

Abstract
Cerebral ischemia and reperfusion increase superoxide anions (O2·−) in brain mitochondria. Manganese superoxide dismutase (Mn-SOD; SOD2), a primary mitochondrial antioxidant enzyme, scavenges superoxide radicals and its overexpression provides neuroprotection. However, the regulatory mechanism of Mn-SOD expression during cerebral ischemia and reperfusion is still unclear. In this study, we identified the signal transducer and activator of transcription 3 (STAT3) as a transcription factor of the mouse Mn-SOD gene, and elucidated the mechanism of O2·− overproduction after transient focal cerebral ischemia (tFCI). We found that Mn-SOD expression is significantly reduced by reperfusion in the cerebral ischemic brain. We also found that activated STAT3 is usually recruited into the mouse Mn-SOD promoter and upregulates transcription of the mouse Mn-SOD gene in the normal brain. However, at early postreperfusion periods after tFCI, STAT3 was rapidly downregulated, and its recruitment into the Mn-SOD promoter was completely blocked. In addition, transcriptional activity of the mouse Mn-SOD gene was significantly reduced by STAT3 inhibition in primary cortical neurons. Moreover, we found that STAT3 deactivated by reperfusion induces accumulation of O2·− in mitochondria. The loss of STAT3 activity induced neuronal cell death by reducing Mn-SOD expression. Using SOD2−/+ heterozygous knock-out mice, we found that Mn-SOD is a direct target of STAT3 in reperfusion-induced neuronal cell death. Our study demonstrates that STAT3 is a novel transcription factor of the mouse Mn-SOD gene and plays a crucial role as a neuroprotectant in regulating levels of reactive oxygen species in the mouse brain.
Introduction
Brain injuries such as ischemia, reperfusion, and stroke increase superoxide anions (O2·−) and reactive oxygen species (ROSs) in mitochondria. These ROSs are critical mediators of mitochondrial-dependent apoptotic pathways (Chan et al., 1996; Green and Reed, 1998; Sugawara et al., 1999; Chan, 2004, 2005). Manganese-containing superoxide dismutase (Mn-SOD or SOD2), a specific antioxidant enzyme for superoxide, is a primary cellular defense enzyme involved in protecting cells from oxidative stress (Chan, 1996). In our previous reports, Mn-SOD-deficient mice (SOD2−/+) have shown increased infarct volume and apoptosis after cerebral ischemia (Murakami et al., 1998; Fujimura et al., 1999; Kim et al., 2002). Overexpression of Mn-SOD provided neuroprotection following cerebral ischemic reperfusion (Maier et al., 2006).
The Mn-SOD gene is highly conserved among many species (Marlhens et al., 1985), and its expression is highly regulated and inducible by numerous stimuli in various cells and tissue, in contrast to constitutively expressed copper/zinc-SOD (Wong and Goeddel, 1988; Visner et al., 1990; Dougall and Nick, 1991). The proximal promoter region of the Mn-SOD gene has multiple CG motifs (without TATA or CAAT boxes) and binding sites for specificity protein 1 (SP-1), which is a transcriptional activator in the constitutive expression of the SOD2 gene (Wan et al., 1994; Yeh et al., 1998; Xu et al., 2002). Also, intronic enhancer activated by NF-κB is responsible for cytokine-induced transcription (Xu et al., 1999; Dhar et al., 2004). However, in brain injuries such as ischemic reperfusion and stroke, regulation of Mn-SOD transcription has not been fully elucidated, although its level of expression is critical for neuroprotection. Interestingly, our promoter analysis revealed the sites most abundant for the potential of signal transducer and activator of transcription 3 (STAT3) binding in the promoter region of mouse Mn-SOD genes.
STAT3 is a transcription factor, as well as an intracellular signal transducer, activated by cytokines, growth factors, and receptor- or nonreceptor-tyrosine kinases (Darnell, 1997; Levy and Lee, 2002). Tyrosine phosphorylation of STAT3 at Y705 is necessary for STAT3 activation. Phosphorylated STAT3 forms dimers, translocates to the nucleus, binds to the specific promoters of target genes, and induces gene expression (Bromberg and Darnell, 2000).
Six STATs have been identified to date (Levy and Darnell, 2002), but only STAT3 is involved in neuroprotection against various types of brain damage, including cerebral ischemia. STAT3 activation causes secretoneurin (Shyu et al., 2008), estradiol (Dziennis et al., 2007), and interleukin-6 (IL-6) (Yamashita et al., 2005) to promote neuroprotection against cerebral ischemic insult and IGF-1 to rescue neurons (Yadav et al., 2005). In contrast, phosphorylation of STAT1 contributes to ischemic brain injury (Takagi et al., 2002). STAT3 activation and Mn-SOD expression are involved in neuroprotection against ischemia, but their relationship is still unclear. In this study, we examined the role of STAT3 as a neuroprotectant, focusing on Mn-SOD expression and O2·− production after reperfusion in mouse cerebral ischemic injury.
Materials and Methods
Animals.
Experiments with animals were performed in accordance with National Institutes of Health guidelines and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. CD1 male mice (30–35 g, 2 months old) were purchased from Charles River Laboratories. The SOD2 gene knock-out mutants (heterozygous) with a CD1 background were backcrossed with CD1 mice for at least 10 generations and genotypes were determined as previously described (Li et al., 1998). Littermate wild-type (WT) mice with the identical genetic background were used in each experiment.
Primary cortical neuron culture.
The protocol followed was adapted from a procedure described previously (Copin et al., 1996) with minor modifications. Cortical neurons were prepared from brains of 16-d-old mouse embryos, plated on coated dishes with poly-d-lysine, and cultured in minimum essential medium (Invitrogen) containing glucose, 5% horse serum, glutamine (2 mm), penicillin (50 U/ml), and streptomycin (50 μg/ml). Two to three days later, the medium was changed with Neurobasal medium containing B-27. Neurons were cultured at 37°C in humidified 5% CO2 atmosphere and used after 7–10 d in vitro.
Focal cerebral ischemia.
Male mice (30–35 g) were subjected to 45 min of transient focal cerebral ischemia and reperfusion. An 11.0 mm 6–0 surgical monofilament nylon suture, blunted at the tip, was introduced into the left internal carotid artery through the external carotid artery stump. The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37°C with a homeothermic blanket. After 45 min of middle cerebral artery occlusion (MCAO), blood flow was restored by withdrawal of the nylon suture. The left common carotid artery was exposed and the left external carotid artery and its branches were electrocoagulated. Sham-operated mice did not undergo surgery. Physiologic parameters were monitored throughout the studies and values were the same as previously reported (Fujimura et al., 1999).
Oxygen–glucose deprivation and reoxygenation.
The primary neuronal cell plates were subjected to oxygen–glucose deprivation (OGD) by replacing the medium with buffered salt solution without glucose and were placed in a gas-tight humidified anoxic chamber (PlasLabs) at 37°C for 150 min. Under these conditions PO2 in the chamber was zero after 10 min and PO2 in the medium was ∼15–20 mmHg. After OGD, the cell medium was changed to a neuronal medium with glucose and reoxygenated for 24 h.
STAT3 inhibitor treatment.
To inhibit phosphorylation of STAT3 pharmacologically, we used the STAT3 inhibitor, AG490, purchased from EMD Biosciences. A lower concentration (50 μm in dimethyl sulfoxide [DMSO] in PBS) was applied in an in vitro primary cortical neuron study, rather than the concentration used in previous studies (100 μm in DMSO in PBS) (Wang et al., 2007; Shyu et al., 2008). Lower-range concentrations of AG490 (5 nmol, 10 nmol, and 20 nmol in 2 μl of 50% DMSO in PBS) were injected intracerebroventricularly (bregma: 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep), rather than the concentration used in a previous study (50 nmol in 5 μl of 50% DMSO in PBS) (Chiba et al., 2009). The vehicle consisted of 50% DMSO in PBS.
IL-6 treatment.
To activate STAT3 with the pharmacological approach, we used IL-6 from mouse recombinant purchased from Sigma-Aldrich. Two injections (i.c.v.) of IL-6 (50 ng in 2 μl of PBS) were administered 30 min before and 15 min after MCAO because the half-life of IL-6 in the brain is short (Loddick et al., 1998).
Small interfering RNA transfection.
To implement a STAT3 knockdown molecular approach, we purchased small interfering RNA (siRNA) probes targeted to mouse STAT3 and nontargeting siRNA for use as a control (Qiagen). The target sequences for the mouse-specific STAT3 siRNA mixture were as follows: TTGGGTGAAATTGACCAGCAA (SI01435301), CAGAGGTTCCTCTTTAAATTA (SI01435308), CAGAGGGTCTCGGAAATTTAA (SI01435287), and CAGGCTGATCATCTATATAAA (SI01435294). Nontargeting siRNA (SI03650318) was used as a control in all siRNA transfection experiments. Primary cortical neurons were transfected with HiPerFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Primary cortical neurons grown on 24-well plates (1 × 105 cells/well) or 6 mm dishes (1 × 106 cells/plate) previously coated with poly-d-lysine were treated with 10 nm siRNA per well, and after 48 h of incubation, they were analyzed for various experiments.
Western blot analysis.
Samples were obtained from the cerebral cortex and caudate–putamen (except the hippocampus). Briefly, whole-cell protein extraction was run on a SDS gel, subsequently transferred to a polyvinylidene difluoride membrane, and incubated with primary antibodies for 24 h at 4°C, and then with secondary antibodies for 1 h at room temperature. The primary antibodies used were monoclonal or polyclonal antibodies against p-STAT3 (Y705), p-STAT3 (Ser-727), p-STAT1 (Y701), p-STAT2 (Y689), and STAT3 (1:1000; Santa Cruz Biotechnology), 3-nitrotyrosine (1:1000; Exalpha Biologicals), β-actin (1:5000; Sigma-Aldrich), and Mn-SOD (1:5000; Stressgen). The signal was then detected with horseradish peroxidase-conjugated IgG using a chemiluminescent kit (Amersham Biosciences).
RT-PCR analysis.
Total RNA was prepared from the ipsilateral hemisphere of each mouse injected with AG490 or the vehicle using the protocol supplied with the Micro-to-Midi Total RNA Purification System (Invitrogen). For RT-PCR analysis, a SuperScript One-Step RT-PCR kit with Platinum Taq (Invitrogen) was used. The following primer sequences (5′–3′) were designed based on GenBank accession number L35525: Mn-SOD, ATGTTGTGTCGGGCGGCG and AGGTAGTAAGCGTGCTCCCACACG. As a control, β-actin was amplified with the following primer sequences (5′–3′): ACCCACACTGTGCCCATCTAA and GCCACAGGATTCCATTACCCAA. The mixtures were subjected to RT-PCR on a thermal cycler (Mastercycler Gradient; Eppendorf). RT-PCR conditions for Mn-SOD were 55°C for 30 min, 94°C for 2 min, 35 cycles of 94°C for 15 s, 60°C for 30 s, 68°C for 1 min, and then 68°C for 5 min.
Chromatin immunoprecipitation assay.
Chromatin isolation and chromatin immunoprecipitation (ChIP) assay were performed according to the manufacturer's protocol using a commercially available kit (EZ-Zyme Chromatin prep kit, EZ-ChIP; Millipore). Briefly, after fixation of brain tissue with 1% formaldehyde, each soluble chromatin was digested and isolated using EZ-Zyme lysis buffer and EZ-Zyme enzymatic cocktail, 4 × 106 cells that were isolated from chopped mouse brain tissue. The chromatin solution was diluted 10-fold with ChIP dilution buffer and precleared with protein G agarose/salmon sperm DNA/preimmune serum. The precleared chromatin solution was divided and used in immunoprecipitation assays with a phospho-STAT3 antibody. Following multiple washes, the antibody-protein-DNA complex was eluted from beads. After reversal cross-link incubation, protein and RNA were removed by proteinase K and RNase A. Purified DNA was subjected to PCR with primers specific for several putative STAT3-binding sites upstream of the transcriptional start site. The sequences of the PCR primers used are as follows: F1 forward, 5′-GAAGACCGCTTTGACATCAGCCAT-3′, R1 reverse, 5′-AATGCTATGGTGGTGTGGTGGCACAT-3′; F2 forward, 5′-ATGTGCCACCACACCACCATAGCATT-3′, R2 reverse, 5′-ATGGCAGTATCCAGGCAGGC-3′; F3 forward, 5′-GCCTGCCTG-GATACTGCCAT-3′, R3 reverse, 5′-GCAGAAGGCAGTTGGCTGAGT-3′; F4 forward, 5′-ACTCAGCCACTGCCTTCTGC-3′, R4 reverse, 5′-TGAGACTGGGGTTGCGGACT-3′; F5 forward, 5′-AGTCCGCAACCCCAGTCTCA-3′, R5 reverse, 5′-AAATTGGTAGAGGCCGCGTGC-TT-3′; F6 forward, 5′-AAGCACGCGGCCTCT-ACCAATTT-3′, R6 reverse, 5′-ATTGAGGTTT-ACACGACCGCTGCT-3′.
Cloning and construction of the mouse Mn-SOD promoter.
A 1779 bp length of the mouse Mn-SOD promoter region in exons 1–2 and 5′ flank of the mouse Mn-SOD gene (GenBank accession number: L35525) was generated by PCR using genomic mouse DNA isolated from brain tissue. The PCR primers used for the (−1779 to −1 bp) region in 5′ flank of the mouse Mn-SOD gene were 5′-GAAGACCGCTTTGACATCAGCCAT-3′ (forward) and 5′-ATTGACGTTTACACGACCGCTGCT-3′ (reverse). Using a TOPO TA Cloning kit (Invitrogen), EcoRI restriction enzyme sites were added on both ends of the PCR product. The pGLu-Basic vector (New England BioLabs) was digested with EcoRI (New England BioLabs) and ligated with the PCR product by T4 DNA Ligase (Fermentas). The final constructs were confirmed by DNA sequencing using pGLu-Basic Sequencing Primers (New England BioLabs). All plasmids were prepared with Qiagen Midiprep kits, and DNA quality and quantity were determined by both spectrophotometry and visual inspection of agarose gels.
Transient transfection and luciferase activity assay.
HEK293T cells or primary cortical neurons were cultured in 24-well plates or 24-well plates previously coated with poly-d-lysine and transfected using Lipofectamine LTX (Invitrogen). The HEK293T cells were transfected when the cultures reached 60–70% confluency, whereas primary neurons were plated and transfected at a density of 1 × 105/well. Two hundred fifty nanograms of pGLu–Mn-SOD promoter reporter DNA were used per well. Transfection took place in the existing media (for neurons, Neurobasal medium with B27; for HEK293T cells, DMEM with 10% fetal bovine serum). After 24 h of incubation, DNA and Lipofectamine LTX were washed off and fresh medium was applied. Then, the cells were treated with 50 μm AG490 or 10 nm siRNA for STAT3 or nontargeting siRNA per well. Cells were incubated for 2 more days after siRNA transfection and subsequently analyzed for luciferase activity. A Gaussia Luciferase Assay kit (New England BioLabs) was used to detect Gaussia luciferase activity from cell culture supernatants, according to the protocol provided by the manufacturer through an Lmax Microplate Luminometer (Molecular Devices). All experiments were performed in quadruplicate and were repeated at least five times.
Nuclear protein extraction and electrophoretic mobility shift assay.
Nuclear extract isolation and electrophoretic mobility shift assay (EMSA) were performed according to the manufacturer's protocol using a commercially available kit (Nuclear Extraction kit; BIOSOURCE International, and EMSA kit; Invitrogen). Briefly, 5 × 106 cells that were isolated from chopped mouse brain tissue or from primary cortical neurons were lysed with hypotonic cell lysis buffer. After washing and centrifugation using complete nuclear wash buffer, the nuclei were extracted using extraction buffer. Normalized nuclear extracts containing ∼5 μg of protein were incubated with a double-stranded oligonucleotide probe using 5× binding buffer. The oligonucleotides containing the putative STAT3 binding sites in the mouse Mn-SOD promoter are as follows: 1-up, 5′-CTGCCTTCTGCTTTCTTAGTGTTT-3′, 1-down, 5′-AAACACTAAGAAAGCAGAAGGCAG-3′; 2-up, 5′-CCCATTAAGTAGAAGAGGAAAGCT-3′, 2-down, 5′-AGCTTTCCTCTTCTACTTAATGGG-3′; 3-up, 5′-GCTGTATAATTGTTATGGAAACAT-3′, 3-down, 5′-ATGTTTCCATAACAATTATACAGC-3′; 4-up, 5′-GATAGCCACTGCTTCTTAGACTAA-3′, 4-down, 5′-TTAGTCTAAGAAGCAGTGGCTATC-3′; 5-up, 5′-CATTTGGTCGAGTGGGTTGGT-3′, 5-down, 5′-ACCAACCCACTCGACCAAATG-3′; 6-up, 5′-CACTGAATGTTGCAGAGTTTA-3′, 6-down, 5′-TAAACTCTGCAACATTCAGTG-3′; 7-up, 5′-GCAGAGTTTAATCAAGAAAGATGA-3′, 7-down, 5′-TCATCTTTCTTGATTAAACTCTGC-3′; 8-up, 5′-TAGTTAACTGGCAAGCTGCACCCG-3′, 8-down, 5′-CGGGTGCAGCTTGCCAGTTAACTA-3′; 9-up, 5′-CCATAATTCTGACCAGCAGCAGAG-3′, 9-down, 5′-CTCTGCTGCTGGTCAGAATTATGG-3′; 10-up, 5′-CCTTGGCTTTCCGGAGGAAAGTCT-3′, 10-down, 5′-AGACTTTCCTCCGGAAAGCCAAGG-3′; 11-up, 5′-CAGGGTTTCCCAGAAGCAGGAGTA-3′, 11-down, 5′-TACTCCTGCTTCTGGGAAACCCTG-3′; 12-up, 5′-GGACCGGGGTTCCCCGAGGCGGGC-3′, 12-down, 5′-GCCCGCCTCGGGGAACCCCGGTCC-3′. At the end of the incubation period, 2 μl of 6× EMSA gel-loading solution was added to each of 10 μl of reaction mixture. DNA–protein complexes were separated by electrophoresis using a nondenaturing polyacrylamide gel. After staining with SYBR Green EMSA, the gel was imaged using an emission filter: 526 SP Fluorescent, Cy 2, Alexa Fluor 488; laser: Green 532, and documented. After washing two times, the gel was stained again with SYPRO Ruby EMSA, after which the gel was imaged using an emission filter: 670 BP 30 Cy 5; laser: Red 633.
Determination of infarction.
Twenty-four hours after reperfusion, the brains were sliced coronally at 1 mm intervals. Individual slices were then incubated in 2% 2,3,5-triphenyltetrazolium chloride in 0.1 mol/L PBS (pH adjusted to 7.4) and warmed to 37°C, followed by 3.7% formalin. General processes and calculation of total infarct volume were performed according to a previous report (Swanson et al., 1990). The infarct area was quantified by an image analysis system (Bio-Rad Laboratories).
Cell death assay.
Cell viability was quantified by a standard measurement of lactate dehydrogenase (LDH) release using a LDH assay kit (BioVision). The amount of extracellular LDH was measured in an aliquot of the medium overlying the cells and a general procedure was followed using the manufacturer's guidelines. Cell viability was also assessed in the medium with a cell proliferation reagent using a WST-1 assay kit (Roche Diagnostics) following the manufacturer's guidelines.
In situ detection of O2·− production.
The early production of O2·− in cerebral ischemia was investigated using hydroethidine (HEt) as in a previously described method (Murakami et al., 1998). HEt solution (200 ml; 1 mg/ml in PBS) was administered intravenously 15 min before induction of ischemia as described. Fluorescence was assessed microscopically at excitation = 355 nm and emission > 415 nm for HEt detection or at excitation = 510–550 nm and emission > 580 nm for ethidium detection.
Statistical analysis.
All results were obtained from 3–5 independent experiments and are presented as mean ± SEM. Data are expressed using Student's t test. Differences were considered statistically significant at a p value <0.05.
Results
Mn-SOD expression is downregulated by ischemic reperfusion in a mouse MCAO model
To investigate the change in Mn-SOD expression after ischemic reperfusion, we examined the protein level of Mn-SOD with an anti-Mn-SOD antibody using Western blot analysis (Fig. 1A). Mice were subjected to 45 min of transient MCAO followed by 1, 3, 6, and 12 h of reperfusion (n = 4 each), and then samples were obtained from the cerebral cortex and caudate–putamen (except the hippocampus). We found that the protein level of Mn-SOD rapidly decreased in the cerebral ischemic cortex, within 1–3 h of reperfusion (Fig. 1A,B), and the mRNA expression level of Mn-SOD also rapidly decreased at early postischemic reperfusion periods (Fig. 1C,D). We also found that the protein level of Mn-SOD was strongly decreased in the primary cortical neurons subjected to 3 h of reoxygenation after OGD for 2.5 h (Fig. 1E). These decreases in mRNA expression of Mn-SOD, as well as the protein level, caused by reperfusion indicate that transcription of the Mn-SOD gene may be downregulated by factors under ischemic reperfusion conditions.
Figure 1.

Mn-SOD expression is significantly reduced by ischemic reperfusion in a mouse MCAO model. A, Western blot analysis of the protein level of Mn-SOD in mouse brains after 1 and 3 h of reperfusion following MCAO with an antibody against anti-Mn-SOD antibody. Whole-cell protein extracts were obtained from the cerebral cortex and caudate–putamen (except the hippocampus). B, Summary graph depicting the change in the Mn-SOD protein level relative to β-actin. *p < 0.05 (n = 4 per group). C, The mRNA level of Mn-SOD in mouse brains after 1 and 3 h of reperfusion following MCAO was determined by RT-PCR using a specific primer against the mouse Mn-SOD gene. Total RNA was prepared from the cerebral cortex and caudate–putamen (except the hippocampus), and equal RNA loading was confirmed using mouse β-actin-specific primers. D, Summary graph depicting the change in Mn-SOD mRNA levels relative to β-actin. *p < 0.05 (n = 4 per group). E, Western blot analysis of the protein level of Mn-SOD in primary cortical neurons subjected to reoxygenation 3 h after OGD for 2.5 h. IR, Ischemic reperfusion; S, sham; O.D., optical density; OR, oxygen–glucose deprivation/reoxygenation; C, control.
STAT3 is significantly downregulated by reperfusion and is involved in Mn-SOD expression in a mouse MCAO model
To investigate which mediator regulates transcription of the Mn-SOD gene during ischemic reperfusion in mouse brains, we explored the promoter region of the mouse Mn-SOD gene. Interestingly, we found many putative STAT3 binding sites in the promoter region (−1779 to −1 bp) of the gene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). It is well known that STAT3 plays the role of transcriptional regulator in the expression of various genes, as well as that of a signal transducer in response to various cellular stresses. To test the possibility of transcriptional regulation by STAT3 in Mn-SOD genes, we first investigated STAT3 activity after reperfusion in the cerebral ischemic mouse brains. It is well known that phosphorylation of STAT3 at Tyr-705 is more important than phosphorylation of STAT3 at Ser-727 for the dimerization and translocation of STAT3 and its role as a transcription factor. We found that phosphorylation of STAT3 at Tyr-705 is significantly decreased after 1 h of reperfusion in the cerebral ischemic brains (Fig. 2A,B). However, phosphorylation of STAT3 at Ser-727 was not changed after reperfusion (Fig. 2A). Also, we detected that STAT3 was localized in the cytoplasm rather than in the nucleus of cortical neurons under ischemic reperfusion conditions, compared with STAT3 in the nucleus under normal conditions in the cerebral cortex (Fig. 2C). Unlike STAT3, phosphorylation of STAT1 (Y701) was increased rather than decreased in response to reperfusion after cerebral ischemia (Fig. 2A). We also found that phosphorylation of STAT2 (Y689) was increased in response to reperfusion (data not shown). Next, to clarify the relationship between reduction in Mn-SOD expression and STAT3 deactivation at early postischemic reperfusion periods, we directly injected (i.c.v.) 5 nmol, 10 nmol, and 20 nmol of AG490, a STAT3 inhibitor, into the mouse brains and examined the level of Mn-SOD expression. Because gene knock-out of STAT3 is lethal to mouse embryos (Takeda et al., 1997), we used a pharmacological approach for STAT3 inhibition using AG490, or a molecular approach for STAT3 knockdown using siRNA transfection. As shown in Figure 3, A and B, the protein level of Mn-SOD and the mRNA expression level of Mn-SOD rapidly decreased after 3 h in cerebral cortices injected with AG490 in a dose-dependent manner. In addition, the protein level of Mn-SOD was strongly decreased in primary cortical neurons transfected with STAT3-specific siRNA or treated with AG490 (Fig. 3C). These results indicate that STAT3 activity is involved in Mn-SOD expression in the cerebral ischemic brain.
Figure 2.

STAT3 is significantly downregulated by ischemic reperfusion in a mouse MCAO model. Whole-cell protein extracts were obtained from the cerebral cortex and caudate–putamen (except the hippocampus) of male mouse brains after 1, 3, 6, and 12 h of reperfusion following MCAO. A Western blot was performed with an antibody against phospho-STAT3 (Y705), phospho-STAT3 (Ser-727), phospho-STAT1 (Y701), and total STAT3. A, Representative phospho-STAT3 (Y705), phospho-STAT3 (Ser-727), phospho-STAT1 (Y701), and total STAT3 blots and a β-actin blot showing equal protein loading. B, Summary graph depicting the change in phospho-STAT3 relative to total protein loading. *p < 0.05 (n = 4 per group). C, Phosphorylation status of STAT3 (red) and exhibition of the neuronal marker NeuN (green) from ischemic regions (cortex) on the contralateral and ipsilateral sides shown by immunohistochemistry analysis. Scale bar, 20 μm. IR, Ischemic reperfusion; S, sham; O.D., optical density; NeuN, neuron-specific nuclear protein.
Figure 3.
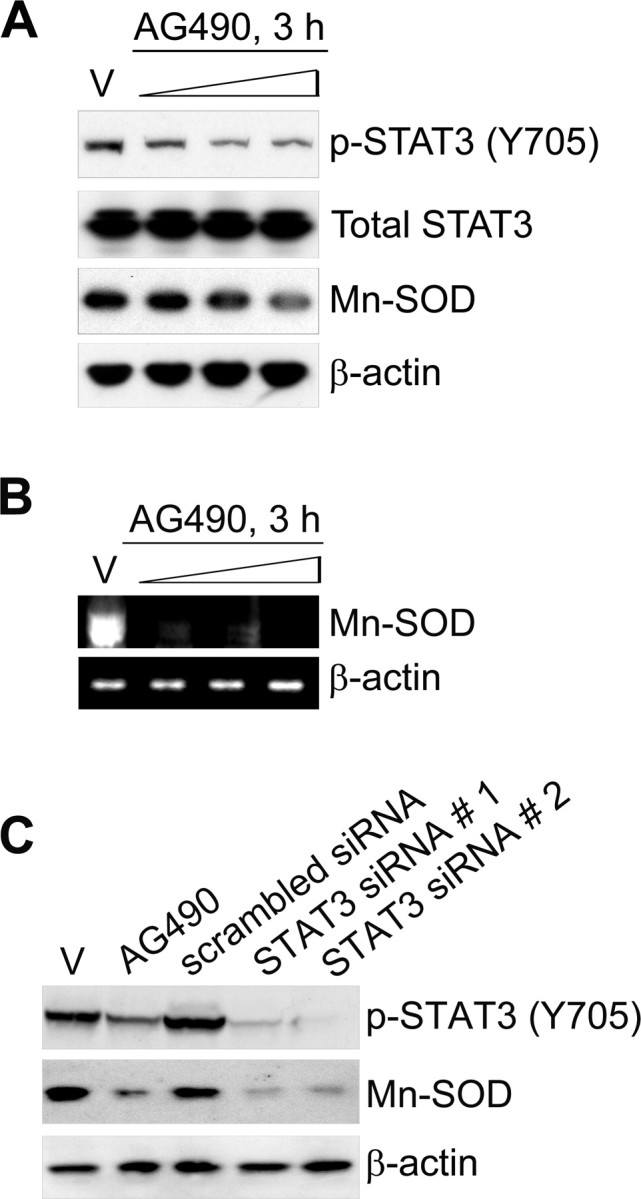
Inhibition of STAT3 reduces Mn-SOD expression in mouse brains and cortical neurons. A, The protein levels of Mn-SOD, total STAT3, and phospho-STAT3 (Y705) in the cerebral cortex and caudate–putamen (except the hippocampus) of male mouse brains injected intracerebroventricularly with the vehicle (V) (50% DMSO in PBS) or AG490 (5 nmol, 10 nmol, and 20 nmol in 50% DMSO in PBS) were determined by Western blot with antibodies against anti-Mn-SOD, anti-STAT3, and anti-phospho-STAT3 (Y705). B, The mRNA level of Mn-SOD in mouse brains injected intracerebroventricularly with the vehicle (50% DMSO in PBS) or AG490 (5 nmol, 10 nmol, and 20 nmol in 50% DMSO in PBS) was determined by RT-PCR using a specific primer against the mouse Mn-SOD gene. Total RNA was prepared from the cerebral cortex and caudate–putamen (except the hippocampus) and equal RNA loading was confirmed using mouse β-actin-specific primers. C, Western blot analysis of the protein levels of Mn-SOD and phospho-STAT3 (Y705) in primary cortical neurons transfected with STAT3-specific siRNA for 24 h or treated with 50 μm AG490 for 3 h.
Recruitment of STAT3 into the promoter of the mouse Mn-SOD gene is diminished by ischemic reperfusion
We tested whether STAT3 plays a role as a transcriptional regulator of Mn-SOD gene expression in the cerebral ischemic brain. First, we examined recruitment of STAT3 into the mouse Mn-SOD promoter using a ChIP assay of chromatin samples from the brain cortices of sham-operated mice or in mice that underwent MCAO and reperfusion using specific pairs of primers spanning the STAT3 binding sites in the Mn-SOD promoter. This promoter (−1779 to −1 bp) was divided into six sections (supplemental Fig. 1, available at www.jneurosci.org as supplemental material): I (−1779 to −1420 bp), II (−1419 to −1122 bp), III (−1121 to −831 bp), IV (−830 to −557 bp), V (−556 to −213 bp), and VI (−212 to −1 bp). We found that in chromatin from the sham-operated mouse brain cortices, STAT3 was strongly recruited into regions IV (−830 to −557 bp) and V (−556 to −213 bp) in the Mn-SOD promoter. However, in chromatin from the MCAO/reperfused cortices, recruitment of STAT3 was completely blocked (Fig. 4). According to many reports, regions IV (−830 to −557 bp) and V (−556 to −213 bp) are critical promoter regions of the mouse Mn-SOD gene, because these regions contain the binding sites of SP-1 and NF-κB, which are transcription factors of Mn-SOD gene expression (Fig. 5A). Our results show that phosphorylated STAT3 is usually recruited into the promoter of mouse Mn-SOD under normal conditions in the brain. However, this recruitment is blocked by reperfusion in cerebral ischemic injury.
Figure 4.
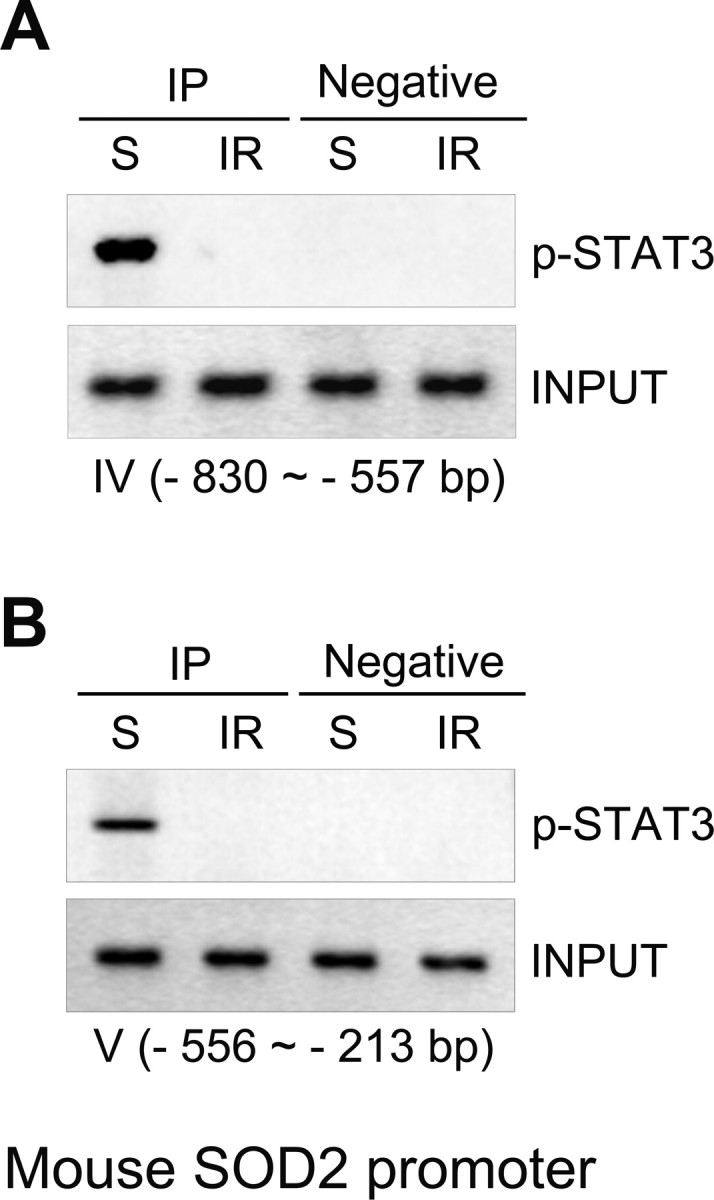
Recruitment of STAT3 into the promoter of the mouse Mn-SOD gene is diminished by ischemic reperfusion. A, B, Recruitment of STAT3 into the promoter of the mouse Mn-SOD gene in chromatin from sham-operated mouse cortices or from mouse cortices subjected to 3 h of reperfusion after MCAO was determined by ChIP analysis using mouse Mn-SOD promoter-specific pairs of primers spanning the STAT3 binding sites. IP, Immunoprecipitation; S, sham; IR, ischemic reperfusion.
Figure 5.

STAT3 is a potential transcriptional regulator in the promoter of the mouse Mn-SOD gene. A, Schematic diagram for putative STAT3 binding sites in the promoter of the mouse Mn-SOD gene. B, Transcriptional activity of the mouse Mn-SOD gene was determined by luciferase assay. The HEK293T cells transfected with pGLu–Mn-SOD luciferase construct were treated with 50 μm AG490 and 50 μm JSI-124 for 24 h and evaluated by luciferase activity. Data are mean ± SEM. *p < 0.001, #p < 0.05 versus control. C, Transcriptional activity of the mouse Mn-SOD gene in response to changes in STAT3 activity was determined by luciferase assay in HEK293T cells transfected with STAT3-specific siRNA. Data are mean ± SEM. *p < 0.001, #p < 0.05 versus control. D, Transcriptional activity of the mouse Mn-SOD gene in response to changes in STAT3 activity was determined by luciferase assay in primary cortical neurons transfected with STAT3-specific siRNA or treated with 50 μm AG490. Data are mean ± SEM. *p < 0.001, #p < 0.05 versus control. E, Western blot analysis of the protein level of total STAT3 in HEK293T cells transfected with STAT3-specific siRNA. F, Western blot analysis of the protein level of total STAT3 in primary cortical neurons transfected with STAT3-specific siRNA.
STAT3 is a potential transcription factor of the mouse Mn-SOD gene
To clarify whether STAT3 recruited into the mouse Mn-SOD promoter regulates transcriptional activity of the Mn-SOD gene, we checked the transcriptional activity of the Mn-SOD promoter using a luciferase assay. First, a length of 1779 bp for the promoter region of the mouse Mn-SOD gene was generated by PCR using genomic mouse DNA isolated from mouse brain tissue. Using a TOPO TA Cloning kit, EcoRI restriction enzyme sites were added on both ends and ligated with the pGLu-Basic vector by T4 DNA ligase. Using the above mouse pGLu–Mn-SOD luciferase construct, we evaluated the transcriptional activity of the mouse Mn-SOD promoter. We transfected the mouse pGLu–Mn-SOD luciferase construct into HEK293T cells, which have a high transfection efficiency. After 24 h of incubation, the cells were treated with 50 μm AG490 and another STAT3 inhibitor, JSI-124 (50 μm), for 24 h. To confirm a more direct effect of STAT3 inhibition, we used JSI-124, which is a novel selective inhibitor of Jak2/STAT3 signaling. As shown in Figure 5B, the luciferase activity of pGLu–Mn-SOD in cells treated with both STAT3 inhibitors was significantly decreased. In addition, with STAT3 inhibition using transfection with STAT3-specific siRNA, the luciferase activity was also significantly decreased in the HEK293T cells transfected with pGLu–Mn-SOD (Fig. 5C). All STAT3-specific siRNA sequences designed to target the mouse gene have >80% homology with the human gene, and two of them, which were used in our study, have 91% and 81% homology with the human gene. In our preliminary screening for efficacy of STAT3-specific siRNA sequences against both mouse neurons and human HEK293T cells, we selected two STAT3-specific siRNA sequences that have high efficacy for STAT3 gene knockdown in both cells. The transfection efficiency of STAT3-specific siRNA in each cell was confirmed by quantification of the total protein level for STAT3 using an anti-STAT3 antibody in Western blot analysis (Fig. 5E). To confirm whether STAT3 also regulates transcriptional activity of the Mn-SOD promoter in cortical neurons, we transfected STAT3-specific siRNA into primary cortical neurons previously transfected with pGLu–Mn-SOD and evaluated the luciferase activity. As shown in Figure 5D, luciferase activity was significantly decreased by STAT3 inhibition even though the level of decrease in the primary cortical neurons was less than in the HEK293T cells. This difference between the primary cortical neurons and HEK293T cells was a result of the low transfection efficiency of primary cortical neurons. The transfection efficiency of STAT3-specific siRNA in each cell was confirmed by quantification of the total protein level for STAT3 using the anti-STAT3 antibody in Western blot analysis (Fig. 5F). These results show that STAT3 regulates transcription activity of the Mn-SOD promoter.
Putative binding sites of STAT3 in the promoter of the mouse Mn-SOD gene
To identify the real STAT3 binding sites of the mouse Mn-SOD promoter, we performed EMSA using nuclear extracts from the sham-operated mouse brain cortices and from the cortices of mice that underwent 45 min of MCAO/3 h of reperfusion. We numbered and designed the oligonucleotide probes for STAT3 putative binding in regions IV and V of the mouse Mn-SOD promoter (Fig. 5A). In the sham-operated cortices, STAT3-DNA bindings on #3, 7, 10, and 12 motifs of the Mn-SOD promoter were detected and were supershifted by an anti-phospho-STAT3 (Y705) antibody (Fig. 6A). However, these bindings were significantly diminished in the cortices of mice subjected to 3 h of reperfusion/MCAO. In the primary cortical neurons, the STAT3-DNA bindings on #3, 7, 10, and 12 motifs were also detected and supershifted by the anti-phospho-STAT3 (Y705) antibody and were diminished by STAT3 inhibition using 50 μm AG490 (Fig. 6C).
Figure 6.

Active STAT3 binds on the putative motifs in the Mn-SOD promoter and its binding is blocked by ischemic reperfusion. A, Activity of STAT3 binding to DNA in the Mn-SOD promoter was determined by EMSA analysis using nuclear extracts from sham-operated mouse brain cortices or from mouse cortices subjected to 3 h of reperfusion after MCAO. Using 12 pairs of the STAT3 putative binding probes, the putative STAT3 binding sites are selected and identified. Using a phospho-STAT3 (Y705) antibody, the supershift (SS) of the STAT3-DNA binding complex was confirmed. B, Quantitative graph depicting the change in STAT3/DNA binding level after ischemic reperfusion relative to shams. *p < 0.05 (n = 3 per group). C, In primary cortical neurons, activity of STAT3 binding to DNA in the Mn-SOD promoter was determined by EMSA analysis using nuclear extracts from cells treated with the vehicle or 50 μm AG490 for 3 h. Using a phospho-STAT3 (Y705) antibody, the super shift of the STAT3-DNA binding complex was confirmed. D, Quantitative graph depicting the change in the STAT3/DNA binding level after STAT3 inhibition relative to control. *p < 0.05 (n = 3 per group). S, Sham; IR, ischemic reperfusion.
Inhibition of STAT3 by ischemic reperfusion increases generation of O2·−
Our results confirm that Mn-SOD expression is downregulated by STAT3 inhibition after cerebral ischemic reperfusion, which increases O2·− in mitochondria (Chan, 1996). Thus, we investigated whether the decrease in Mn-SOD expression may enhance the increase in superoxide radical generation after ischemic reperfusion. As shown in Figure 7A, oxidized HEt signals were strongly observed in the ischemic cortices of mice subjected to 45 min of transient MCAO followed by 3 h of reperfusion. In our results, STAT3 was significantly downregulated at early postischemic reperfusion periods (Fig. 2). Therefore, we first examined the relationship between STAT3 deactivation and O2·− generation after reperfusion in cerebral ischemia. We directly treated cortical primary neurons with AG490 and determined O2·− production using HEt staining. As shown in Figure 7B, oxidized HEt signals were strongly observed in the neurons treated with AG490 for 3 h. This result indicates that STAT3 inhibition induces O2·− formation. Next, to clarify whether the increase in O2·− in mitochondria after ischemic reperfusion was caused by a decrease in Mn-SOD expression by STAT3 deactivation, we quantified the level of protein nitrosylation, a reaction product of peroxynitrite, using a 3-nitrotyrosine antibody after STAT3 inhibition. As shown in Figure 7, C and D, the protein nitrosylation level in mouse cerebral cortices injected with AG490 was strongly increased, and such increase was dose dependent. Moreover, in primary cortical neurons transfected with STAT3-specific siRNA, the protein nitrosylation level was strongly increased (Fig. 7E,F). These results indicate that the increase in O2·− generation under ischemic reperfusion was caused by the decrease in Mn-SOD expression via reperfusion-induced STAT3 deactivation.
Figure 7.
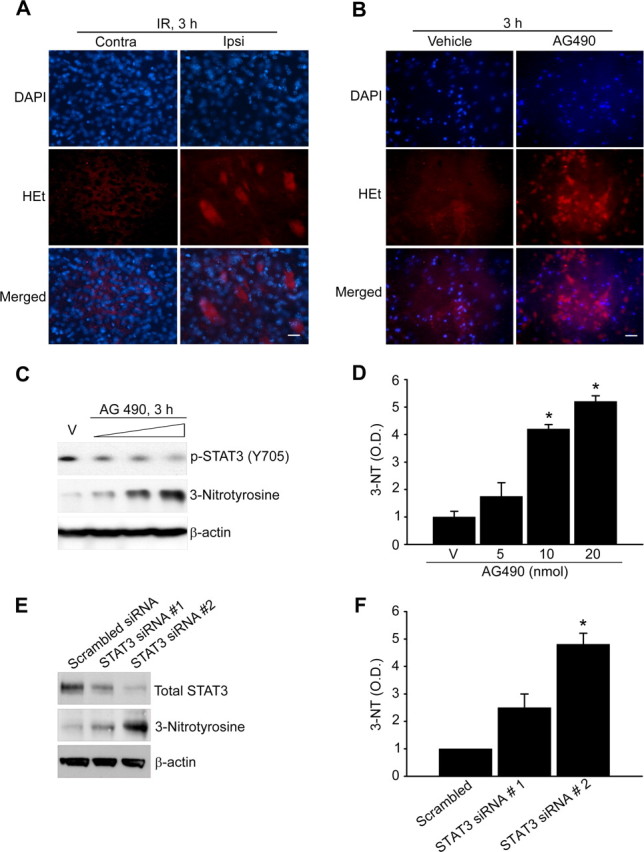
Inhibition of STAT3 by ischemic reperfusion increases generation of O2·−. A, ROS production shown by HEt (red) and 4′,6-diamidino-2-phenylindole (DAPI, blue) staining of ischemic regions (cortex) on the contralateral and ipsilateral sides. Scale bar, 20 μm. B, ROS production shown by HEt (red) and DAPI (blue) staining of primary cortical neurons treated with 50 μm AG490 for 3 h. Scale bar, 20 μm. C, Protein nitrosylation was quantified from Western blots using a 3-nitrotyrosine antibody. Whole-cell protein extracts (30 μg) were obtained from the cerebral cortex and caudate–putamen (except the hippocampus) of male mouse brains injected intracerebroventricularly with the vehicle (V) (50% DMSO in PBS) or AG490 (5 nmol, 10 nmol, and 20 nmol in 50% DMSO in PBS). D, Summary graph depicting changes in 3-nitrotyrosine relative to total protein loading. *p < 0.05 (n = 4 per group). E, Protein nitrosylation was quantified from Western blots using a 3-nitrotyrosine antibody. Whole-cell protein extracts (30 μg) from primary cortical neurons were transfected with STAT3-specific siRNA for 24 h. Transfection efficiency of STAT3-specific siRNA was determined by Western blot analysis using a total STAT3 antibody. F, Summary graph depicting the changes in 3-nitrotyrosine relative to total protein loading. *p < 0.05 (n = 4 per group). O.D., Optical density.
Mn-SOD is a critical neuroprotectant and is a direct target of STAT3 in cell death caused by cerebral ischemic damage
It is well known that excessive ROSs are implicated in brain injury and are mediators of signaling that may lead to apoptosis after cerebral ischemia (Chan, 2001; Dröge, 2002). In our study, the decrease in Mn-SOD expression resulted in overproduction of superoxide radicals during cerebral ischemic reperfusion. To confirm how critical the decrease is in Mn-SOD expression by STAT3 inhibition in reperfusion-induced cell death linked to superoxide overproduction, we measured cell viability related to changes in the Mn-SOD level and superoxide production using primary cortical neurons from SOD2−/+ mice.
In the cerebral cortices of the SOD2 WT mice injected with 10 nmol of AG490, Mn-SOD was significantly downregulated (Fig. 8A,B). However, in the cerebral cortices of the SOD2−/+ heterozygous knock-out mice injected with 10 nmol of AG490, the level of Mn-SOD was not changed. We also compared the level of change in protein nitrosylation for detection of superoxide radical production in response to STAT3 inhibition. Protein nitrosylation was not changed by STAT3 inhibition in the cortices of the SOD2−/+ mice injected with 10 nmol of AG490, compared with a significant increase in the cerebral cortices of the SOD2 WT mice (Fig. 8A,C). We also found the basal level of protein nitrosylation in control cortices of the SOD2−/+ mice was high, even though they were not injected with AG490 (Fig. 8A,C). This was caused by a deficiency in Mn-SOD expression in SOD2−/+ mice. Interestingly, we did not find a significant difference in LDH release between the primary cortical neurons treated with the vehicle and those treated with AG490 for 24 h in the SOD2−/+ group, compared with a significant increase in LDH release in the neurons treated with AG490 in the SOD2 WT group (Fig. 8D). Since the cortical neurons of the SOD2−/+ mice had no significant change in the level of Mn-SOD expression and protein nitrosylation in response to STAT3 inhibition, a difference in the cell death rate between the control and STAT3-inhibition groups was not detected. In addition, we could not detect any significant difference in LDH release between vehicle and AG490 treatment in the primary cortical neurons subjected to 24 h of reoxygenation after 2.5 h of OGD in the SOD2−/+ group, compared with a significant increase in LDH release in the SOD2 WT cortical neurons treated with AG490 and subjected to 24 h of reoxygenation after 2.5 h of OGD (Fig. 8E). We also found that the basal level of LDH release in the control cortical neurons of the SOD2−/+ group was high, even though they were not treated with AG490 (Fig. 8D,E). This was also caused by a deficiency in Mn-SOD expression and a high level of protein nitrosylation in these mice. Our results show that reduced Mn-SOD expression in response to STAT3 inhibition plays a critical role in neuronal cell death linked to superoxide production and that Mn-SOD is a direct target of STAT3 in reperfusion-induced neuronal cell death in ischemic mouse brains.
Figure 8.

STAT3 is an upstream regulator of Mn-SOD in mouse brains and Mn-SOD is a direct target of STAT3 in reperfusion-induced cell death. A, Protein nitrosylation was quantified from Western blots using a 3-nitrotyrosine antibody. The protein level of Mn-SOD and the level of phospho- (Y705) or total STAT3 were determined by Western blots using an anti-Mn-SOD antibody and an anti-phospho- (Y705) or a total STAT3 antibody. Whole-cell protein extracts (30 μg) were obtained after 3 h from the cerebral cortex and caudate–putamen (except the hippocampus) of WT or SOD2−/+ (KO) male mouse brains injected intracerebroventricularly with the vehicle (50% DMSO in PBS) or AG490 (10 nmol in 50% DMSO in PBS). B, Summary graph depicting changes in Mn-SOD relative to total protein loading. *p < 0.05 (n = 4 per group). C, Summary graph depicting changes in 3-nitrotyrosine relative to total protein loading. *p < 0.05 (n = 4 per group). D, Cell death assessed by LDH activity in medium of primary cortical neurons treated with 50 μm AG490 for 24 h from WT or KO mouse brains. Data are mean ± SEM. *p < 0.05 versus control. E, LDH activity in medium of primary cortical neurons of WT or KO mouse brains with or without 50 μm AG490 before exposure to OGD for 2.5 h and reoxygenation for 24 h. Data are mean ± SEM. *p < 0.05 versus control. NS, Not significant.
Inhibition of STAT3 by ischemic reperfusion enhances brain damage and neuronal cell death
To examine the influence of STAT3 dephosphorylation on cerebral ischemic brain damage, we injected 10 nmol (i.c.v.) of the STAT3 inhibitor, AG490, into the mouse brains before MCAO and measured brain infarction volume after 24 h of reperfusion following 45 min of MCAO. We found that the ischemic lesion in the MCA territory of the AG490-treated mice (n = 11) was larger than in the vehicle-treated mice (infarct volume [in mm3]; vehicle vs AG490: 36.6 ± 3.1 versus 64.6 ± 10.07, n = 9), using 2,3,5-triphenyltetrazolium chloride staining (Fig. 9A). Also, we evaluated LDH release from the primary cortical neurons subjected to 24 h of reoxygenation after 2.5 h of OGD with or without AG490. In the neurons pretreated with 50 μm AG490 and subjected to 24 h of reoxygenation after OGD for 2.5 h, LDH release significantly increased approximately twice as much as in the cortical neurons pretreated with the vehicle and subjected to OGD/reoxygenation (Fig. 9B). In addition, we transfected STAT3-specific siRNA into primary cortical neurons and evaluated LDH release after 24 h of incubation. As shown in Figure 9C, LDH release significantly increased in neurons transfected with STAT3-specific siRNA or treated with 50 μm AG490, compared with neurons transfected with scrambled siRNA or treated with the vehicle. In the cell viability assay using the WST-1 reagent, the viability of neurons pretreated with 50 μm AG490 and subjected to 24 h of reoxygenation after OGD for 2.5 h was also significantly decreased by approximately one-half the time for the neurons pretreated with the vehicle and subjected to OGD/reoxygenation (Fig. 9D). Viability was significantly decreased in the neurons transfected with STAT3-specific siRNA or treated with 50 μm AG490, compared with the neurons transfected with scrambled siRNA or treated with the vehicle (Fig. 9E).
Figure 9.
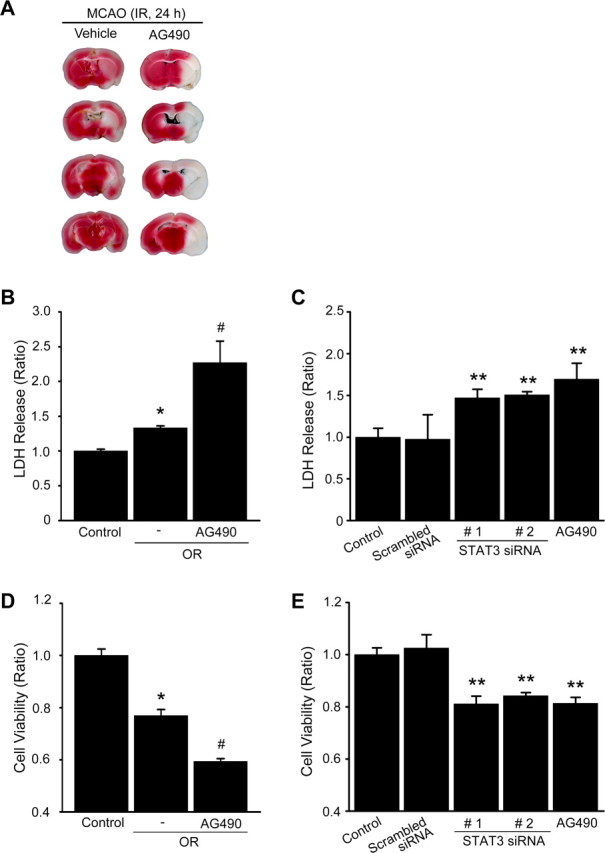
Inhibition of STAT3 by ischemic reperfusion enhances brain damage and neuronal cell death. A, The vehicle (50% DMSO in PBS) and AG490 (10 nmol in 50% DMSO in PBS) were injected intracerebroventricularly before 1 h onset of 45 min of MCAO. The animals were killed after 24 h, and individual brain slices were analyzed to determine infarction using 2,3,5-triphenyltetrazolium chloride staining. B, Cell death assessed by LDH activity in medium of primary cortical neurons with or without 50 μm AG490 before exposure to OGD for 2.5 h and reoxygenation for 24 h. Data are mean ± SEM. *p < 0.01 versus control, #p < 0.05 versus absence of AG490. C, LDH activity in medium of primary cortical neurons transfected with STAT3-specific siRNA or treated with 50 μm AG490 for 24 h. Data are mean ± SEM. **p < 0.05 versus control. D, Cell viability assessed by the WST-1 reagent in medium of primary cortical neurons with or without 50 μm AG490 before exposure to OGD for 2.5 h and reoxygenation for 24 h. Data are mean ± SEM. *p < 0.01 versus control, #p < 0.05 versus absence of AG490. E, Cell viability assay assessed by WST-1 reagent in medium of primary cortical neurons transfected with STAT3-specific siRNA or treated with 50 μm AG490 for 24 h. Data are mean ± SEM. **p < 0.05 versus control. IR, Ischemic reperfusion; OR, oxygen–glucose deprivation/reoxygenation.
Inhibition of STAT3 by ischemic reperfusion induces mitochondrial-dependent apoptosis in mouse brains
To clarify which signal pathway is involved in neuronal cell death by STAT3 inhibition after ischemic reperfusion, we examined the signal cascade in primary cortical neurons subjected to 24 h of reoxygenation after OGD for 2.5 h with or without AG490. It is well known that oxidative stress induces mitochondrial-dependent neuronal cell death in cerebral ischemic injury. First, we checked whether STAT3 deactivation influences cytochrome c release from mitochondria under OGD/reoxygenation conditions. In the cortical neurons pretreated with 50 μm AG490 and subjected to 24 h of reoxygenation after OGD for 2.5 h, cytochrome c release significantly increased in neurons pretreated with the vehicle and subjected to OGD/reoxygenation (Fig. 10A). To examine the effect of STAT3 deactivation on cytochrome c release, we treated the primary cortical neurons with 50 μm AG490 in a time-dependent manner and evaluated cytochrome c release after incubation for each time period. As shown in Figure 10B, in the neurons treated with AG490, cytochrome c release increased significantly after 12 h of incubation, compared with the control cells. Next, we investigated whether STAT3 deactivation induces the mitochondrial-dependent apoptotic pathway after ischemic reperfusion. As shown in Figure 10C, phosphorylation of STAT3 was decreased in the cortical neurons subjected to 12 h of reoxygenation after OGD for 2.5 h and was more significantly decreased in the primary cortical neurons pretreated with AG490 and subjected to OGD/reoxygenation. By enhancing STAT3 deactivation with AG490 under the OGD/reoxygenation systems, apoptotic signaling pathways, such as induction of Bax and cleavage of spectrin, were more strongly enhanced (Fig. 10C). To confirm the effect of STAT3 deactivation on the mitochondrial-dependent apoptotic pathway, we treated the primary cortical neurons with a 50 μm concentration of AG490 in a time-dependent manner. STAT3 deactivated by AG490 also induced Bax and spectrin in a time-dependent manner (Fig. 10D).
Figure 10.

Inhibition of STAT3 by ischemic reperfusion induces mitochondrial-dependent apoptosis in mouse brains. A, Evaluation of the amount of released cytochrome c (Cyto. c) from mitochondria in the cytosolic fraction of primary cortical neurons with or without 50 μm AG490 before exposure to OGD for 2.5 h and reoxygenation for 24 h. Data are mean ± SEM. *p < 0.01 versus control, #p < 0.05 versus absence of AG490. B, Cytochrome c release from mitochondria in medium of primary cortical neurons treated with 50 μm AG490 for 1, 3, 6, and 12 h. Data are mean ± SEM. **p < 0.05 versus control. C, Western blot analysis of the apoptotic signaling pathways Bax and spectrin in primary cortical neurons with or without 50 μm AG490 before exposure to OGD for 2.5 h and reoxygenation for 12 h. D, Western blot analysis of the apoptotic signaling pathways Bax and spectrin in primary cortical neurons treated with 50 μm AG490 for 1, 3, 6, and 12 h. OR, Oxygen–glucose deprivation/reoxygenation.
Discussion
This study demonstrates the role of STAT3 as a transcriptional regulator of Mn-SOD gene expression, as well as a neuroprotectant, and elucidates the molecular mechanism of ROS overproduction after cerebral ischemic injury via Mn-SOD reduction caused by STAT3 inhibition. Although the neuroprotective properties of STAT3 have been studied, this is the first report that shows how STAT3 regulates continuous Mn-SOD expression in neuronal cells and how critical STAT3 activity is for sustaining the cellular defense systems such as regulation of cellular ROS levels.
Our results show that STAT3 and Mn-SOD were significantly downregulated at early postischemic reperfusion periods in mouse cerebral ischemic brains (Figs. 1, 2). STAT3 deactivation gave rise to a decrease in Mn-SOD mRNA levels, as well as Mn-SOD protein levels (Fig. 3). Interestingly, the phosphorylation level of STAT3 (Y705) in mouse cerebral cortices and in primary cortical neurons was very high, even though they were under normal physiological conditions (Figs. 2, 3A,C). This means that STAT3 activity is pivotal in neuronal cell survival as a neuroprotectant. Indeed, we showed that STAT3 inhibition by STAT3-specific siRNA or treatment with AG490 induced neuronal cell death (Fig. 9C,E). Also, the high phosphorylation of STAT3 in normal neuronal cells indicates that STAT3 may regulate an essential gene that is expressed like a housekeeping gene. Mn-SOD is highly expressed like a housekeeping gene in neuronal cells, even though it is an expressed enzyme, highly inducible by various cellular stimuli. This means that the continuous functioning of Mn-SOD is critical for sustaining the defense system against oxidative stress in neuronal cells. It is well known that overexpression of Mn-SOD is neuroprotective and that changes in Mn-SOD expression cause neuronal cell death in response to oxidative stress (Murakami et al., 1998; Fujimura et al., 1999; Kim et al., 2002). However, the detailed mechanism underlying regulation of Mn-SOD expression during cerebral ischemic insults is not fully elucidated. In fact, the relationship between continuously activated STAT3 and highly expressed Mn-SOD in mouse brains has been distinctly revealed in this study.
We found that transcription of Mn-SOD is significantly downregulated by STAT3 inhibition in mouse primary cortical neurons (Fig. 5D). In the analysis of the mouse Mn-SOD promoter, we found the most abundant putative binding motifs of STAT3 (−1779 to −1 bp), which contain many SP-1 motifs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We found that phosphorylated STAT3 is usually recruited into the region (−828 to −210 bp) of the mouse Mn-SOD promoter and upregulates transcription of the Mn-SOD gene under normal physiological conditions (Figs. 4–6). However, STAT3 deactivated by reperfusion after cerebral ischemic injury could not be recruited into the promoter of the Mn-SOD gene and could not sustain the upregulation of Mn-SOD transcription (Figs. 4–6). These phenomena led to a decrease in Mn-SOD expression during cerebral ischemic reperfusion. Our findings strongly suggest that STAT3 is a novel transcriptional activator in the constitutive expression of Mn-SOD as a neuroprotectant, and its activity affects dynamic change in Mn-SOD expression during ischemic reperfusion injury. The relationship between SP-1 and STAT3 in the regulation of Mn-SOD transcription will be elucidated in a future study. Recently, it was reported that STAT3 activation leads to its interaction with SP-1/SP-3 and that their recruitment to the proximal sodium-hydrogen exchanger 3 promoter increased transcription (Su et al., 2009). Our preliminary study showed that phosphorylated STAT3 interacts with SP-1 in the normal mouse cerebral cortex. However the interaction was completely diminished after cerebral ischemic reperfusion (data not shown). This finding leads us to hypothesize that STAT3 phosphorylation under normal conditions may sustain recruitment of SP-1 to the promoter region of the Mn-SOD gene, interacting with SP-1, forming a transcriptional unit like SP-1/STAT3, and then activating Mn-SOD gene transcription. In our EMSA and ChIP analyses, a real STAT3 binding site (−285 to −277 bp) exists close to the next site of SP-1 binding motif (−275 to −266 bp) in the mouse Mn-SOD promoter. Thus, a plausible interaction exists between SP-1 and STAT3 in regulation of Mn-SOD transcription.
Interestingly, in our study, the dynamic change in Mn-SOD expression by STAT3 inhibition after reperfusion resulted in overproduction of O2·− in cerebral ischemic insults. We found that generation of O2·−] after reperfusion was strongly increased in mouse cerebral ischemic injury (Fig. 7A) and that STAT3 inhibition increased the superoxide level in primary cortical neurons (Fig. 7B). Superoxide can rapidly react with nitric oxide, and the byproduct of this reaction is the formation of 3-nitrotyrosine on tyrosine residues of proteins or free tyrosine (Ischiropoulos, 1998). We found a strong increase in protein nitrosylation by STAT3 inhibition in primary cortical neurons, as well as in cerebral cortices (Fig. 7C,E). In addition, we found that the overproduction of superoxide (Fig. 7B) and an increase in 3-nitrotyrosine were caused by inhibition of Mn-SOD expression via STAT3 deactivation (Figs. 3A,C, 7C,E). These findings were confirmed using SOD2−/+-deficient mice, quantifying for 3-nitrotyrosine by STAT3 inhibition (Fig. 8A–C). We could not detect any significant change in the 3-nitrotyrosine level by STAT3 inhibition in the cerebral cortices of the SOD2−/+ heterozygous knock-out mice, compared with a distinct increase in the SOD2 WT mice (Fig. 8C). These results strongly indicate that overproduction of O2·− after ischemic reperfusion injury was caused by Mn-SOD reduction via STAT3 deactivation.
It is well known that ROSs generated from mitochondria are critical intermediaries in oxidative stress-induced neuronal cell death (Chan, 2001). We found a significant increase in cell death, as well as protein nitrosylation caused by STAT3 inhibition, in the primary cortical neurons from the SOD2 WT mice. However, we could not detect any significant difference in cell death and 3-nitrotyrosine between the vehicle-treated group and the STAT3-inhibition group in the primary cortical neurons from the SOD2−/+ mice (Fig. 8D,E). These results strongly indicate that Mn-SOD is a target of STAT3 in reperfusion-induced neuronal cell death linked to O2·− production, and that STAT3 activity is involved in regulation of ROS levels, which trigger neuronal cell death under ischemic reperfusion. With STAT3 inhibition, we also observed the additional increase in infarct volume caused by transient focal cerebral ischemia (Fig. 9A) and the additional increase in neuronal cell death caused by OGD/reoxygenation (Fig. 9B,D). Moreover, STAT3 inhibition caused by STAT3-specific siRNA alone significantly increased neuronal cell death (Fig. 9C,E). We found that STAT3 inhibition increased the release of cytochrome c in primary cortical neurons (Fig. 10B) and that it induced Bax and cleaved spectrin (Fig. 10D). Moreover, a more strongly enhanced induction of Bax, release of cytochrome c, and cleaved spectrin caused by OGD/reoxygenation were observed when we inhibited STAT3 pharmacologically (Fig. 10A,C). These results indicate that deactivated STAT3 triggers mitochondrial-dependent neuronal cell death under ischemic reperfusion and also suggest that STAT3 can be a target of clinical therapy in brain injury such as ischemic reperfusion and stroke. In fact, many recent reports support the neuroprotective effects of STAT3 activation in stroke. STAT3 dephosphorylation caused by administration of an IL-6 receptor antibody increased apoptosis and enlarged infarct size in mice after MCAO (Yamashita et al., 2005). Estradiol-induced STAT3 phosphorylation reduced infarct size in rats after MCAO (Dziennis et al., 2007). Secretoneurin promoted neuroprotection against OGD in primary cortical cells via STAT3-induced antiapoptotic protein expression, as well as reducing infarct size in rats after MCAO via STAT3 activation, and promoting neuronal plasticity (Shyu et al., 2008). However, several studies reported that STAT3 is activated at time points as late as 24 h or even several days after ischemia (Dziennis and Alkayed, 2008), in contrast to our study, which showed immediate STAT3 deactivation within 1–3 h after ischemia. This early response of STAT3 to stimuli can be explained as a typical transcriptional factor reaction. The response of STAT3 at early and late time points is likely to have different roles in ischemic reperfusion. In our study, reduction in Mn-SOD expression by STAT3 inhibition after cerebral ischemic reperfusion was critical in reperfusion-induced neuronal cell death. However, we found that recovery of STAT3 activity using IL-6 rescues Mn-SOD expression after cerebral ischemic reperfusion in mouse brains (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This result implies that STAT3 recovery rescues neuronal cells after cerebral ischemic injury.
In conclusion, we have demonstrated for the first time that STAT3 is a novel transcription regulator that upregulates expression of the Mn-SOD gene as a neuroprotectant under normal conditions in the mouse brain and that STAT3 loses its activity under cerebral ischemic reperfusion conditions and cannot sustain expression of Mn-SOD. Moreover, this study clearly elucidates the mechanism of ROS overproduction after cerebral ischemic injury via a decrease in Mn-SOD expression caused by STAT3 deactivation and provides an explanation for the model of neuronal cell death via STAT3 deactivation after cerebral ischemic injury. Therefore, our study suggests that recovery of STAT3 activity at an early time after reperfusion following cerebral ischemic damage is a potential new approach for molecular targeting therapy in various types of ischemic brain damage including stroke.
Footnotes
This work was supported by Grants P50 NS014543, R01 NS025372, R01 NS036147, and R01 NS038653 from the National Institute of Neurological Disorders and Stroke to P.H.C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health. We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.
References
- Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- Chan P, Niu CS, Cheng JT, Tsao CW, Tsai SK, Hong CY. Trilinolein preserves mitochondria ultrastructure in isolated rat heart subjected to global ischemia through antioxidant activity as measured by chemiluminescence. Pharmacology. 1996;52:216–225. doi: 10.1159/000139386. [DOI] [PubMed] [Google Scholar]
- Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S. Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry. 2009;14:206–222. doi: 10.1038/mp.2008.105. [DOI] [PubMed] [Google Scholar]
- Copin J-C, Reola LF, Chan TYY, Li Y, Epstein CJ, Chan PH. Oxygen deprivation but not a combination of oxygen, glucose, and serum deprivation induces DNA degradation in mouse cortical neurons in vitro: attenuation by transgenic overexpression of CuZn-superoxide dismutase. J Neurotrauma. 1996;13:233–244. doi: 10.1089/neu.1996.13.233. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Dhar SK, Lynn BC, Daosukho C, St. Clair DK. Identification of nucleophosmin as an NF-κB co-activator for the induction of the human SOD2 gene. J Biol Chem. 2004;279:28209–28219. doi: 10.1074/jbc.M403553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougall WC, Nick HS. Manganese superoxide dismutase: a hepatic acute phase protein regulated by interleukin-6 and glucocorticoids. Endocrinology. 1991;129:2376–2384. doi: 10.1210/endo-129-5-2376. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Alkayed NJ. Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev Neurosci. 2008;19:341–361. doi: 10.1515/revneuro.2008.19.4-5.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziennis S, Jia T, Rønnekleiv OK, Hurn PD, Alkayed NJ. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J Neurosci. 2007;27:7268–7274. doi: 10.1523/JNEUROSCI.1558-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice. Implications for the production and role of superoxide radicals. Stroke. 2002;33:809–815. doi: 10.1161/hs0302.103745. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Copin JC, Reola LF, Calagui B, Gobbel GT, Chen SF, Sato S, Epstein CJ, Chan PH. Reduced mitochondrial manganese-superoxide dismutase activity exacerbates glutamate toxicity in cultured mouse cortical neurons. Brain Res. 1998;814:164–170. doi: 10.1016/s0006-8993(98)01082-8. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1998;18:176–179. doi: 10.1097/00004647-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- Marlhens F, Nicole A, Sinet P-M. Lowered level of translatable messenger RNAs for manganese superoxide dismutase in human fibroblasts transformed by SV 40. Biochem Biophys Res Commun. 1985;129:300–305. doi: 10.1016/0006-291x(85)91437-8. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Chiang MF, Chen DC, Su CY, Wang HJ, Liu RS, Tsai CH, Li H. Secretoneurin promotes neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest. 2008;118:133–148. doi: 10.1172/JCI32723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HW, Wang SW, Ghishan FK, Kiela PR, Tang MJ. Cell confluency-induced Stat3 activation regulates NHE3 expression by recruiting Sp1 and Sp3 to the proximal NHE3 promoter region during epithelial dome formation. Am J Physiol Cell Physiol. 2009;296:C13–C24. doi: 10.1152/ajpcell.00263.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19 doi: 10.1523/JNEUROSCI.19-22-j0002.1999. RC39(1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Yu ACH, Chan PH, Sharp FR. Glutamate increases glycogen content and reduces glucose utilization in primary astrocyte culture. J Neurochem. 1990;54:490–496. doi: 10.1111/j.1471-4159.1990.tb01898.x. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. J Cereb Blood Flow Metab. 2002;22:1311–1318. doi: 10.1097/01.WCB.0000034148.72481.F4. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visner GA, Dougall WC, Wilson JM, Burr IA, Nick HS. Regulation of manganese superoxide dismutase by lipopolysaccharide, interleukin-1, and tumor necrosis factor. Role in the acute inflammatory response. J Biol Chem. 1990;265:2856–2864. [PubMed] [Google Scholar]
- Wan XS, Devalaraja MN, St Clair DK. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 1994;13:1127–1136. doi: 10.1089/dna.1994.13.1127. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cai D, Brendel C, Barett C, Erben P, Manley PW, Hochhaus A, Neubauer A, Burchert A. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109:2147–2155. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- Wong GHW, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St. Clair DK. An intronic NF-κB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-α and interleukin-1β. DNA Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- Xu Y, Porntadavity S, St Clair DK. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2) Biochem J. 2002;362:401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav A, Kalita A, Dhillon S, Banerjee K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem. 2005;280:31830–31840. doi: 10.1074/jbc.M501316200. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. 2005;94:459–468. doi: 10.1111/j.1471-4159.2005.03227.x. [DOI] [PubMed] [Google Scholar]
- Yeh CC, Wan XS, St Clair DK. Transcriptional regulation of the 5′ proximal promoter of the human manganese superoxide dismutase gene. DNA Cell Biol. 1998;17:921–930. doi: 10.1089/dna.1998.17.921. [DOI] [PubMed] [Google Scholar]