

Abstract
The interaction of an amphiphilic, 40-amino acid β-amyloid (Aβ) peptide with liposomal membranes as a function of sterol mole fraction (Xsterol) was studied based on the fluorescence anisotropy of a site-specific membrane sterol probe, dehydroergosterol (DHE), and fluorescence resonance energy transfer (FRET) from the native Tyr-10 residue of Aβ to DHE. Without Aβ, peaks or kinks in the DHE anisotropy versus Xsterol plot were detected at Xsterol ≈ 0.25, 0.33, and 0.53. Monomeric Aβ preserved these peaks/kinks, but oligomeric Aβ suppressed them and created a new DHE anisotropy peak at Xsterol ≈ 0.38. The above critical Xsterol values coincide favorably with the superlattice compositions predicted by the cholesterol superlattice model, suggesting that membrane cholesterol tends to adopt a regular lateral arrangement, or domain formation, in the lipid bilayers. For FRET, a peak was also detected at Xsterol ≈ 0.38 for both monomeric and oligomeric Aβ, implying increased penetration of Aβ into the lipid bilayer at this sterol mole fraction. We conclude that the interaction of Aβ with membranes is affected by the lateral organization of cholesterol, and hypothesize that the formation of an oligomeric Aβ/cholesterol domain complex may be linked to the toxicity of Aβ in neuronal membranes.
Introduction
The presence of extracellular amyloid plaques on neuronal membranes is a major histological hallmark of Alzheimer's disease (AD), a progressive, neurodegenerative disorder that severely impairs memory and cognitive capability (1,2). A major component of these plaques is an aggregated form of β-amyloid (Aβ), a short amphiphilic peptide of 39–42 residues with a molecular mass of ∼4 kDa (2). The monomeric Aβ is released upon a sequential proteolytic cleavage of the 100 kDa transmembrane amyloid precursor protein by two proteases, i.e., ββ and gamma secretases (2). The normal, nonpathological concentration of Aβ in the extracellular matrix is subnanomolar (2).
In vitro studies on Aβ in aqueous solution have revealed that monomeric Aβ adopts a globular, largely random-coil structure (3–5). However, at nonphysiological, micromolar concentrations monomeric Aβ unfolds and self-aggregates to form protofibrils or oligomers, which are toxic to neurons (1). The detailed structure of these Aβ oligomers is still unknown, but it is believed to consist of a cross-β sheet stack of Aβ monomers involving the hydrophobic C-terminal segment, which is similar to the Aβ aggregate found in the core of the amyloid plagues in AD patients (6). According to the amyloid cascade hypothesis (1,7), self-aggregation of Aβ into these toxic oligomers represents an early, critical event in the pathogenesis of AD. The key factors that modulate the interaction of monomeric or oligomeric Aβ with the neuronal membrane, a major site of the Aβ deposit formation (7), remain unclear.
Recent studies have indicated that Aβ can interact with lipid bilayers peripherally or insert into the hydrocarbon region, consequently adopting either a β-sheet or α-helix arrangement, respectively (3–5). Such interactions may alter the physiochemical properties of the neuronal membranes by altering, e.g., the lipid dynamics, the lateral organization of lipids, and the functionality of membrane proteins. In the presence of divalent ions, the formation of Aβ/ion complexes may also promote oxidation of membrane components via the production of H2O2 (8), or form ion channels (9). Of interest, insertion of monomeric Aβ may play a protective role by preventing the formation of toxic Aβ oligomers in the aqueous phase (4,5). However, the interaction of monomeric Aβ on the membrane surface may create a two-dimensional template or seed for promoting Aβ oligomer formation on the membrane surface (3). The detailed mechanisms underlying the interactions of monomeric or oligomeric Aβ with a well-defined liposomal system that mimics the neuronal membrane remain to be elucidated.
Recent theoretical and experimental studies have revealed that lipid molecules are not randomly distributed but form lipid domains in model lipid bilayers (10–12) and probably in cell membranes as well (13). Up to now, the regulatory role of such lipid domains, including cholesterol superlattice domains (10–12), in the membrane associations of Aβ had not been fully explored. Using multicomponent phospholipid/cholesterol liposomal bilayers, we studied the effect of cholesterol content on the membrane interactions of Aβ by measuring the fluorescence anisotropy of dehydroergosterol (DHE), a site-specific fluorescent sterol probe with structure similar to cholesterol, and fluorescence resonance energy transfer (FRET) from the native Tyr-10 of Aβ to DHE versus the sterol content of the bilayer. In the absence or presence of monomeric Aβ, several peaks or kinks in DHE fluorescence anisotropy and FRET were observed at sterol concentrations that fall close to the superlattice compositions predicted by the cholesterol superlattice model (10–12). These data suggest that binding of monomeric Aβ does not significantly perturb the natural lateral arrangement of cholesterol. In contrast a single, prominent DHE anisotropy peak at Xsterol ≈ 0.38 was observed in the presence of oligomeric Aβ, indicating major changes in the lateral arrangement of lipids or the formation of an oligomeric Aβ/cholesterol domain complex. We hypothesize that modulation of the membrane interaction of Aβ by the lateral arrangement of cholesterol is a critical factor in the pathogenesis of AD.
Materials and Methods
Lipid, protein, and other reagents
1-Palmitoyl-2-oleoyl-phosphatidylcholine (POPC) and 1-palmitoyl-2-oleoyl-phosphatidylserine (POPS) were purchased from Avanti Polar Lipids (Alabaster, AL), cholesterol (CHOL) was obtained from Nu Chek Prep (Elysian, MN), and DHE and 1,4-bis[2-(5-phenyloxazolyl)]benzene (POPOP) were obtained from Sigma (St. Louis, MO). POPC and POPS have identical acyl chain compositions but different polar headgroups (the former is neutral and the latter is negative at neutral pH). Also, CHOL and DHE have an identical 3-β OH polar headgroup and a similar nonpolar body. The use of an identical acyl chain composition of phospholipids allowed us to focus on the effect of sterol content on the protein interaction with membranes.
Lipid purity (>99%) was confirmed by thin layer chromatography on washed, activated silica gel plates (Alltech Associates, Deerfield, IL) and developed with chloroform/methanol/water (65:25:4; v/v) for phospholipid analysis, or with petroleum ether/ethyl ether/chloroform (7:3:3) for cholesterol analysis. All solvents were of HPLC grade. The concentrations of all stock phospholipid solutions were determined by means of a phosphate assay (14). A PIPES buffer (pH 7.0, 5 mM PIPES, 200 mM KCl, 1 mM NaN3) was prepared in deionized water (∼18 MΩ) and filtered through a 0.1 μm filter before use.
Synthetic Aβ, 40 amino acids long (Aβ1−40, primary sequence: H-Asp-Ala-Glu-Phe-Arg-His-Asp-Ser-Gly-Tyr-Glu-Val-His-His-Gln-Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-Gly-Gly-Val-Val-OH), was obtained from Anaspec (San Jose, CA) and stored as lyophilized powders in sealed vials at −30°C before use.
Preparation of monomeric or oligomeric Aβ protein solution
Aβ powders in sealed vials were first slowly warmed to room temperature (RT) inside a dried box and then completely dissolved in dried DMSO by vortexing for 10 min at RT. For experiments with monomeric Aβ, this freshly prepared Aβ-solution was added directly into the liposomes to reach a protein concentration of 10 or 20 μM and a lipid concentration of 200μM, followed by vortexing. For experiments with oligomeric Aβ, a 115μM Aβ in aqueous solution was first prepared by adding Aβ (in DMSO) into PIPES buffer. The protein solution was sealed with argon and incubated at RT in the dark for 48 h to allow the peptide to form oligomers (15). This oligomeric Aβ-solution was then mixed with liposomes and the mixture was appropriately diluted to reach the identical protein and lipid concentrations indicated above. The DMSO concentration was 1.2 and 0.2 mol % in the monomeric and oligomeric Aβ solutions, respectively. Because of the propensity of Aβ to aggregate (15,16), and to maintain a consistent thermal history of protein preparation (16), all Aβ stock solutions were consumed within 3 h after preparation and an identical protein preparation protocol was strictly followed for all replicated preparations.
The 42-amino acid Aβ1−42 is more neurotoxic and exhibits faster aggregation in solution (6) than the Aβ1−40 used in this study. However, previous spectroscopic studies (3–5,17,18) focused on Aβ1−40 because it aggregates more slowly in solution, allowing interactions of monomeric Aβ with lipid membrane to be monitored and carefully characterized in a timely fashion. We believe that the molecular mechanisms underlying the interactions of Aβ1−42 with lipid membranes are probably similar to those of Aβ1−40; therefore, our results provide useful insights into the interaction of Aβ peptide with neuronal membranes.
Preparation of multicomponent liposomes
Compositionally homogeneous and uniform POPC/POPS/CHOL/DHE liposomes were prepared by means of a rapid solvent exchange (RSE) method (19). Details of the RSE liposome preparation procedure have been presented elsewhere (20,21). In contrast to other conventional liposomal preparation methods, such as dry film hydration, sonication, and extrusion (19,22), RSE is a convenient method with no binding, contamination, or sample recovery concerns. In addition, the accessible external surface in RSE samples is ∼33% of the total lipids, which is quite close to the 50% theoretical value for true unilamellar vesicles (19,22).
The mole fraction of the fluorescent sterol DHE was fixed at 0.01, and the sterol mole fraction (Xsterol), i.e., total sterol (DHE and cholesterol) divided by total lipid (sterol and POPS and POPC) in moles, was varied from 0 to 0.55. The structures of DHE and cholesterol are similar, so we expect DHE to minimally perturb the lipid matrix and mix well with cholesterol in the lipid membranes. As a result, the effect of small cholesterol composition increment on the protein interaction with lipid bilayers can be examined. POPS has been shown to promote the binding and subsequent interactions of Aβ to the lipid bilayer (17,18). It also prevents aggregation of RSE liposomes during their preparation, Since we focused only on the cholesterol effect, the POPS mole fraction was therefore fixed at 0.11, and a finer cholesterol increment of 0.02 was used to cover the physiologically relevant cholesterol mole fractions (0.30–0.55) found in cell membranes (12,23). These multicomponent RSE liposomes were purged in argon and allowed to incubate at RT for 15–20 days in the dark to ensure equilibration of lipid components in the liposomes before fluorescence measurements were obtained. The lipid/protein molar ratio in all our samples was 10 or 20. A similar ratio was also used in previous fluorescence studies that employed the diphenylhexatriene (DPH) probe (15,16) in multicomponent liposomes containing a fixed content of cholesterol.
Fluorescence measurements
Steady-state fluorescence anisotropy (r) measurements of DHE-containing liposomes inside a 150μL quartz curvette (Starna Cells, Atascadero, CA) in the absence and presence of Aβ were performed on a QuantaMaster C61/2000 spectrofluorimeter (PTI, Lawrenceville, NJ) using a T-mode single-photon-counting configuration. The fluorescence excitation was at 325 nm and emission was at 375 nm. Here, r is defined by
| (1) |
where I|| and I⊥ are the fluorescence intensities from the parallel and perpendicular polarized emissions upon a parallel polarized excitation, respectively, and g is a factor associated with the relative sensitivity of the two emission channels and can be determined when the excitation is set to perpendicular. The same PTI spectrofluorimeter was used to collect the fluorescence spectra of DHE and Aβ.
To ensure consistency in the thermal history of liposome and protein, as well as their interactions, fluorescence measurements were acquired exactly 1 h after protein was added to the liposomes at RT. At least 11 min were required to collect both the steady-state anisotropy and spectra from each protein/liposome sample. Since the protein stock solutions have to be used within 3 h (see above), 16 different sterol compositions were covered in each set of fluorescence measurement. The static light scattering of our samples was similar and independent of sterol contents.
Fluorescence intensity decay measurements of DHE were performed on a GREG 200 fluorimeter (ISS, Champaign, IL) equipped with digital multifrequency cross-correlation phase and modulation acquisition electronics (24,25). A 4240NB cw UV He-Cd laser (Liconix, Santa Clara, CA) emitting at 325 nm was used for fluorescence excitation. An excitation polarizer with its transmission axis set at 35° and no emission polarizer on the emission channel were used to eliminate the rotational diffusion effect of the sample to the measurements (26). Phase delay and demodulation values of the DHE fluorescence were compared with those of a standard (POPOP in ethanol) that has a fluorescence decay lifetime (τ) of 1.34 ns (24), with modulation frequency varied from 5 to 200 MHz. DHE exhibited multiexponential decay in liposomes (25), with an average τ of ∼0–3 ns depending on the lipid composition. Due to the low fluorescence signal and bleaching of DHE, fewer than 10 frequency-domain data points were acquired within 12 min for each sample. The limited number of data points did not allow us to perform an accurate multiexponential analysis of the DHE decay in this study. Therefore, a single-exponential decay was employed to analyze the frequency-domain data. The rotational correlation time (ρ) of DHE in the liposomes was obtained from the measured values of τ and r of DHE using the Perrin equation (25,27)
| (2) |
The value of ρ is inversely proportional to the average rate of rotation of DHE. Note that the rotational dynamics of DHE is best described by a hindered rotation model (28) involving at least one rotational rate and an angular freedom (order) parameter obtained from the anisotropy decay measurement of DHE. Again, due to the signal and bleaching issues, we were not able to collect the anisotropy decay of DHE, and our calculated ρ is a combination of both rotational and order parameters.
The critical FRET distance Ro between the donor (Try-10 of Aβ) and acceptor (DHE) at which the transfer efficiency is 50% was estimated from the following equation (29,30):
| (3) |
where κ, ΦD, and n are the dipole orientation factor, quantum yield of the donor, and refractive index of the medium, respectively. Also, I(λ) and ɛ(λ) represent the emission spectrum of the donor and the molar absorption spectrum of the acceptor as a function of wavelength λ. Assuming an isotropic distribution (29) of dipoles of the acceptor and donor (κ2 = 2/3), the value of Ro was estimated to be ∼20 Å for our donor-acceptor pair in this study.
Statistical significance of the peaks, dips, and kinks of spectroscopic data
The statistical significance of the peaks, dips, and kinks of spectroscopic data (DHE anisotropy or FRET versus Xsterol plots) was analyzed using an undirectional t-test of unequal variances as described previously (31). Here, peaks or dips were defined as the composition points at which the spectroscopic values were significantly higher or lower than the baseline spectroscopic values at both the adjacent lower (left-handed) and higher (right-handed) compositions. Kinks were defined as the composition points at which the spectroscopic values reach a plateau, and were significantly higher than the values at adjacent lower compositions. Briefly, the statistical mean, defined as the averaged spectroscopic measurement of anisotropy or FRET from N independently prepared parallel samples, at the critical sterol composition (X∗) was compared with that at the adjacent baseline sterol composition on the left-handed side (XL) or the right-handed side (XR) based on the t-test. The probability, or p-value, of accepting the null hypothesis of the two means being identical, was then calculated (32). The probability P of rejecting the null hypothesis, or probability of significance of the difference of the two means, is defined as (1 − p) × 100%. In our case, the P-value of the biphasic behavior at X∗ as compared with that at XL or XR is given by PL or PR, respectively. Peaks or dips with both PL and PR < 70% were ignored. Although the 70% cutoff is arbitrary, the P-values provide an objective and standard gauge to evaluate and cross-compare biphasic behavior of our spectroscopic data versus lipid composition plot (31).
Results
Fluorescence anisotropy of DHE
The DHE anisotropy in POPS/POPC/CHOL/DHE liposomes was measured as a function of Xsterol. At zero cholesterol content, DHE anisotropy, averaged over several independently prepared samples, was 0.310 ± 0.002 (mean ± SE; N = 24). To allow comparison of different sample sets, normalized DHE anisotropy, calculated by subtracting the DHE anisotropy at Xsterol = 0.01 (internal control) from the DHE anisotropy at each sterol content, was used. Fig. 1 shows the averaged normalized DHE anisotropy as a function of Xsterol. With increasing cholesterol content, several kinks or peaks (“critical” sterol mole fractions, X∗) were observed, including a sharp peak at X∗ = 0.25, an asymmetric peak at 0.33, and a kink at 0.53. The statistical significance of these deviations was evaluated (see Materials and Methods) and the results are summarized in Table 1. The values of the probability of significance [PL, PR] of the deviations were [99, 96], [99, 98], and [98, ND] at X∗ = 0.25, 0.33, and 0.53, respectively. Similar DHE anisotropy peaks were also observed in other DHE-containing phospholipids/sterol liposomes by Liu et al. (33).
Figure 1.
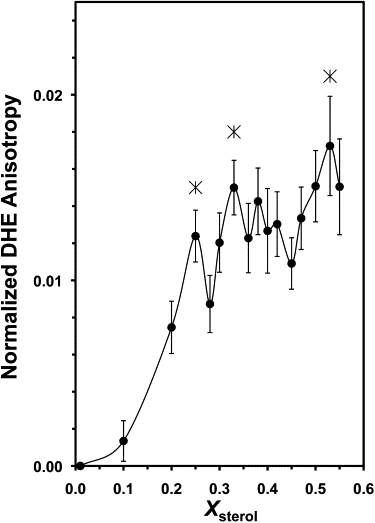
Normalized fluorescence anisotropy of DHE in POPS/POPC/DHE/CHOL versus Xsterol. The three asterisks indicate the locations of the critical changes in the DHE anisotropy. Bars indicate SEs of the averages of fluorescence measurements from independently prepared liposomes. All measurements were performed at RT.
Table 1.
Comparison of critical sterol mole fractions X∗ from DHE anisotropy measurements with X∗CHOL values predicted by the superlattice model
| X∗CHOL | Aβ (μM) | (XL, X∗, XR) | [PL, PR] | N |
|---|---|---|---|---|
| 0.25 | 0 | (0.20, 0.25p, 0.28) | [99, 96] | 24 |
| 10m | (0.20, 0.25p, 0.28) | [92, 80] | 8 | |
| 20m | (0.20, 0.25p, 0.28) | [80, 69] | 12 | |
| 10° | (0.20, 0.25p, 0.28) | [94, 86] | 11 | |
| 20° | ||||
| 0.33 | 0 | (0.28, 0.33p, 0.45) | [99, 98] | 24 |
| 10m | (0.28, 0.33p, 0.45) | [85, 81] | 8 | |
| 20m | (0.28, 0.33p, 0.45) | [89, 78] | 12 | |
| 10° | ||||
| 20° | ||||
| 0.40 | 0 | |||
| 10m | ||||
| 20m | ||||
| 10° | ||||
| 20° | (0.30, 0.38p, 0.47) | [95, 91] | 12 | |
| 20o§ | (0.30, 0.38p, 0.47) | [93, 93] | 12 | |
| 0.50 | 0 | (0.45, 0.53k, ND) | [98, ND] | 24 |
| 10m | (0.45, 0.53k, ND) | [75, ND] | 8 | |
| 20m | (0.45, 0.53k, ND) | [87, ND] | 12 | |
| 10° | ||||
| 20° |
The probability of significance [PL, PR] of the peak or kink (denoted by superscripts p and k, respectively), and the number of independent DHE anisotropy measurements (N) in the absence or presence of monomeric (superscript m) or oligomeric (superscript o) Aβ based on a t-test comparing the anisotropy at X∗ with the adjacent values at XL or XR (see Materials and Methods) are given. The statistical significance of the peak of the DHE anisotropy difference (superscript §) at X∗ = 0.38 is also given.
The effect of monomeric Aβ on DHE anisotropy was examined (Fig. 2). Monomeric Aβ systematically reduced the DHE anisotropy at all sterol contents, with the effect being stronger for 20 μM than for 10 μM Aβ. Also, the anisotropy reduction was much stronger at low sterol than at high sterol contents at each Aβ concentration. Notably, the deviations in DHE anisotropy versus Xsterol observed in the absence of Aβ (Fig. 1) were largely preserved in the presence of monomeric Aβ. The probability of significance [PL, PR] for each X∗ was also determined (Table 1). Except for the PR of 69% at X∗ = 0.25 for 20 μM Aβ, P > 70% for all X∗ values at both Aβ concentrations.
Figure 2.
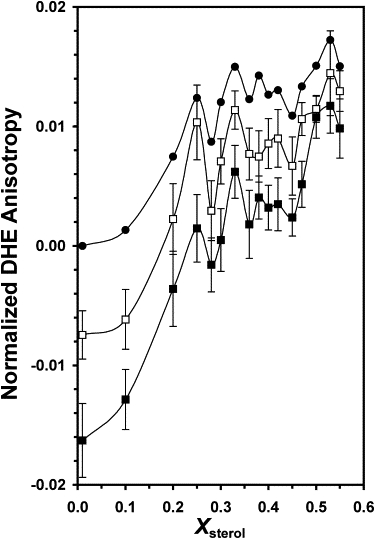
Normalized fluorescence anisotropy of DHE in POPS/POPC/DHE/CHOL versus Xsterol in the absence (solid circle) and presence of 10 μM (open square) or 20 μM (solid square) of monomeric Aβ. See the legend of Fig. 1 for other details.
The effect of oligomeric Aβ on DHE anisotropy was also examined (Fig. 3). Oligomeric Aβ increased the DHE anisotropy at all sterol contents, with the effect being much stronger for 20 μM than for 10 μM Aβ. Note that a similar increase in DPH anisotropy in multicomponent liposomes containing cholesterol in the presence of oligomeric Aβ and of similar lipid/protein ratio was also reported in a previous study (15). At 10 μM Aβ, only the peak at X∗ = 0.25 was still evident, as judged by the probability of significance of [PL, PR] = [94, 86] as shown in Table 1. However, at 20 μM Aβ, the deviations at X∗ = 0.25, 0.33, and 0.53 were suppressed, but a statistically significant peak at X∗ = 0.38 with [PL, PR] = [95, 91] was detected.
Figure 3.
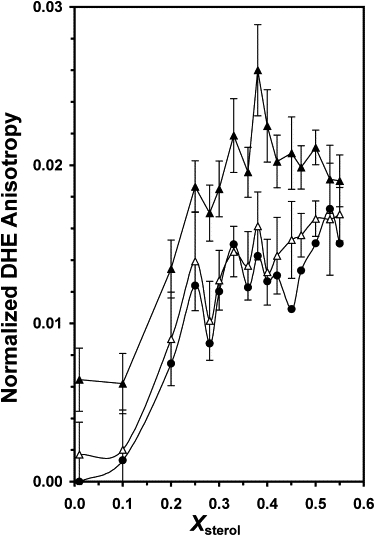
Normalized fluorescence anisotropy of DHE in POPS/POPC/DHE/CHOL versus Xsterol in the absence (solid circle) and presence of 10 μM (open triangle) or 20 μM (solid triangle) of oligomeric Aβ. See the legend of Fig. 1 for other details.
Since the DHE anisotropy varied with Xsterol even in the absence of Aβ, the anisotropy difference (obtained by subtracting the anisotropy in the absence of Aβ from that in the presence of Aβ) was calculated to directly assess the effect of Aβ. Fig. 4 shows the averaged anisotropy difference as a function of Xsterol for monomeric and oligomeric Aβ. A gradual increase in the anisotropy difference with increasing Xsterol was observed for monomeric Aβ, with the slope being greater for 20 μM than for 10 μM. On the other hand, a flat response, or a nearly zero slope, in the anisotropy difference versus Xsterol was found for 10 μM oligomeric Aβ. However, with 20 μM, a significant peak was again detected at X∗ = 0.38 with [PL, PR] = [93, 93].
Figure 4.
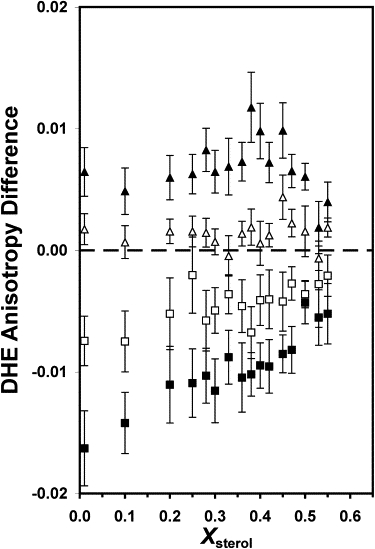
DHE anisotropy difference versus Xsterol in the presence of 10 μM of monomeric Aβ (open square), 10 μM of oligomeric Aβ (open triangle), 20 μM of monomeric Aβ (solid square), or 20 μM of oligomeric Aβ (solid triangle). See the legend of Fig. 1 for other details.
Time-resolved fluorescence of DHE
The time-resolved fluorescence intensity decay of DHE in POPS/POPC/CHOL/DHE liposomes was studied for selected sterol compositions at Xsterol = 0.01, 0.30, 0.36, 0.40, and 0.45, and the results are summarized in Table 2. In the absence of cholesterol, the average DHE fluorescence decay lifetime (τ) was 1.2 ns, but it decreased slightly to 1.1 and 1.0 ns in the presence of monomeric and oligomeric Aβ, respectively. In the presence of cholesterol, τ was somewhat higher but nearly independent of Xsterol. However, addition of monomeric Aβ increased τ to 1.5 ns, whereas in the presence of oligomeric Aβ τ decreased to 0.8–1.1 ns.
Table 2.
Comparison of time-resolved fluorometric parameters of DHE in lipid bilayers of different sterol mole fractions in the absence and presence of Aβ
| Xsterol | Aβ (μM) | τ (ns) | ρ (ns) |
|---|---|---|---|
| 0.01 | 0 | 1.22 ± 0.01 | 7.30 ± 0.39 |
| 20m | 1.10 ± 0.02 | 5.00 ± 0.22 | |
| 20° | 1.00 ± 0.01 | 6.87 ± 0.40 | |
| 0.30 | 0 | 1.28 ± 0.01 | 10.58 ± 0.92 |
| 20m | 1.46 ± 0.02 | 8.20 ± 0.55 | |
| 20° | 1.12 ± 0.01 | 12.24 ± 1.96 | |
| 0.36 | 0 | 1.28 ± 0.01 | 10.27 ± 0.96 |
| 20m | 1.50 ± 0.01 | 8.05 ± 0.57 | |
| 20° | 1.04 ± 0.01 | 12.72 ± 1.61 | |
| 0.40 | 0 | 1.33 ± 0.01 | 11.30 ± 1.11 |
| 20m | 1.54 ± 0.01 | 9.22 ± 0.69 | |
| 20° | 1.15 ± 0.01 | 15.25 ± 2.02 | |
| 0.45 | 0 | 1.27 ± 0.02 | 10.21 ± 0.72 |
| 20m | 1.52 ± 0.01 | 10.17 ± 0.65 | |
| 20° | 0.84 ± 0.02 | 10.27 ± 1.36 |
The average fluorescence decay lifetime (τ) and rotational correlation time (ρ) of DHE in the absence and presence of monomeric or oligomeric Aβ (denoted by superscript m or o, respectively) are shown.
The rotational correlation time (ρ) of DHE was calculated from the steady-state anisotropy and fluorescence lifetime data using Eq. 2, and the results are summarized in Table 2. In the absence of cholesterol, the value of ρ was 7.3 ns in neat liposomes (no protein), but it decreased to 5.0 and 6.9 ns in the presence of monomeric and oligomeric Aβ, respectively. In the presence of cholesterol, the value of ρ in neat liposomes was ∼11 ns and independent of Xsterol. In the presence of monomeric Aβ, ρ decreased to 8–9 ns at Xsterol = 0.30, 0.36, and 0.40, whereas the presence of oligomeric Aβ increased ρ to 12–15 ns at those same concentrations. Of interest, at Xsterol = 0.45, no change in ρ was detected in the presence of either monomeric or oligomeric Aβ.
FRET from Tyr-10 of Aβ to DHE
We next examined the effect of Xsterol on FRET from the native Tyr-10 residue of Aβ to the DHE in POPS/POPC/CHOL/DHE liposomes. As shown in Fig. 5, monomeric Aβ in solution showed a typical tyrosine emission peaking at ∼300 nm, and the excitation spectrum of DHE overlapped the tyrosine emission spectrum. These results indicate that FRET from Tyr-10 of Aβ to DHE could take place when Aβ binds to the POPS/POPC/CHOL/DHE liposomes, provided that the tyrosine-to-DHE distance is in the range of 2 nm, the critical FRET distance for this donor-to-acceptor pair (Eq. 2). Indeed, in the presence of DHE-containing liposomes, the emission spectrum of the protein/liposome mixture exhibited a shoulder at 330–450 nm coinciding with the emission spectrum of DHE in the absence of Aβ, as shown in Fig. 5.
Figure 5.
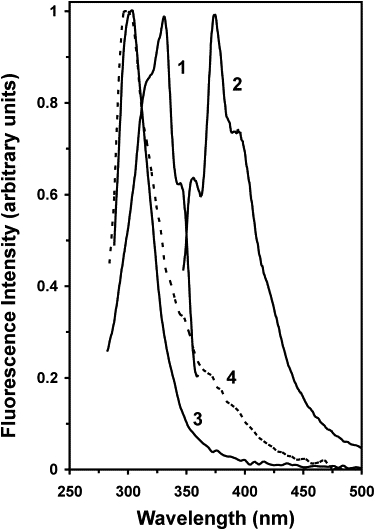
Representative spectra for demonstrating FRET from Tyr-10 of Aβ to DHE in liposomes (POPS/POPC/DHE molar fractions = 0.11:0.88:0.01). The fluorescence excitation (line 1) and emission (line 2) spectra of DHE in liposomes are shown. The emission and excitation wavelengths for collecting the above DHE spectra were 420 and 325 nm, respectively. The Tyr-10 of Aβ fluorescence emission spectra of a 20 μM monomeric Aβ solution in the absence (line 3) and presence (line 4) of DHE-containing liposomes with the excitation wavelength at 275 nm are also shown. All spectra were recorded at room temperature.
The efficiency of FRET from Tyr-10 of Aβ to DHE was estimated from the ratio of the acceptor emission intensity at 376 nm to the donor emission intensity at 300 nm upon excitation of the sample at 275 nm. To eliminate the contribution of the direct excitation of DHE, intensities at 376 and 300 nm for the neat liposomes were subtracted from those of the liposomes in the presence of Aβ. In the absence of cholesterol, the values of FRET for the monomeric and oligomeric Aβ were 0.092 ± 0.024 (mean ± SE; N = 8) and 0.049 ± 0.005 (mean ± SE; N = 12), respectively. Fig. 6 shows a plot of the FRET versus Xsterol. A peak at X∗ = 0.38 with [PL, PR] = [88, 91], and two dips at X∗ = 0.33 with [PL, PR] = [91, 89] and 0.50 with [PL, PR] = [93, 90] were found in the presence of 20 μM monomeric Aβ (see Table 3). On the other hand, a dip at X∗ = 0.28 with [PL, PR] = [96, 96] and a broad peak centered at X∗ ≈ 0.40 with [PL, PR] = [96, 92] were found in the presence of 20 μM oligomeric Aβ (see Table 3).
Figure 6.
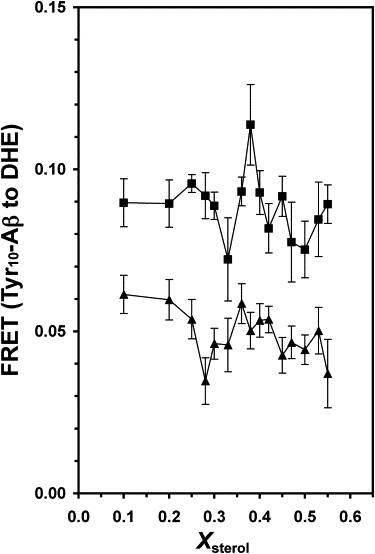
FRET from Tyr-10 of Aβ to DHE in POPS/POPC/DHE/CHOL liposomes versus Xsterol in the presence of 20 μM of monomeric (solid square) and oligomeric (solid triangle) Aβ.
Table 3.
Comparison of critical sterol mole fractions X∗ from FRET (Tyr-10-Aβ to DHE) spectroscopic measurements with X∗CHOL values predicted by the superlattice model
| X∗CHOL | Aβ (μM) | (XL, X∗, XR) | [PL, PR] | N |
|---|---|---|---|---|
| 0.25 | 20m | |||
| 20° | (0.20, 0.28d, 0.36) | [96, 96] | 8 | |
| 0.33 | 20m | (0.25, 0.33d, 0.36) | [91, 89] | 12 |
| 20° | ||||
| 0.40 | 20m | (0.36, 0.38p, 0.42) | [88, 91] | 12 |
| 20° | (0.28, 0.36p, 0.45) | [96, 92] | 8 | |
| 0.50 | 20m | (0.45, 0.50d, 0.55) | [93, 90] | 12 |
| 20° |
The probability of significance of the peak and dip (denoted by superscripts p and d, respectively) is given. See the legend of Table 1 for other details.
Discussion
The role of the lateral organization of cholesterol in modulating the membrane association of protein is a subject of great interest (13). Notably, the activities of several membrane surface-acting proteins, such as cholesterol oxidase (10,34) and phospholipase (35), exhibited abrupt changes at certain “critical” XCHOL values of the target bilayers. These critical XCHOL values coincide favorably with those predicted by the cholesterol superlattice model (11,12,34), which suggests that cholesterol tends to adopt energetically favorable, regular, superlattice-like lateral distributions or domains in bilayers. Thus, cholesterol superlattice domain formation may be actively involved in modulating the membrane activities of those proteins. We set out to investigate whether this might also apply to Aβ.
Sterol critical compositions and Aβ-membrane interactions
In the absence of Aβ, our DHE anisotropy versus Xsterol plot revealed statistically significant deviations at X∗ ≈ 0.25, 0.33, and 0.53 (Fig. 1). These critical sterol compositions agree favorably with the superlattice compositions predicted by the cholesterol superlattice model (11,12,34), suggesting that the sterols adopt a regular lateral distribution at those particular compositions in our multicomponent liposomes. Note that previous fluorescence measurements on site-specific diphenylhexatriene chain-labeled PC (DPH-PC) (31), free DPH (36), and surface-sensitive 6-lauroyl-2-(dimethylamino)naphthalene (Laurdan) probes (36), as well as probe-free FTIR measurements on the O=P=O and C=O vibrational bands of native phospholipids in less complicated POPC/CHOL liposomes (31), also revealed similar critical changes at XCHOL ≈ 0.33 and 0.50. We therefore conclude that the presence of POPS in the liposomes does not preclude the formation of sterol superlattices, as expected at those superlattice compositions.
The superlattice composition at 0.40 was not observed in our DHE anisotropy measurements on neat POPS/POPC/CHOL liposomes, yet it was observed in the Laurdan (36), DPH-PC (31), and FTIR (31) measurements on POPC/CHOL liposomes. This may be explained by the lack of significant changes in the fluorescence lifetime and/or rotational dynamics of DHE at that superlattice composition in the presence of POPS. Also, a longer liposome incubation time (i.e., >20 days) may be required to reveal this sterol critical composition of 0.40, as demonstrated by a recent study on the kinetics of formation of the cholesterol superlattice (36). Further work is required to identify the effect of POPS on the stability of superlattice formation.
We also examined whether Aβ supports or suppresses the lateral distribution of sterol at X∗ = 0.25, 0.33, and 0.53. Significant DHE anisotropy deviations at those X∗ values were preserved upon addition of monomeric Aβ (Fig. 2). This finding suggests that the lateral distribution of cholesterol in POPC/POPS/CHOL/DHE liposomes is not markedly affected by the binding of monomeric Aβ. However, oligomeric Aβ suppressed the above X∗ values and created a new DHE anisotropy peak at X∗ ≈ 0.38 (Fig. 3). Notably, the deviation at 0.38 is close to the superlattice composition at XCHOL = 0.40 (11,12,34). Domain boundaries and/or abrupt changes in membrane physical properties are predicted to occur at values close to the predicted superlattice mole fractions (11,12,34). Thus, binding of oligomeric Aβ to the bilayers may perturb the existing putative superlattice organization of cholesterol and lead to the formation of an oligomeric Aβ/cholesterol domain complex at the predictable superlattice composition (≈0.40 in our case).
Microenvironment and dynamics of DHE
Since the fluorescence anisotropy of DHE depends on both the fluorescence decay lifetime and the rotational dynamics of the probe in the bilayer as depicted in Eq. 2, time-resolved measurements were performed to identify the fluorescence decay lifetime and rotational contributions to the DHE anisotropy. The fluorescence decay lifetime of DHE reports the fluorescence quenching microenvironment of the sterol at the fused ring region of the sterol where the DHE emission dipole is located. The rotational correlation time ρ of DHE is sensitive to the packing of lipids in the hydrocarbon region of the bilayer.
In the presence of cholesterol, a modest increase and a decrease in the fluorescence decay lifetime of DHE were observed in the presence of monomeric and oligomeric Aβ, respectively. These results suggest that the interactions of different forms of Aβ perturbed the microenvironments of DHE differently in the membranes. Specifically, relatively lower and higher fluorescence quenching environments of DHE were created in the presence of a monomeric and oligomeric Aβ, respectively. The latter may be associated with an increase in water permeability (28) in the hydrocarbon region of the lipid bilayer due to the presence of oligomeric Aβ, especially at high sterol contents (e.g., Xsterol = 0.45; Table 2).
Monomeric Aβ increased the rotational rate of DHE for Xsterol up to 0.40. However, this increase vanished at a higher sterol content (i.e., Xsterol = 0.45). This change in the rotational dynamics behavior explains the observation that a large DHE anisotropy difference prevailed at low sterol contents but vanished at high sterol contents (Fig. 4). We propose that this increase in the sterol rotation rate could be due to the insertion of monomeric Aβ into the hydrocarbon region of the lipid bilayer, which disrupted the packing of sterols near the lipid/peptide interface at low sterol content. At higher sterol content (Xsterol > 0.45), no significant insertion of monomeric Aβ occurred, probably because of the tighter packing of the lipids, and therefore resulted in no change in the rotational rate of sterol. In this respect, the monomeric Aβ might prefer the inserted state at low cholesterol contents, but the surface interaction state at high cholesterol contents.
Oligomeric Aβ decreased, rather than increased, the rotational rate of DHE at low sterol contents. Similar to the case of monomeric Aβ, the perturbation of the rotational rate of DHE vanished at a higher sterol content of 0.45. These findings indicate that the oligomeric Aβ increased the packing of the sterol, probably by interacting with the headgroups of the phospholipids or even the sterols. We observed the highest decrease in the rotational rate of DHE (i.e., a significant 35% increase in the value of ρ) at Xsterol = 0.40 (Table 2). This abrupt decrease in the DHE rotation rate further agrees with the single peak in the DHE anisotropy difference plot (Fig. 4). Our rotational dynamics results therefore indicate that an “ordered” oligomeric Aβ/cholesterol domain complex formed near the critical sterol composition of 0.40, and that the rotation of the sterols within that complex was highly hindered due to the presence of the oligomeric Aβ. In addition, the decrease in the DHE fluorescence lifetime further signifies an increase in the water permeability (28) accompanied by the formation of the putative ordered oligomeric Aβ/cholesterol domain complex.
Implications of Aβ interaction with cholesterol domains at the molecular level
The FRET from the native Tyr-10 of Aβ to DHE in the multicomponent liposomes provides noninvasive information about the distance between the hydrophilic N-terminal of Aβ with the sterols located in the hydrophobic region of the lipid bilayer. The significant FRET peak at Xsterol ≈ 0.38 for both monomeric and oligomeric Aβ indicates a closer proximity of the hydrophilic region of Aβ to the sterols, or deeper penetration of the peptide to the lipid membrane, at the critical composition than at the other noncritical compositions. We propose that the presence of the putative, center-rectangular cholesterol superlattice domains at XCHOL = 0.40 enhance the interactions of the peptide to the lipid bilayer. Also, the average distance of the N-terminal segment of the peptide to the sterols is significantly longer for the oligomeric Aβ than for the monomeric Aβ, as evidenced by the higher FRET for the monomeric Aβ than the oligomeric Aβ (Fig. 6) at all sterol contents. The FRET dips at the critical Xsterol ≈ 0.28, 0.33, and 0.50 may be interpreted as relatively weaker associations of the Aβ with the hydrocarbon region of the membranes, or a lack of significant insertion of the protein, at those critical sterol contents. Of interest, those sterol contents also coincide favorably with the superlattice compositions predicted by the cholesterol superlattice model (11,31), as well as those from the DHE anisotropy measurements above. Therefore, the deviations in the proximity of Aβ to sterol based on FRET at Xsterol ≈ 0.28, 0.33, 0.38, and 0.50 agree favorably with those from the DHE anisotropy measurements, further supporting the role of cholesterol lateral distribution, or the formation of cholesterol superlattice domains, in modulating the Aβ interactions with sterols in the lipid bilayer.
It is not clear whether Aβ might extract the lipids from the liposomes, as does phospholipase A2 (37), and create new “Aβ-lipid” particles that may be structurally and compositionally different from the protein-liposome complexes. Our preliminary scanning electron microscopy measurements revealed that the lipid vesicles remained intact in the presence Aβ. Also, the static light scattering of our samples was independent of sterol contents. Yet, no conclusive evidence is available to preclude the existence of these putative Aβ-lipid particles. However, the initial binding of Aβ to the liposome surface preceding the formation of the putative Aβ-lipid particles may also be regulated by the cholesterol superlattice domain, similar to the observed activities of other membrane-active peptides, cholesterol oxidase (10,34), and phospholipase (35). More work is required to explore the presence of these interesting lipid-protein particles.
In conclusion, our results, obtained with the use of a site-specific sterol, DHE, and FRET from the native Aβ to DHE, suggest that monomeric Aβ preserves the small but significant deviations of DHE anisotropy at certain critical sterol contents, and therefore supports the formation of cholesterol superlattice domains in multicomponent lipid bilayers. In addition, we hypothesize that the prominent deviation of oligomeric Aβ interaction with DHE at X∗ ≈ 0.38 is due to the formation of a putative ordered oligomeric Aβ/cholesterol domain complex that may be associated with the critical Aβ-membrane association event leading to the neurotoxicity of oligomeric Aβ in vivo.
Acknowledgments
The suggestions of Martin Hermansson of the Institute of Biomedicine, University of Helsinki, are gratefully acknowledged.
This work was supported by a grant from the Robert A. Welch Research Foundation (D-1158).
References
- 1.Roberson E.D., Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe D.J. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Bokvist M., Lindstrom F., Watts A., Grobner G. Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 4.Ji S.R., Wu Y., Sui S.F. Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (A β 1–40), which may potentially inhibit the fibril formation. J. Biol. Chem. 2002;277:6273–6279. doi: 10.1074/jbc.M104146200. [DOI] [PubMed] [Google Scholar]
- 5.Terzi E., Holzemann G., Seelig J. Self-association of β-amyloid peptide (1–40) in solution and binding to lipid membranes. J. Mol. Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 6.Luhrs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy J.A., Higgins G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 8.Nelson T.J., Alkon D.L. Oxidation of cholesterol by amyloid precursor protein and β-amyloid peptide. J. Biol. Chem. 2005;280:7377–7387. doi: 10.1074/jbc.M409071200. [DOI] [PubMed] [Google Scholar]
- 9.Lin H., Bhatia R., Lal R. Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 10.Cheng K.H., Cannon B., Metze J., Lewis A., Huang J. Lipid headgroup superlattice modulates the activity of surface-acting cholesterol oxidase in ternary phospholipid/cholesterol bilayers. Biochemistry. 2006;45:10855–10864. doi: 10.1021/bi060937y. [DOI] [PubMed] [Google Scholar]
- 11.Chong P.L. Evidence for regular distribution of sterols in liquid crystalline phosphatidylcholine bilayers. Proc. Natl. Acad. Sci. USA. 1994;91:10069–10073. doi: 10.1073/pnas.91.21.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Somerharju P., Virtanen J.A., Cheng K.H. Lateral organisation of membrane lipids. The superlattice view. Biochim. Biophys. Acta. 1999;1440:32–48. doi: 10.1016/s1388-1981(99)00106-7. [DOI] [PubMed] [Google Scholar]
- 13.Simons K., Vaz W.L. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 14.Kingsley P.B., Feigenson G.W. The synthesis of a perdeuterated phospholipid: 1,2-dimyristoyl-sn-glycero-3-phosphocholine-d72. Chem. Phys. Lipids. 1979;24:135–147. [Google Scholar]
- 15.Kremer J.J., Pallitto M.M., Sklansky D.J., Murphy R.M. Correlation of β-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry. 2000;39:10309–10318. doi: 10.1021/bi0001980. [DOI] [PubMed] [Google Scholar]
- 16.Kremer J.J., Sklansky D.J., Murphy R.M. Profile of changes in lipid bilayer structure caused by β-amyloid peptide. Biochemistry. 2001;40:8563–8571. doi: 10.1021/bi010417x. [DOI] [PubMed] [Google Scholar]
- 17.Arispe N., Doh M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer's disease AβP (1–40) and (1–42) peptides. FASEB J. 2002;16:1526–1536. doi: 10.1096/fj.02-0829com. [DOI] [PubMed] [Google Scholar]
- 18.Curtain C.C., Ali F.E., Smith D.G., Bush A.I., Masters C.L. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J. Biol. Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 19.Buboltz J.T., Feigenson G.W. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim. Biophys. Acta. 1999;1417:232–245. doi: 10.1016/s0005-2736(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 20.Cannon B., Weaver N., Pu Q., Thiagarajan V., Liu S. Cholesterol modulated antibody binding in supported lipid membranes as determined by total internal reflectance microscopy on a microfabricated high-throughput glass chip. Langmuir. 2005;21:9666–9674. doi: 10.1021/la0502645. [DOI] [PubMed] [Google Scholar]
- 21.Cannon B., Lewis A., Metze J., Thiagarajan V., Vaughn M.W. Cholesterol supports headgroup superlattice domain formation in fluid phospholipid/cholesterol bilayers. J. Phys. Chem. B. 2006;110:6339–6350. doi: 10.1021/jp0558371. [DOI] [PubMed] [Google Scholar]
- 22.Ali M.R., Cheng K.H., Huang J. Assess the nature of cholesterol-lipid interactions through the chemical potential of cholesterol in phosphatidylcholine bilayers. Proc. Natl. Acad. Sci. USA. 2007;104:5372–5377. doi: 10.1073/pnas.0611450104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waschuk S.A., Elton E.A., Darabie A.A., Fraser P.E., McLaurin J.A. Cellular membrane composition defines A β-lipid interactions. J. Biol. Chem. 2001;276:33561–33568. doi: 10.1074/jbc.M103598200. [DOI] [PubMed] [Google Scholar]
- 24.Gratton E., Jameson D.M., Hall R.D. Multifrequency phase and modulation fluorometry. Annu. Rev. Biophys. Bioeng. 1984;13:105–124. doi: 10.1146/annurev.bb.13.060184.000541. [DOI] [PubMed] [Google Scholar]
- 25.Cheng K.H., Virtanen J., Somerharju P. Fluorescence studies of dehydroergosterol in phosphatidylethanolamine/phosphatidylcholine bilayers. Biophys. J. 1999;77:3108–3119. doi: 10.1016/S0006-3495(99)77141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spencer R.D., Weber G. Influence of Brownian rotation and energy transfer upon the measurements of fluorescence lifetime. J. Chem. Phys. 1970;52:1654–1663. [Google Scholar]
- 27.Perrin F. Mouvement Brownien d'un ellipsoide. II. Rotation libre et dipolarisation des florescences. Transaction et diffusion de molecules ellipsoidoles. J. Phys. Radium. 1936;71:1–44. [Google Scholar]
- 28.Schroeder F., Barenholz Y., Gratton E., Thompson T.E. A fluorescence study of dehydroergosterol in phosphatidylcholine bilayer vesicles. Biochemistry. 1987;26:2441–2448. doi: 10.1021/bi00383a007. [DOI] [PubMed] [Google Scholar]
- 29.Wu P., Brand L. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 1992;31:7939–7947. doi: 10.1021/bi00149a027. [DOI] [PubMed] [Google Scholar]
- 30.Cheng K.H., Wiedmer T., Sims P.J. Fluorescence resonance energy transfer study of the associative state of membrane-bound complexes of complement proteins C5b-8. J. Immunol. 1985;135:459–464. [PubMed] [Google Scholar]
- 31.Cannon B., Heath G., Huang J., Somerharju P., Virtanen J.A. Time-resolved fluorescence and fourier transform infrared spectroscopic investigations of lateral packing defects and superlattice domains in compositionally uniform cholesterol/phosphatidylcholine bilayers. Biophys. J. 2003;84:3777–3791. doi: 10.1016/S0006-3495(03)75106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferguson G.A. McGraw-Hill; New York: 1971. Statistical Analysis in Psychology and Education. [Google Scholar]
- 33.Liu F., Sugar I.P., Chong P.L. Cholesterol and ergosterol superlattices in three-component liquid crystalline lipid bilayers as revealed by dehydroergosterol fluorescence. Biophys. J. 1997;72:2243–2254. doi: 10.1016/S0006-3495(97)78868-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M.M., Olsher M., Sugar I.P., Chong P.L. Cholesterol superlattice modulates the activity of cholesterol oxidase in lipid membranes. Biochemistry. 2004;43:2159–2166. doi: 10.1021/bi035982+. [DOI] [PubMed] [Google Scholar]
- 35.Liu F., Chong P.L. Evidence for a regulatory role of cholesterol superlattices in the hydrolytic activity of secretory phospholipase A2 in lipid membranes. Biochemistry. 1999;38:3867–3873. doi: 10.1021/bi982693q. [DOI] [PubMed] [Google Scholar]
- 36.Venegas B., Sugar I.P., Chong P.L. Critical factors for detection of biphasic changes in membrane properties at specific sterol mole fractions for maximal superlattice formation. J. Phys. Chem. B. 2007;111:5180–5192. doi: 10.1021/jp070222k. [DOI] [PubMed] [Google Scholar]
- 37.Stepaniants S., Izrailev S., Schulten K. Extraction of lipids from phospholipid membranes by steered molecular dynamics. J. Mol. Model. 1997;3:473–475. [Google Scholar]