§: 57% | 3.0 (0.3) | 3.0 (0.3) | — | — |
| 2: TA-HB and N3A-O2B: 14%† | 2.8 (0.2) | 3.1(0.3) | — | — | |
| 3: TA-HB and N3A-O1B: 12%† | 2.9 (0.2) | 3.2 (0.4) | — | — | |
| 4: N2A-O2B and O2A-HB: 6%‡ | 3.4 (0.4) | 3.1(0.4) | — | — | |
| 5: HA-O2B and O1A-N2B: 5% | 2.6 (1.0) | 2.6 (1.0) | — | — | |
| 6: N2A-2B and O2A-N2B: 4%‡ | 3.2 (0.4) | 3.2 (0.4) | — | — | |
| other / 4% | - | — | |||
| Notation | FF | FFF | |||
The analysis was performed for the two-stranded sheets listed in Table 2. The atom notation is explained in the last panel of the table; A and B refer to the two strands of the sheet.
∗,¶In FF, a subset of sheets satisfy simultaneously the criteria of families 2 and 3, or 3 and 5.
†,‡In FFF, a subset of sheets satisfy simultaneously the criteria of families 2 and 3, or 4 and 6.
§Eight percent of conformations have two additional head-tail interactions; 24% of conformations have one additional head-tail interaction.
Figure 5.
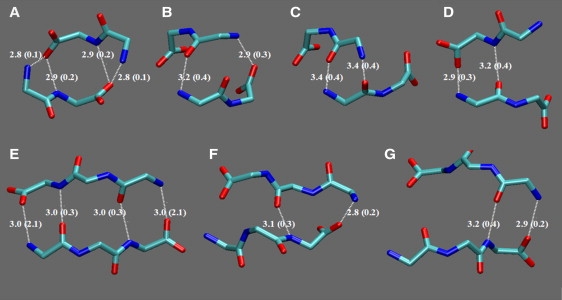
Hydrogen-bonding patterns of the most important two-stranded β-sheet structures observed in the FF (top) and FFF (bottom) 300 K simulations. Only the main-chain heavy atoms of the structures are shown in licorice representation. Plots A–C correspond to families 1, 3, and 5, and plot D corresponds to family 4. Plots E–G correspond to FFF families 1–3. The average distances and SDs (in parentheses) correspond to the values in Table 3.
In the case of FFF, the majority of observed sheets (families 1, 3, and 6) contain aligned strands. The three most important families are shown in Fig. 5, E–G. The conformations of the first family always contain the two hydrogen-bonding interactions of the internal peptide groups; in 24% and 8% of conformations, respectively, one or two head-tail interactions are also present.
The FFF nanoplates and FF nanotubes are likely to contain β-sheet interactions, as suggested by the spectroscopic results of this work and a previous study (9). It is plausible that the individual strands of these sheets consist of peptide networks that interact via some of the hydrogen-bonding patterns observed in the conformations of Table 3 and Fig. 5. Fig. S4 shows an example of an aligned antiparallel sheet created by the periodic placement on the plane of a conformation with the hydrogen-bonding pattern of the second FF family (Fig. 5 D). Fig. S4 also includes an example of a partly aligned/partly unaligned sheet with hydrogen-bonding patterns of the first, third, and second FF families (Fig. 5, A, C, and D).
Side-chain interactions
The stabilities of the aggregates and the observed structural patterns depend on the interactions of the aromatic side chains.
Probability distributions of pseudodihedral angles, describing the relative orientation of side chains within the FF and FFF monomers, are shown in Fig. S5. A characteristic feature of the FF peptides in FF crystals, also observed in our simulations, is the placement of both side chains at the same side of the intermediate peptide bond; in this way, all side chains face toward the exterior of the rings comprising the crystals. In the case of FFF rings, side chains can be in the ring interior, without interfering with the head-tail interactions (presumably due to the larger FFF length). This variation between the behavior of the FF and FFF systems may point to a different organization of the FF and FFF nanostructures.
The geometry of interacting phenylalanine side-chain pairs can be described by a probability distribution involving the distance (r) between the aromatic ring centers, the angle (χ) between the rings, and the two angles (ϕ1, ϕ2) between the vector connecting the ring centers and the normals to the aromatic-ring planes (Fig. S6). In the FF and FFF simulations, the distance between the rising centers of interacting side-chain pairs was <7 Å. A large fraction of inter- and intramolecular pairs (47% and 51%, respectively) adopted approximately T-shaped orientations (χ = 60–90°), as in the crystals. Fig. S7 shows a representative six-member ring from the 300 K FF simulations, which contains several T-shaped side-chain interactions.
Energetic analysis of the networks
The above analysis indicates that peptide networks are likely to be important ingredients of the FF and FFF nanostructures. To gain insight into the network stabilities, we computed the network association energies (see Eq. S1 in the Supporting Material and the accompanying discussion). The total values (at 300 K) are plotted against the network size (the number of participating peptides) in the left panel of Fig. 6; nonpolar and polar contributions are shown in the middle and right panels.
Figure 6.
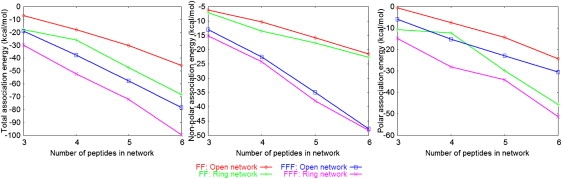
Association energies of the FF and FFF networks in the 300 K simulations, plotted as a function of network size (number of peptides). The panels from left to right show, respectively, the total values, the nonpolar (vdW + SA) and polar (Coulomb + GB) contributions.
The polar and nonpolar components are negative, i.e., both stabilize the networks. The stability increases with the number of constituent peptides. Within each system, ring networks are more stable energetically relative to open networks of the same size. This is mainly due to stronger polar interactions arising from the larger number of head-tail contacts in the rings.
FFF networks are more stable relative to FF networks of the same topology (open or closed) and number of monomers, mainly due to the stronger intermolecular van der Waals interactions (the accessible SA contribution to the nonpolar association energy is ∼8–9% in the FF and FFF networks).
The polar interactions are modulated by the surrounding environment (e.g., the network exposure to solvent). In the generalized Born (GB) implict-solvent approximation, the polar interactions depend on the surrounding medium through the magnitudes of the atomic solvation radii, which reflect the atomic distances from the solvent (53). To investigate the impact from the surrounding aggregate on the network stabilities, we computed the GB components of the network association energies, assuming that all peptides that did not participate in the network were replaced by water. The relative GB network association energies (in the solvated-aggregate environment, relative to a water-only environment) are plotted in Fig. 7.
Figure 7.
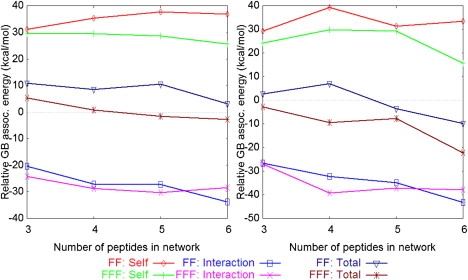
Impact of the surrounding medium on the network stabilities: change in the GB component of the network association energy when the network is transferred from water to the environment of the aggregate (see text). (Left) Open networks. (Right) Ring networks.
The positive self-energies show that the formation of networks is disfavored in the aggregate, relative to water, due to the burial of polar groups. The negative interaction energies are due to the reduced screening of electrostatic interactions in the aggregate, compared to water. Overall, open FF networks are less stable in the aggregate, but ring FF networks can be more stable provided they are sufficiently large (N > 4). FFF networks have higher relative stabilities compared to FF, i.e., ring FFF networks are more stable in the aggregate for all sizes, and open networks become more stable for sizes N > 4. The differences in the relative stabilities of the FFF and FF networks originate mainly from the self-energy terms, which are less positive for FFF. Presumably, this is due to the larger relative size of the FFF networks in the FFF aggregate, which enables them to remain more solvent-exposed.
The above analysis suggests that the formation of peptide networks can be promoted by the surrounding aggregate, depending on the network type and size. Thus, the formation of FF and FFF nanostructures could proceed via an initial coalescence of peptides into unstructured aggregates, followed by the development of structural order through specific hydrogen-bonding interactions.
Discussion and Conclusions
The aromatic dipeptide building blocks represent major structural elements for future technological applications. However, despite their great potential and proven chemical, physical, and biological properties, their mechanism of self-assembly and organization on the nanoscale are not fully understood.
FF nanotubes have vibrational spectra and birefringence similar to those of amyloid fibrils, which suggests that they share some structural properties (9). On the other hand, work by Görbitz (36) suggests that FF nanotubes may have close structural similarity to FF crystals. Our simulations produced several structural features reminiscent of FF crystals (e.g., ring networks with distances and orientations of contact side chains, as in the crystals). Some of these features may be present in FF nanotubes, or reflect early structural formations during FF and FFF self-assembly.
The experimental spectra (Fig. 2) (9) suggest that FF and FFF nanostructures are rich in β-sheet. Conformations with two- and (less often) three-stranded β-sheets were also formed in the simulations. More extended sheets were not observed, presumably due to the small system size and limited duration of the runs. Nevertheless, some of the observed hydrogen-bonding patterns (Table 3 and Fig. 5) are likely to be present in nanostructures. Some examples of planar conformations, which are periodic in two dimensions and contain some of the hydrogen-bonding patterns observed in the FF sheets of the simulations, are shown in Fig. S4.
An important question is whether the morphologies and stabilities of the peptide nanostructures are determined mostly by charged-termini or side-chain interactions. The impressive variety of crystals formed by dipeptides with hydrophobic (1) and hydrophilic (2) side chains suggests that the relative importance of these interactions is system-specific. The nanostructures of FF analogs do depend on the chemical nature of the termini (3,27,41,54). In FF crystals, the channels seem to be stabilized by both head-tail interactions and extensive side-chain contacts, as discussed above. In line with this, the peptide networks are stabilized energetically by both polar and nonpolar interactions.
A second question is related to the role of aromaticity in the systems presented here. In the FF crystals and simulations, intermolecular pairs of interacting side chains adopt low-energy orientations (T-shaped or parallel displaced; see the Supporting Material) (55). The abundance of aromatic residues in amyloidogenic peptide fragments, as well as results from experimental (6,7,56–58) and computational (12,59,60) studies and phenomenological models (61,62) suggest that aromatic interactions promote amyloid formation. Aromatic amino acids have been estimated to have the highest amyloid-forming propensity (63). The exact mechanism for aromatic stabilization is still unknown. Stacking interactions could contribute to stability of the nanostructure (13,57). Other studies have suggested that the self-assembly of aromatic peptides is due to their hydrophobic character (14,64–66).
The FF and FFF systems are similar in that they both self-organize into networks and β-sheet conformations. On the other hand, the FFF aggregates are more stable and the FFF networks have a higher propensity and are somewhat more stable than the FF networks. In the β-sheets of the simulations (Table 3 and Fig. 5), FF and FFF show a tendency for unaligned and aligned strands, respectively. These differences stem from the different sizes of the FF and FFF monomers, due to which FFF have twice as many internal hydrogen-bonding groups compared to FF and an additional side chain. It is difficult to link the differences observed in the small systems of the simulations to the geometrical properties of the much larger FF nanotubes and FFF nanoplates.
In summary, we explored the self-assembly of FF and FFF peptides in aqueous solution by experiments and molecular-dynamics simulations. The FF peptides formed nanotubes in the experiments, as shown previously (9). Under similar experimental conditions, the FFF peptides self-assembled into plate-like nanostructures. Both types of nanostructures are rich in β-sheet content. In the simulations, the peptides often formed open and ring-like peptide networks, as well elementary and network-containing structures with β-sheet characteristics. The FFF networks were somewhat more stable than FF networks, mainly due to nonpolar interactions. An energetic analysis showed that the FFF and FF networks can have lower association energies in the aggregate compared to a water-surrounding medium, depending on the network topology and size. This suggests that the initial formation of aggregates could be promoted by hydrophobic interactions. Backbone hydrogen-bonding interactions and more ordered structures could then be facilitated due to the reduced screening of electrostatic interactions in the presence of the aggregates. This is in line with previous observations regarding the self-assembly of the dipeptide Ile-Phe (66).
Supporting Material
Details of the employed methods, definitions of the peptide networks, and additional results, including some tables, figures, and references, are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00767-X.
Supporting Material
Acknowledgments
We thank Professor Carl H. Görbitz for providing us with the coordinates of the FF crystals, Dr. Yacov Delarea for help with the TEM experiments, Dr. Zahava Barkay for help with the SEM experiments and ESEM analysis, Dr. Alexander Barbul for confocal microscopy analysis, and members of the Gazit laboratory for helpful discussions. We also thank the anonymous referees for constructive remarks. All simulations were performed on Linux clusters of the Biophysics Group at the University of Cyprus.
This work was funded by a grant from the A. G. Leventis Foundation (to G.A. and Ph.T). L.A.A. gratefully acknowledges the support of the Colton Foundation.
Footnotes
Phanourios Tamamis and Lihi Adler-Abramovich contributed equally to this work.
Contributor Information
Ehud Gazit, Email: ehudg@post.tau.ac.il.
Georgios Archontis, Email: archonti@ucy.ac.cy.
References
- 1.Görbitz C.H. Microporous organic materials from hydrophobic dipeptides. Chem. Eur. J. 2007;13:1022–1031. doi: 10.1002/chem.200601427. [DOI] [PubMed] [Google Scholar]
- 2.Görbitz C.H., Hartviksen L.M. The monohydrates of the four polar dipeptides L-seryl-L-asparagine, L-seryl-L-tyrosine, L-tryptophanyl-L-serine and L-tyrosyl-L-tryptophan. Acta Crystallogr. C. 2008;64:o171–o176. doi: 10.1107/S0108270108004228. [DOI] [PubMed] [Google Scholar]
- 3.Mahler A., Reches M., Rechter M., Cohen S., Gazit E. Rigid, self-assembled hydrogel composed of a modified aromatic dipeptide. Adv. Mater. 2006;18:1365–1370. [Google Scholar]
- 4.Gazit E. Self-assembled peptide nanostructures: the design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 2007;36:1263–1269. doi: 10.1039/b605536m. [DOI] [PubMed] [Google Scholar]
- 5.Chiti F., Dobson C.M. Protein misfolding, functional amyloid and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 6.Gazit E. Mechanisms of amyloid fibril self-assembly and inhibition. Model short peptides as a key research tool. FEBS J. 2005;272:5971–5978. doi: 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- 7.Gazit E. Self-assembly of short aromatic peptides into amyloid fibrils and related nanostructures. Prion. 2007;1:32–35. doi: 10.4161/pri.1.1.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colombo G., Soto P., Gazit E. Peptide self-assembly at the nanoscale: a challenging target for computational and experimental biotechnology. Trends Biotechnol. 2007;25:211–218. doi: 10.1016/j.tibtech.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Reches M., Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X.J., Zhang S.G. Designer self-assembling peptide materials. Macromol. Biosci. 2007;7:13–22. doi: 10.1002/mabi.200600230. [DOI] [PubMed] [Google Scholar]
- 11.Knowles T.P., Fitzpatrick A.W., Meehan S., Mott H.R., Vendruscolo M. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 12.Zanuy D., Porat Y., Gazit E., Nussinov R. Peptide sequence and amyloid formation: molecular simulations and experimental study of a human islet amyloid polypeptide fragment and its analogs. Structure. 2004;12:439–455. doi: 10.1016/j.str.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Sawaya M.R., Sambashivan S., Nelson R., Ivanova M.I., Sievers S.A. Atomic structures of amyloid cross-b spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 14.Bemporad F., Galloni G., Campioni S., Plakoutsi G., Taddei N. Sequence and structural determinants of amyloid fibril formation. Acc. Chem. Res. 2006;39:620–627. doi: 10.1021/ar050067x. [DOI] [PubMed] [Google Scholar]
- 15.Iconomidou V.A., Chryssikos G.D., Gionis V., Galanis A.S., Cordopatis P. Amyloid fibril formation propensity is inherent into the hexapeptide tandemly repeating sequence of the central domain of silkmoth chorion proteins of the A-family. J. Struct. Biol. 2006;156:480–488. doi: 10.1016/j.jsb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Mousseau N., Derreumaux P. Exploring the early steps of amyloid peptide aggregation by computers. Acc. Chem. Res. 2005;38:885–891. doi: 10.1021/ar050045a. [DOI] [PubMed] [Google Scholar]
- 17.Cecchini M., Rao F., Seeber M., Caflisch A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J. Chem. Phys. 2004;121:10748–10756. doi: 10.1063/1.1809588. [DOI] [PubMed] [Google Scholar]
- 18.Hills R.D., Brooks C.L. Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 2007;368:894–901. doi: 10.1016/j.jmb.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarus B., Straub J.E., Thirumalai D. Structures and free energy landscapes of the wild type and mutants of the A β(21–30) peptide are determined by an interplay between intrapeptide electrostatic and hydrophobic interactions. J. Mol. Biol. 2008;379:815–829. doi: 10.1016/j.jmb.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumketner A., Shea J.E. The structure of the Alzheimer amyloid β 10–35 peptide, probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007;366:275–285. doi: 10.1016/j.jmb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Hall C.K., Wagoner V.A. Computational approaches to fibril structure and formation. Methods Enzymol. 2007;412:338–365. doi: 10.1016/S0076-6879(06)12020-0. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen P.H., Li M.S., Stock G., Straub J.E., Thirumalai D. Monomer adds to preformed structured oligomers of Aβ-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma B.Y., Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Paci E., Gsponer J., Salvatella X., Vendruscolo M. Molecular dynamics studies of the process of amyloid aggregation of peptide fragments of transthyretin. J. Mol. Biol. 2004;340:555–569. doi: 10.1016/j.jmb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Song W., Wei G., Mousseau N., Derreumaux P. Self-assembly of the β2-microglobulin NHVTLSQ peptide using a coarse-grained protein model reveals a β-barrel species. J. Phys. Chem. B. 2008;112:4410–4418. doi: 10.1021/jp710592v. [DOI] [PubMed] [Google Scholar]
- 26.Reference deleted in proof.
- 27.Adler-Abramovich L., Gazit E. Controlled patterning of peptide nanotubes and nanospheres using inkjet printing technology. J. Pept. Sci. 2008;14:217–223. doi: 10.1002/psc.963. [DOI] [PubMed] [Google Scholar]
- 28.Panda J.J., Mishra A., Basu A., Chauhan V.S. Stimuli responsive self-assembled hydrogel of a low molecular weight free dipeptide with potential for tunable drug delivery. Biomacromolecules. 2008;9:2244–2250. doi: 10.1021/bm800404z. [DOI] [PubMed] [Google Scholar]
- 29.Reches M., Gazit E. Formation of closed-cage nanostructures by self-assembly of aromatic dipeptides. Nano Lett. 2004;4:581–585. [Google Scholar]
- 30.Singh G., Bittner A.M., Loscher S., Malinowski N., Kern K. Electrospinning of diphenylalanine nanotubes. Adv. Mater. 2008;20:2332–2336. [Google Scholar]
- 31.Song Y., Challa S.R., Medforth C.J., Qiu Y., Watt R.K. Synthesis of peptide-nanotube platinum-nanoparticle composites. Chem. Commun. 2004;9:1044–1045. doi: 10.1039/b402126f. [DOI] [PubMed] [Google Scholar]
- 32.Han T.H., Kim J., Park J.S., Park C.B., Ihee H. Liquid crystalline peptide nanowires. Adv. Mater. 2007;19:3924–3927. [Google Scholar]
- 33.Toledano S., Williams R.J., Jayawarna V., Ulijn R.V. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 2006;128:1070–1071. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- 34.Tsai C.J., Zheng J., Nussinov R. Designing a nanotube using naturally occurring protein building blocks. PLoS Comput. Biol. 2006;2:311–319. doi: 10.1371/journal.pcbi.0020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kol N., Abramovich L., Barlam D., Shneck R.Z., Gazit E. Self-assembled peptide nanotubes are uniquely rigid bioinspired supramolecular structures. Nano Lett. 2005;5:1343–1346. doi: 10.1021/nl0505896. [DOI] [PubMed] [Google Scholar]
- 36.Görbitz C.H. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer's amyloid polypeptide. Chem. Commun. 2006;22:2332–2334. doi: 10.1039/b603080g. [DOI] [PubMed] [Google Scholar]
- 37.Görbitz C.H. Nanotube formation by hydrophobic dipeptides. Chem. Eur. J. 2001;7:5153–5159. doi: 10.1002/1521-3765(20011203)7:23<5153::aid-chem5153>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 38.Yemini M., Reches M., Gazit E., Rishpon J. Peptide nanotube-modified electrodes for enzyme-biosensor applications. Anal. Chem. 2005;77:5155–5159. doi: 10.1021/ac050414g. [DOI] [PubMed] [Google Scholar]
- 39.Yemini M., Reches M., Rishpon J., Gazit E. Novel electrochemical biosensing platform using self-assembled peptide nanotubes. Nano Lett. 2005;5:183–186. doi: 10.1021/nl0484189. [DOI] [PubMed] [Google Scholar]
- 40.Reches M., Gazit E. Controlled patterning of aligned self-assembled peptide nanotubes. Nat. Nanotechnol. 2006;1:195–200. doi: 10.1038/nnano.2006.139. [DOI] [PubMed] [Google Scholar]
- 41.Reches M., Gazit E. Self-assembly of peptide nanotubes and amyloid-like structures by charged-termini capped diphenylalanine peptide analogues. Isr. J. Chem. 2005;45:363–371. [Google Scholar]
- 42.Nymeyer H., Gnanakaran S., Garcia A. Atomic simulations of protein folding, using the replica exchange algorithm. Methods Enzymol. 2004;30:119–149. doi: 10.1016/S0076-6879(04)83006-4. [DOI] [PubMed] [Google Scholar]
- 43.Hukushima K., Nemoto K. Exchange Monte Carlo method and application to spin glass simulation. J. Phys. Soc. Jpn. 1996;65:1604–1608. [Google Scholar]
- 44.Sanbonmatsu K.Y., Garcia A.E. Structure of Met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins. 2002;46:225–234. doi: 10.1002/prot.1167. [DOI] [PubMed] [Google Scholar]
- 45.Im W., Lee M.S., Brooks C.L. Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 46.Chen J., Im W., Brooks C.L. Balancing solvation and intramolecular interactions: towards a self-consistent generalized born force field. J. Am. Chem. Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacKerrell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 48.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 49.Ghadiri M.R., Granja J.R., Milligan R.A., McRee D.E., Khazanovich N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature. 1993;366:324–327. doi: 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]
- 50.Holmes T.C., de Lacalle S., Su X., Liu G.S., Rich A. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc. Natl. Acad. Sci. USA. 2000;97:6728–6733. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 52.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Bashford D., Case D.A. Generalized Born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 54.Smith A.M., Williams R.J., Tang C., Coppo P., Collins R.F. Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based on π-π interlocked β-sheets. Adv. Math. 2008;20:37–41. [Google Scholar]
- 55.Sinnokrot M.O., Sherrill C.D. High-accuracy of quantum mechanical studies of π-π interactions in benzene dimers. J. Phys. Chem. A. 2006;110:10656–10668. doi: 10.1021/jp0610416. [DOI] [PubMed] [Google Scholar]
- 56.Gazit E. Global analysis of tandem aromatic octapeptide repeats: the significance of the aromatic-glycine motif. Bioinformatics. 2002;18:880–883. doi: 10.1093/bioinformatics/18.6.880. [DOI] [PubMed] [Google Scholar]
- 57.Gazit E. A possible role for stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 58.Jack E., Newsome M., Stockley P.G., Radford S.E., Middleton D.A. The organization of aromatic side groups in an amyloid fibril probed by solid-state 2H and 19F NMR spectroscopy. J. Am. Chem. Soc. 2006;128:8098–8099. doi: 10.1021/ja0581898. [DOI] [PubMed] [Google Scholar]
- 59.Zanuy D., Nussinov R. The sequence dependence of fiber organization. A comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J. Mol. Biol. 2003;329:565–584. doi: 10.1016/s0022-2836(03)00491-1. [DOI] [PubMed] [Google Scholar]
- 60.Wu C., Lei H., Duan Y. The role of Phe in the formation of well-ordered oligomers of amyloidogenic hexapeptide (NFGAIL) observed in molecular dynamics simulations with explicit solvent. Biophys. J. 2005;88:2897–2906. doi: 10.1529/biophysj.104.055574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tartaglia, G., A. Cavalli, R. Pellarin and A., Caflisch. Prediction of aggregation rate and aggregation-prone segmens in polypeptide sequences. Protein Sci. 14:2723–2734. [DOI] [PMC free article] [PubMed]
- 62.Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr. Opin. Chem. Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 63.Pawar A.P., Dubay K.F., Zurdo J., Chiti F., Vendruscolo M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005;350:379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 64.Tracz S.M., Abedini A., Driscoll M., Raleigh D.P. Role of aromatic interactions in amyloid formation by peptides derived from human amylin. Biochemistry. 2004;43:15901–15908. doi: 10.1021/bi048812l. [DOI] [PubMed] [Google Scholar]
- 65.Bemporad F., Taddei N., Stefani M., Chiti F. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Protein Sci. 2006;15:862–870. doi: 10.1110/ps.051915806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanchez de Groot N., Parella T., Aviles F.X., Vendrell J., Ventura S. Ile-Phe dipeptide self-assembly: clues to amyloid formation. Biophys. J. 2007;92:1732–1741. doi: 10.1529/biophysj.106.096677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.