

Abstract
The macroscopic ion-selective behavior of K+ channels is mediated by a multitude of physiological factors. However, considering the carbonyl-lined binding site of a conductive K+ channel as a canonical eightfold coordinated construct can be useful in understanding the principles that correlate the channel's structure with its function. We probe the effects of structure and chemical composition on the K+/Na+ selectivity provided by a variety of simplified droplet-like ion binding site models. We find that when carbonyl- and water-based models capture the qualitative structural features of the K+ channel binding site, a selective preference for K+ emerges. Thus our findings suggest that the preference for K+ over Na+ exhibited by such models is principally built-in, and is not due to a unique K+-selective property of carbonyl functional groups. This suggestion is confirmed by a general thermodynamic assessment, which provides a basis for using simplified models to study the design principles underlying the molecular evolution of K+ channels.
Introduction
The locality of a ligated ion at a site within a solvated protein may be characterized as a system in which the degrees of freedom R of available ligands are influenced by ion-ligand and ligand-ligand interactions and an external field, provided by the surrounding solvated proteinaceous environment (see prior work (1–5) and references therein). An analogous characterization may be applied to an ionic complex in a liquid or gas of ligating molecules. From the definition of Helmholtz free energy, F, it follows that the density function, ρ(R), describing the configurational space available to the ligands is uniquely determined by this external field, Wext(R), and vice versa (1–5,7–9). For example, one may write (1–4,7–9)
| (1) |
where, δF/δW represents a functional derivative of F with respect to W. (Note: if ion-ligand and ligand-ligand interactions are represented by the potential, V(R), the external mean field is represented by Wext(R), and K represents the kinetic energy, we may write the canonical partition function for a solvated protein with an ion bound at a specific site as Q = Tr[exp(K + V + Wext)], where Tr is the classical trace operator. The degrees of freedom, R, are written with respect to the position of the ion. Thus, dependence of Q on the position of the ion is implicit. The Helmholtz free energy of the system is given by F = −RTlnQ, where R is the gas constant and T is the temperature.)
This tautology states that the free energy of an ion at a binding site cannot be considered independently from the structure of the site as represented by ρ(R), which is uniquely determined by the solvated protein/host (or surrounding environment), Wext(R). (Note: the thermodynamic implications of an equilibrium configurational ensemble cannot generally be captured with a single configuration. For this reason, we employ a more general usage of the word, structure, than sometimes inferred. Throughout our work, the word, “structure,” and its derivatives are meant to imply the configurational space available to a system at equilibrium, along with its statistical thermodynamic implications. With this interpretation, one may address structure as it applies to the atoms or molecules of gases, liquids, and solids, alike. The interpretation, of course, also extends to nuclear degrees of freedom in proteins and toy models.) Given this, the relative affinity of the site for ion A with respect to ion B is also determined by such topological control (1–5). This relative affinity leads to the selective free energy, ΔΔFA→B = −RTln(KB/KA) (KX is the equilibrium binding constant of ion X for a given host in a given medium—usually aqueous solution), which is positive if the site is selective for A and negative if it is selective for B. Interpreting observables in K+ channels in terms of such relative affinity typically indicates ∼10-fold to ∼1000-fold K+ selectivity over Na+, depending on the specific channel and type of measurement (see Table S1 in the Supporting Material).
It is possible to design hypothetical constructs that isolate and probe the selective effect of topologically controlling structural properties—order parameters such as coordination number, radius, or the orientation of the coordinating ligands—of a complex composed of a particular type of ligand (1,2,11). According to Eq. 1, such forms of control amount to placing constraints on ρ, and imply a specialized form of Wext, representing a hypothetical host. A thermodynamic deconstruction also allows one to discriminate the topological contribution to ΔΔFA→B, created by coupling the external field, , to the ligands available to form the site, from the contribution yielded by uncoupled ligands alone (1,2,11).
A different hypothetical construct has been introduced, referred to as a simplified “toy” binding site model, in an effort to understand the determinants of selectivity in a K+ selective binding site of a K+ channel (12–14). The toy replaces (representing a solvated K+ channel) with a model potential function, , that acts on some number, Ntoy, of dipolar ligands in the vicinity of a central K+/Na+ cation (Fig. 1 A). The potential, Utoy, was intended to qualitatively reproduce characteristics of the so-called S2 site of a conductive KcsA channel filter as represented by ion-carbonyl oxygen radial distributions from molecular dynamics (MD) simulations (Fig. 1 B) of membrane-embedded KcsA (12–14). In principle, Utoy could possess any one of many functional forms. In past work (12–14), Utoy has been chosen to confine the included ligands around K+/Na+, but not prevent radial collapse of the ligands' oxygen atoms to accommodate the central cation's size.
Figure 1.
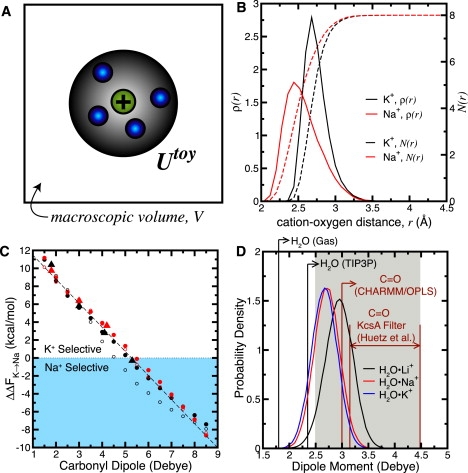
Structure and electrostatics in toy binding site models. (A) Schematic of the toy construct. Ligands are coupled to a potential, Utoy, around a cation, and surrounded by a vacuous macroscopic volume. (B) Linear probability density of finding a carbonyl oxygen atom (ρ(r) − solid lines) and cumulative number of carbonyl oxygen atoms (N(r) − dashed lines) as a function of radial distance, r, from K+/Na+ in site S2 derived from MD simulations of KcsA using the CHARMM force field (25) (structural data provided by P. W. Fowler and M. S. P. Sansom). (C) Dependence of ΔΔFK→Na on the dipole moment of linear model ligands (possessing a carbonyl-like structure) in previously described toys ((12), red triangles; (18), black triangles). Additional data (circles), calculated with standard (black) or modified (red, Table S2) CHARMM parameters, extend the existing trend. These toys used a 3.5 Å radial ion-ligand half-harmonic (solid circles) or LJ (open circles) restraint (see Methods). The general trend (dashed lines) is independent of these different flavors of Utoy. (D) Summary of dipole moments obtained from different models of water and the carbonyl functional group. Distributions of water dipole moment (black, red, and blue curves) around various cations (in a bulk aqueous environment—derived from previous simulations (1,2)) as represented by the polarizable AMOEBA force field are shown. Also demarcated, along the abscissa, are the dipole moments of various models of water and carbonyl species, including gas phase water (20), TIP3P water, CHARMM and OPLS carbonyl moieties, and the range of carbonyl dipole moments represented by the ab initio calculations of Huetz et al. (∼3.15–4.48 D; see Huetz et al. (27)). The gray region (2.5–4.5 D) spans the range of dipole moments suggested for ligands to be intrinsically selective for K+ over Na+ (12–14).
When simulated, the ligands of site S2 in KcsA and the 8-carbonyl toy fluctuate and radially adapt to accommodate K+ or Na+ (12–14). Describing this configurational behavior as “freely fluctuating” and “liquid-like”, K+ selectivity in these environments was attributed to the intrinsic electrostatic properties of the carbonyl ligands comprising the site. It was hypothesized that as long as the coordinating ligands possessed a large carbonyl-like dipole in the range of ∼2.5–4.5 D, a sufficiently unfavorable collapse of the coordination radius around Na+ would ensure positive (K+) selective free energy (12). Moreover, it was suggested that water molecules, with a lesser dipole moment, do not possess the capability of displaying selectivity for K+ over Na+ in the eightfold coordinated construct (13,14).
This field-strength/carbonyl-repulsion requirement (12–14) for K+ selectivity was suggested in the spirit of the electrostatic field strength concept, outlined by Eisenman et al. (15–17). However, Thomas et al. point out that Eisenman's field strength concept indicates smaller ions prefer to complex with ligands of higher field strength than larger ions (18) (Fig. 1 C). Thus, although the field-strength/carbonyl-repulsion hypothesis might seem to explain the absence of selectivity in bulk aqueous solution or in a binding site exposed largely to water, it immediately presents a dichotomy—namely, the notion that ligands possessing a large dipole moment are required for selecting the larger K+ ion over the smaller Na+ ion would imply the Eisenman concept's inverse in the low field strength limit (18).
If we assume that a dipole moment in the range of ∼2.5–4.5 D is required for the toy model to be K+-selective, then an aqueous water molecule, with a dipole moment in the range of ∼2.6–3.1 D, meets this criterion (19–22). Although many classical models of water using pairwise effective potentials capture important physical properties of aqueous solution with an effective molecular dipole moment <2.5 D, models that account for electronic degrees of freedom are able to reproduce such properties while simultaneously representing more realistic electrostatic behavior (20,21,23) (Fig. 1 D). If one takes, together, the dependence of toy selectivity on the ligand dipole moment (Fig. 1C) and the suggested range of dipole moment required for K+ selectivity (∼2.5–4.5 D) (12), why then should a toy model composed of water molecules not be K+-selective? Contrariwise, one could also pose the following question: if a water-based toy model captures the “essential” (13) features of the eightfold coordinated construct while simultaneously providing a liquid-like (12–14) environment, then how could a water-based toy be selective for any ion (because liquid water is definitively nonselective—ΔΔFK→Na ≡ 0)?
Recent theoretical works (1–4,18,24) have provided routes toward resolving the incongruities that give rise to such questions. None of these works suggest carbonyl repulsion does not contribute toward yielding positive ΔΔFK→Na in the eightfold construct. Rather, they point out the simple implication of Eq. 1—that the eightfold binding site, providing nonrigid “molecular complementarity of the selectivity filter with K+” (24), yields a preference for K+ over Na+ precisely because it is not, in any usual sense, liquid-like nor freely fluctuating (1–4,18,24). This complementarity is not encoded into the ligands, but due to topological forces derived from the architecture of the protein acting on the degrees of freedom of the site (1–4,18). There are many ways such forces might modulate selectivity for a given ion. For example, to select K+, they can provide the context for ligand repulsion on binding Na+. They can also provide stiffness in the site to cause thermodynamic cost on binding Na+. Although one may posit many other forms of topological control over selectivity, it has been shown that, in the absence of other constraints, enhancement of the coordination number around both K+ and Na+ above that provided by liquid water is sufficient, although not necessary, to achieve K+ selectivity (ΔΔFK→Na > 0) whether the coordinating moieties are carbonyl groups or water molecules (1–4,18). This does not suggest that enhancement, by the host, of the number of carbonyl moieties ligating K+/Na+ is unrelated to modulation of their mutual repulsion. At this point, we would like to clarify that the above interpretation of the word “host” is identical to what we refer to as “topological control” (1–5) over the coordinated state of a given ion. As such, this interpretation represents the information encoded within the external field, Wext or Utoy imposed on any ion-ligating system, irrespective of the functional form or structural implications of such fields. Thus, throughout this study, we will speak of topological control, the host, and Wext or (for a toy) Utoy, and their free-energetic implications as interchangeable concepts.
This study explores structure and thermodynamics in a variety of toy models where the included cation-ligating species are taken to be water molecules or fictitious linear dipolar moieties. We have previously given analytical and computational evidence that, by design; following from Eq. 1, a toy does not necessarily allow one to isolate the contribution to selectivity arising from ligands whose degrees of freedom are unmodulated by topological control (1). Thus, any given toy model may not necessarily be used to show the previous conclusion (12–14) that selectivity is encoded into the ligands. However, one may use a toy to show that the eightfold coordinated construct is K+-selective irrespective of whether the coordinating ligands are taken to be water molecules or carbonyl moieties. This would be sufficient, although not necessary, to show (because in liquid water, ΔΔFK→Na ≡ 0), that positive (K+) selectivity in such a construct is encoded into, or caused by, the host , which modulates interactions within the complex. In addition, we see that an appropriately designed toy can illustrate what has been observed in a variety of other analyses (1–4,18)—that, in the absence of other constraints on the cationic complex, K+ selectivity may be achieved if a hypothetical host enhances the number of K+/Na+-coordinating dipolar ligands relative to that provided by the bulk aqueous environment. We wish to make it clear that, in this study, an instance of K+ selectivity is meant to imply the condition, ΔΔFK→Na > 0, and not a specific positive value of ΔΔFK→Na. Any instance where a specific value or extent in K+ selectivity is under discussion will state so explicitly.
Methods
We conducted Langevin dynamics simulations (at a temperature of 298 K) on a series of toy binding site models composed of a cation surrounded by Ntoy ≤ 8 dipolar ligands under the influence of an applied external field. The ligands were taken to be either water molecules or fictitious linear dipolar moieties. Fictitious linear carbonyl-like moieties were described by either standard or modified CHARMM carbonyl parameters (Table S2). Water molecules were described by the CHARMM variant of the TIP3P model. Ligand dipole moment was varied in toys using linear ligands by reassigning partial charges on the nuclear sites. To investigate the effect of linear ligand shape versus the bent shape of a water molecule on toy selectivity (as in Fig. 3 B), we investigated a crippled water molecule in which the TIP3P dipole (2.3 D) and dispersive oxygen and hydrogen interactions were maintained, but where one of the hydrogen sites was removed.
Figure 3.
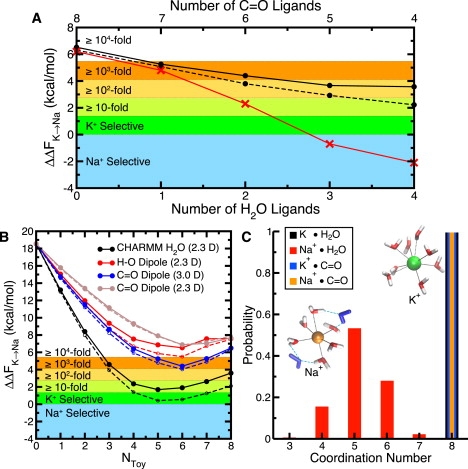
Selectivity and structure in naive toy models using water molecules and/or linear dipoles. (A) Selective free energy as a function of composition (number of included water and carbonyl moieties; force field parameters in Table S2) in 8-ligand toys implementing either a LJ (black solid lines/circles) or a half-harmonic (black dashed lines; open circles) restraining potential. Similar calculations of selectivity from Table 2 of previous works by Noskov and Roux (13,14) (implementing a half-harmonic restraining potential) are shown (red data). (B) Selective free energy as a function of the number of included ligands (Ntoy) where the ligands are taken to be TIP3P water molecules (black), fictitious crippled TIP3P ligands (red), standard fictitious carbonyl-like ligands (blue), and fictitious carbonyl-like ligands with a reduced dipole of 2.3 D (brown). Results from toys using either a half-harmonic (dashed lines; open circles) or LJ (solid lines; solid circles) restraint are shown. (Note also Fig. 6 of Thomas et al. (18), showing similar curves for linear ligands of various dipole moment.) (C) Coordination number (quasicomponent) probability distributions (for ligands around K+ and Na+) in 8-ligand naive toy models using TIP3P water molecules or standard fictitious carbonyl moieties. Results for toys using a LJ restraint for Utoy are shown (results for the half-harmonic restraint are similar; see Fig. S2 and Fig. S3). Inset shows exemplary configurations for K+ (green) and Na+ (orange) in 8-water naive toys. Second-shell water molecules and hydrogen bond interactions are blue.
Three classes of spherically symmetric potential, , were used to probe the effects of different structural constraints on K+/Na+ selectivity. The first of these classes, explained previously (1,12–14), incorporated a volume confining restraint that prevents included ligands from traveling significantly >3.5 Å away from the central cation. Because this toy aims to capture the essential features of the canonical eightfold binding site via a uniformly prescribed volume confinement, we warmly refer to it as the naive toy. We used two types of confining potential. One type (12–14) was a half-harmonic restraint acting on the oxygen atoms of the included ligands, and the other (1) was a Lennard-Jones (LJ) restraint acting on all nuclear sites of the included ligands. The second class of toy used the same functional forms for (half-harmonic or LJ), but the restraint was radially adjusted (depending on the identity of the central cation—K+ or Na+) to suppress formation of secondary solvation shells; thereby enhancing Ntoy-fold coordination. The third class of toy used a harmonic ion-oxygen restraint to enforce structure comparable with site S2 of KcsA as depicted by cation-carbonyl oxygen pair correlation function analyses (12,14,24,25). For each toy, we used free energy methods to carry out the alchemical transformation, K+→Na+. The resulting free energy, , and the difference in K+ and Na+ hydration free energy, , were used to obtain the selective free energy, . (See the Supporting Material for further details pertaining to the calculations of this work.)
Results
Dependence of selectivity on dipole moment in volume-confined models composed of fictitious linear ligands
We extended the ligand dipole-dependent trend in selectivity suggested by Thomas et al. (18). Fig. 1 C shows a scan of ΔΔFK→Na as a function of assigned dipole moment in naive toys incorporating eight linear ligands of carbonyl-like structure. This scan yields the selectivity sequence, K+ > Na+, for fictitious ligands of low dipole moment (≲4.5 D) and the selectivity sequence, Na+ > K+, for very high dipole moment. Ironically, studies positing the field-strength/carbonyl-repulsion hypothesis (12–14), despite suggesting ligands possessing a low dipole should be less selective for K+, also included selective free energy data from 8-ligand toy models that indicate the opposite trend. Considering these data, the trend in Fig. 1 C is in broad agreement with previous observations (12,18).
In Fig. 2 the calculated free energy, , for the alchemical transformation, K+→Na+, within such toys is decomposed into internal energy, ΔU, and entropic, −TΔS, components. The decomposition supports the previous assertion (12–14) that the dominant contribution to arises from the internal energy (Fig. 2 A). If one neglects the entropic contribution, then the scan of dipole moment suggests a range (∼2–3 D) where favorable ion-ligand interactions (ΔUIL) roughly compensate for K+/Na+ dehydration (Fig. 2 B). In this range, unfavorable ligand-ligand interactions (ΔULL) provide for net positive ΔΔFK→Na (Fig. 1 C). At lower dipole moments, the unfavorable ΔULL is diminished, however, ΔΔFK→Na increases with concomitant increase in ΔUIL (Figs. 1 C and 2 B). For highly dipolar or charged ligands, the unfavorable ΔULL is augmented, but ΔΔFK→Na decreases with concomitant decrease in ΔUIL (Figs. 1 C and 2 B). Thus, as suggested by Noskov et al. (12), dipole moments in the range ∼2.5–4.5 D broadly display unfavorable ΔULL to an extent that yields positive net ΔΔFK→Na in the 8-ligand toy. In particular, given standard carbonyl moieties (∼3 D) in the context of eightfold coordination, unfavorable ligand-ligand interaction contributes toward yielding positive ΔΔFK→Na (1,2,4,12–14). However, it does not follow that dipole moments smaller than ∼2.5 D yield ΔΔFK→Na ≤ 0. Nor does it necessarily follow that unfavorable ΔULL must always be the discriminating term yielding positive ΔΔFK→Na for all ligand types.
Figure 2.
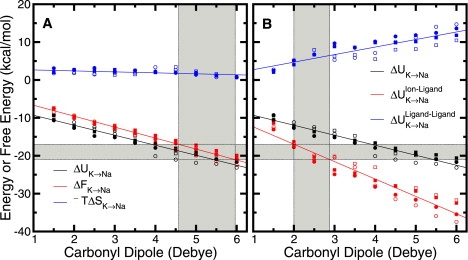
Thermodynamic breakdown for K+ → Na+ in naive 8-ligand toy models as a function of the dipole moment of the carbonyl-like ligands. Shown are results from calculations using standard (circles) and modified (squares, Table S2) CHARMM parameters for a range of assigned dipole moment, where the absolute value of partial charge on C or O is ≲ e. The calculations used either a half-harmonic (solid shapes) or a LJ (open shapes) restraint (see Methods). The gray shaded area along the ordinate axes demarcates a liberal range of values for the difference in K+ and Na+ hydration free energy. The lower and upper bounds of this range were taken from a combination of experiment (28) and model calculations (24). (A) Total internal energy, ΔU, and entropic, −TΔS, contributions to the net free energy, ΔF, for the alchemical reaction as a function of the dipole moment assigned to the fictitious ligands. The shaded gray region along the abscissa indicates a range of dipole moment for which ΔF is roughly equal to the difference in K+ and Na+ hydration free energy. (B) Ion-ligand and ligand-ligand interaction contributions to the net total internal energy as a function of the dipole moment assigned to the fictitious ligands. The shaded gray region along the abscissa indicates a range of dipole moment for which the energy from ion-ligand interactions is roughly equal to the difference in K+ and Na+ hydration free energy.
Structure and selectivity in naive toy models
As observed previously (1), a water-based naive toy model, with a 3.5 Å volume-confining restraint, yields nonzero ΔΔFK→Na for all tested values of Ntoy (Fig. 3, A and B, and Fig. S3 A, black curves). Because liquid bulk water is, by definition, nonselective, this is sufficient evidence to conclude that the model is incapable of isolating the effect of ligands in what may be considered a liquid-like environment. Previously, this finding (using a polarizable water model) prompted us to turn our attention toward population analyses of the coordinative environment provided to K+/Na+ solvated in unquestionably bulk liquid media (1). These analyses provided a mapping of the dependence of ΔΔFK→Na (nK,nNa) on the coordination number of K+, nK, and Na+, nNa, for polarizable and pairwise-additive water models and fictitious carbonyl ligands. The findings generally agreed for both types of water model. However, previous work by Noskov and Roux (13,14), using naive TIP3P water-based toys, asserted that “… progressively replacing the carbonyls by water molecules leads to a loss of selectivity” (ΔΔFK→Na ≤ 0) (Fig. 3 A). As an explanation for such “systematic loss of selectivity (∼1.8 kcal/mol) for each water molecule that replaces a carbonyl group” (13,14), we hypothesized that the confined environment of naive toy models “… may have taken the conventional water model (TIP3P) on which they were based out of the context for which it was designed” (1). The data of Fig. 3, A and B, do not necessarily support this explanation.
Further comparison of results from different water models (Fig. S2) shows a dependence of ΔΔFK→Na on Ntoy that agrees with our previous calculations (1). For Ntoy = 8 water molecules, we obtained ΔΔFK→Na in the range of ∼3.1–4.6 kcal/mol and ∼1.5–3.3 kcal/mol on using LJ and half-harmonic volume-confining restraints, respectively (Fig. S2 and Fig. S3). Although both forms of restraint produce invariably K+-selective environments, the LJ-based toys are seen to provide a greater extent of K+ selectivity (Fig. 3, A and B, and Fig. S3 A). This is due to a small basin in the LJ restraining potential of −0.5 kcal/mol located at 4 Å from the central cation. Despite the quantitative differences seen in the selective free energies, with respect to the naive half-harmonic restraint, the configurational ensembles sampled with either restraint are qualitatively similar (Fig. S2). As we will discuss, neither restraint reproducibly yields an eightfold coordinated construct. When the toy is modified to suppress formation of a second solvation shell with these different flavors of restraint, the differences in selective free energy as a function of Ntoy diminish (Fig. 4).
Figure 4.
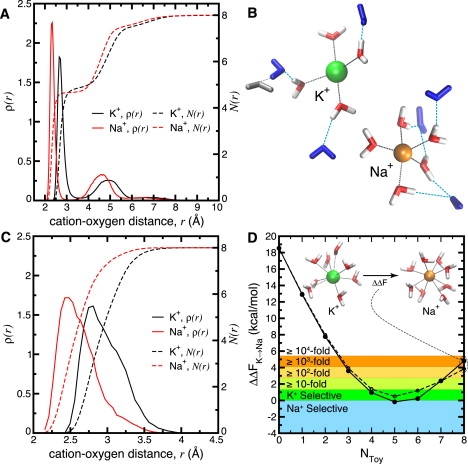
Structure of eight water molecules around K+/Na+ resulting from different applied external fields. (A) Linear probability density, ρ(r), and average cumulative number, N(r), of water oxygen atoms as a function of distance, r, from K+/Na+ in a gas phase cluster environment . In this host-uncoupled environment, both ions prefer direct coordination by 4–5 water molecules, 2–3 water molecules in the second solvation shell, and ∼1 water molecule in the third shell. (B) Representative snapshots of K+ (green) and Na+ (orange) with eight surrounding water molecules in the special case where Utoy = 0 (gas phase). Second coordination shell water molecules and hydrogen bonds are blue, and third shell water molecules and hydrogen bonds are silver. (C) Linear probability density, ρ(r), and average cumulative number, N(r), of water oxygen atoms around K+/Na+ in a toy model where is a volume constraint designed to suppress secondary solvation shells. The result shown is derived from LJ volume confining restraints (results from half-harmonic restraints are shown in Fig. S6). (D) Selectivity in toys where Utoy suppresses formation of more than one solvation shell. Data for toys using a LJ (solid lines; solid circles) or half-harmonic (dashed lines; open circles) potential are shown. Inset shows representative snapshots of K+ (green) and Na+ (orange) surrounded by eight coordinating water molecules provided by the 8-ligand toy.
As seen previously (1), irrespective of the confining potential (LJ or half-harmonic), fictitious carbonyl ligands yield larger K+-selectivity than water molecules in the context of a naive toy (Fig. 3 B). However, this does not seem to be due to the larger magnitude of the carbonyl dipole (∼3.0 D) in comparison to that of the water model (∼2.3 D). When the TIP3P dipole moment is imparted to the fictitious carbonyl ligand or when a crippled TIP3P (linear dipolar) molecule is used (see Methods), we see that ΔΔFK→Na is greater than that arising from the original (∼3.0 D) carbonyl model (Fig. 3 B). As one can expect, we observed differences in the structure (as depicted by the ion-oxygen radial probability density function, ρ(r)) and internal energy contributions, ΔUIL and ΔULL, for naive toys incorporating TIP3P water molecules, crippled TIP3P molecules, and carbonyl-like ligands with a dipole of 2.3 D (data not shown). Irrespective of these differences, Fig. 1 C and Fig. 3 B suggest that, among the different properties possessed by the water and fictitious carbonyl models, the linear shape of the carbonyl model, not necessarily the larger dipole moment, provides for generally larger selectivity in the carbonyl-based versus the water-based toys.
In addition to this finding, we also observed that, although the confines of the naive 8-water toy causes a K+-selective deviation from liquid behavior, it does not actually probe the effect of eightfold Na+-coordination (Fig. 3 C). Taking the standard definition for the number of nearest neighbors (i.e., the coordination number)—the number of oxygen atoms falling beneath the radial position of the first minimum in the ion-oxygen density function, ρ(r)—we find average coordination numbers of 8 and 5–6 are prevalent for the cases of K+ and Na+, respectively (Fig. 3 C, Fig. S2, and Fig. S3). As evidenced by observation of a second maximum in the Na+-oxygen radial probability density (Fig. S2 and Fig. S3), the lower coordination number of Na+ is accompanied by buckling of the complex to form a second solvation shell composed of two to three water molecules, shown pictorially in Fig. 3 C and Fig. S2 C. Occurrence of a marked second maximum is not observed for Na+ in the carbonyl-based naive toys (Fig. 3 C and Fig. S4). Digressively, it is interesting to note that such relaxation in coordination number qualitatively mirrors observations in computational studies of KcsA and NaK (25,26), where bound Na+ elicits either distortions in the selectivity filter or relocation of Na+ such that its preference for lower coordination numbers may be satisfied (in a manner dependent on the particular binding site probed or force field used).
One might suspect that the qualitative differences in ligand packing in the naive carbonyl-based and water-based toys (most notably, the inability of the water-based toy to enforce eightfold Na+ coordination) is due to the larger dipole moment of the carbonyl model versus the water model. However, this is not necessarily (or entirely) the case, because imparting lesser dipole moments to the carbonyl-like ligands does not cause a second maximum in the Na+-oxygen radial density function to appear (Fig. S1). Rather, we find it more likely, as a hypothesis, that the differences in observed coordination number are due to the (nearly 2 times) larger molecular volume and linear geometry of the fictitious carbonyl in comparison to the smaller water molecule, which has a bent geometry (Fig. S2, Fig. S3, Fig. S4, and Fig. S5). Because this issue is tangential, we do not pursue these possibilities further in the naive toy model. It will suffice to say that, for reasons other than the difference in molecular dipole moment, naive water- and carbonyl-based toys (with Ntoy = 8) do not equivalently probe the effect of coordination number on selectivity.
A water-based toy that suppresses formation of secondary solvation shells
The original intention of the naive toy model was to “… include only the ion surrounded by the ligands in the first coordination shell” (14). Therefore, we designed a class of simplified toy model that suppresses formation of a second solvation shell (see Methods). In Fig. 4 the implications of Eq. 1 are demonstrated by showing the drastic structural ramifications of coupling such a host to eight water molecules around K+/Na+. In the special case where (Fig. 4, A and B), coordination of K+ and Na+ by four to five water molecules is most preferable. The remaining water molecules flee the first coordination shell to form second and third solvation shells. An analogous experiment in the case of eight model carbonyl ligands (with ) shows similar behavior, with a preference for five to six K+/Na+-coordinating ligands (Fig. S4, D and E). We note, however, that although these environments allow relaxation in coordination number, they also (like the other toys investigated in this study) do not represent what one would normally refer to as liquid-like environments. By virtue of Eq. 1, an external field representing a liquid-like environment would necessarily, although not sufficiently, reproduce ion-oxygen pair structural analyses of K+/Na+ solvated in liquids (Fig. S5) composed of these model ligands. The gas phase structure implied by Fig. 4, A and B, and Fig. S4, D and E, also make it clear that naive toy models, whether composed of water molecules (Fig. 3 C or Fig. S2) or fictitious carbonyl groups (Fig. 3 C or Fig. S4, A and B), do not represent what might be considered freely moving environments for ligands, as suggested previously (13,14). As such, analyses derived from naive toy models should not be compared to analyses of gas phase droplets.
On coupling eight water molecules to this new class of toy, we see that it inhibits formation of outer second and third solvation shells (Fig. 4 C and Fig. S6). Fig. 4 D displays ΔΔFK→Na as a function of Ntoy for the toy. Prior studies (1) gleaned the selective free energy as a function of the number of water molecules directly coordinating K+ and Na+ in a bulk liquid water environment. Owing to the differences between such (bulk water) analyses and the context provided by this class of toy, we expect quantitative differences in their respective results. Nonetheless, in comparison with naive toy models (Fig. 3), the trend in Fig. 4 D more closely resembles our prior bulk water analyses (1). This trend suggests minimal selectivity in a host that, in the absence of other constraints, enforces five- to sixfold coordination of K+/Na+. Fig. 4 D also suggests a host that enforces sevenfold coordination or greater, in the absence of other constraints, will yield selectivity for K+ over Na+.
Finally, in the case of fictitious carbonyl ligands, this class of toy produced nearly identical selective free energy to the naive toy (3.5 Å restraint) for all Ntoy (Fig. S4, A and B). This agreement may be attributed to the fact that, for carbonyl ligands, both the naive toy and this new class of toy ensure all included Ntoy moieties are in coordination with the central ion (Fig. S4).
A water-based toy that mimics an MD model of KcsA site S2
We designed an 8-ligand TIP3P water-based toy model that mimics the shape of the ion-carbonyl oxygen radial density of site S2 as computed from recent MD studies (25) (Fig. 5 A). Fig. 5 B shows combinatorially optimized spatial distribution functions (1) describing the ensemble of eightfold coordinated configurations provided by the model. As suggested (1), despite the spherically symmetric external field of the toy, the enforced eightfold coordination yields an emergent coordination geometry for K+/Na+, which minimizes the internal strain of the complex. The total internal energy, ΔU (−13.8 kcal/mol), and entropic contribution, −TΔS (+0.9 kcal/mol), to the free energy, ΔFK→Na (−12.9 kcal/mol) imply a very large preference for K+.
Figure 5.
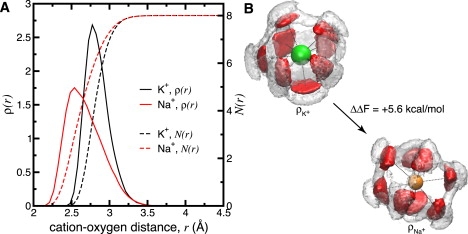
(A) Structural properties of K+/Na+ in a TIP3P water-based 8-ligand toy where Utoy is designed to mimic the shape of the MD-derived ion-oxygen density of site S2 (Fig. 1B; see Methods). Ion-ligand oxygen linear probability density, ρ(r), and cumulative number distributions, N(r), are shown. (B) Combinatorially optimized spatial distribution functions (1) depicting the structure of the K+/Na+-bound water-based toy.
We note that neither ΔFK→Na, nor any set of subdivided internal energy or entropic components obtained from this model can be expected to be identical to that obtained from MD simulations of site S2 in KcsA (12,14,24,25). although the K+/Na+ ion-oxygen radial density and overall structure of this simplified binding site model is qualitatively similar to that derived from a MD simulation of KcsA (Fig. 5), these observables are only projections of the full configurational density, ρK/Na, from the simulated channel. Obviously, the exact configurational ensemble and selectivity of the site cannot be reproduced by solely mimicking such ion-oxygen density functions with water molecules. Given the observed trends for water molecules versus fictitious carbonyl moieties (Fig. 3 B), we might expect the selectivity of this toy to be lower than what would be obtained with fictitious carbonyls. Irrespective of this, the finding that a water-based mimic (Fig. 5) provides such selectivity for K+ implies the principles underlying the design of such a site.
Discussion
Our analyses do not support equivalence among ligand configurational ensembles in what might be considered liquid-like environments (Fig. S5), freely fluctuating/moving environments (Fig. 4, A and B, and Fig. S4, D and E), naive toy model environments (Fig. 3, Fig. S2, and Fig. S4, A and B), or the canonical K+ channel binding site (Fig. 1 B). One must therefore consider that, more generally, ΔΔFK→Na, depends not only on the composition of bulk water (water molecules) versus the composition of the binding site (carbonyl ligands), but also on the cation-coordinative configurational ensemble sampled in either of these media. For a given composition, this ensemble is uniquely determined by the surrounding environment or system remainder or host (those elements of the system that are not ligands), which may be represented in the real channel system by the external field, Wext, and in the toy model by the external field, Utoy (Eq. 1). This external field is different in aqueous solution from that of the host at a K+-selective site (or toy), and therefore, produces different K+/Na+-ligated structure, , than that of the site, (or toy, ). A naive toy model, by design, imposes an external field that results in nonaqueous cation-ligated structure . Thus, by design, it does not isolate the effect of ligand composition (carbonyl groups versus water molecules) on selectivity (1). In the case of water molecules, however, a naive toy does isolate the effect of imposing a prescribed hypothetical ligand-modulating external field, or host (i.e., topological control (1,2)), and therefore (by Eq. 1) configurational ensemble, on selectivity.
Our results indicate that the external field of the naive water-based toy does not reproduce all of the qualitative structural properties of the naive carbonyl-based toy or the canonical construct of a K+ channel binding site. Among other things, the naive 8-water toy does not provide eightfold coordination of both K+ and Na+, and therefore, does not capture the “essential” (13) features of the eightfold coordinated construct. Despite this, in agreement with previous work (1), the volume confinement offered by the water-based toy provides a K+-selective environment (ΔΔFK→Na>0) (Fig. 3, A and B, and Fig. S2) for all Ntoy. A naive 8-carbonyl toy provides eightfold coordination of both K+ and Na+, and larger ΔΔFK→Na than a naive water-based toy (Fig. 3). However, in agreement with the suggestions of Thomas et al. (18) and Eisenman's field-strength concept (15–17), this does not seem to be due to the larger dipole moment of the carbonyl ligand in comparison to the water molecule. We infer this because linear fictitious dipolar moieties of the same size and shape, but with lower dipole moments (<2.3 D) provide larger ΔΔFK→Na than standard (∼3 D) fictitious carbonyl moieties (Figs. 1 and 3). Given this, our results suggest that, in the context of the naive toy, the lower selectivity of water is due to its bent geometry and, to some extent, its smaller volume. These differences in shape, volume, and charge distribution lead to different ion-ligand and ligand-ligand interactions, and different modulation by of both the selective free energy and the sampled configurational ensemble than manifested in the case of carbonyl-like ligands with the same assigned dipole moment (∼2.3 D).
Scanning the dipole moment in naive carbonyl-based models (Figs. 1 C and 2) further informs the suggestion (12–14) that the eight ligands of a naive toy must possess a dipole moment of ∼2.5–4.5 D to yield K+ selectivity. In the case of linear dipolar carbonyl-like ligands, this might only be the case if we assume for all dipole moments (Fig. 2). Our analysis suggests, however, that ΔUIL depends on the dipole moment (Fig. 2). When considering ligands of differing shape, volume, and charge distribution, not only do we observe different packing tendencies in naive toys, but ΔUIL and ΔULL can, in principle, be different. For example, the naive (3.5 Å half-harmonic restraint) toy model using eight TIP3P water molecules (∼2.3 D) yields ΔUIL = −29.5 kcal/mol and ΔULL = 10.7 kcal/mol. Other water models might yield different decompositions of ΔU. However, despite the lesser dipole moment of TIP3P, the unfavorable ΔULL is similar to (or greater than) that of the analogous 8-carbonyl (∼3.0 D) toy model (Fig. 2 B), but ΔUIL far undershoots .
If we choose to reproduce qualitative aspects of an eightfold binding site, as represented by ion-oxygen radial distribution analyses from simulations (12,14,24,25) of KcsA, we find that K+ selectivity is achieved even if the ligands of the toy are chosen to be TIP3P water molecules (Figs. 4, C and D, and 5). In agreement with prior studies (1,2,4,18) the toy calculations of this work suggest that, to achieve such selectivity, it is sufficient, but not necessary, for a host to solely enforce sevenfold coordination or greater for both K+ and Na+ (Fig. 4, C and D).
Encoded information in Wext: topological control as an evolutionary fitness function
The toy models presented in this study illustrate a variety of ways in which the structure, ρK/Na, and selectivity, ΔΔFK→Na, of a K+/Na+ binding site might be influenced. Among the toy attributes we have probed, the ligand type—its volume, shape, and charge distribution—and the functional form of the external field , representing the host, all play roles. From the standpoint of statistical thermodynamics, this finding is a mundane consequence of the definition of the free energy, F, which, for a toy model system with a given type of ion, is a functional of both the potential, V(R), describing ion-ligand and ligand-ligand interactions, and the external field, Utoy(R) or Wext(R). Thus we require no calculations to conclude that the free energy of a system of coordinators, FK/Na, is affected by varying the potential, VK/Na, or equivalently, by varying the identity or composition of the ligands. We also require no calculations to conclude that FK/Na is affected by varying or . However, for a set of ligands of specified identity and in the context of a host, the free energy, FK/Na, and configuration, ρK/Na, are uniquely determined by or arising from the system remainder (7,8). Following from Eq. 1, such external fields dictate the structural design of a binding site.
In a real protein, topological control arising from the system remainder, Wext, determines the number, chemical identity, and particular configurational ensemble with which a given ion is directly coordinated at a site. The molecular evolution of an ion-selective protein may therefore be considered a learning process whereby information about the selected ion is encoded within Wext (i.e., within the folded protein host). From this evolutionary standpoint, the role of the ligands is obviously important, but desultory. Given a set of coordinators, VA/B is defined, and may be designed, in principle, to yield selectivity for either ion A or ion B to an extent required by evolutionary pressure. The interest of a protein engineer or synthetic chemist is in learning what design or encoded information would yield selectivity for ion A or ion B.
In a toy model, a human designer prescribes the degrees of freedom, R, their interactions with the ion and one another, VA/B(R), and the host, (R). For a particular choice in VA/B(R) (ligand composition), the designer encodes ion-selective information into (R). To understand the topological encoding for a particular choice in Utoy(R), let us consider, for a given ion, the intrinsic free energy of the toy (7,8):
| (2) |
The internal energy component, USH, arises from system ion and ligand interactions, with the host, Utoy(R). Perhaps, at a glance, it might seem that, by subtracting this component from the total free energy, F, the remaining component F is caused or controlled by the composition of the binding site—the specified ion and ligands described by the potential V(R). This would be an incomplete assessment. Although F = UIL + ULL − TS contains the internal energy components from ion-ligand and ligand-ligand interactions, such components are, in fact, uniquely determined by Utoy(R) (7,8). For example, given Eqs. 1 and 2, one may show the following (7,8):
| (3) |
Thus, for a particular choice in ion and ligand composition [V(R)], to say that the intrinsic free energy (and, via Eqs. 1–3, the structure—coordination number and configurational ensemble) of a toy model is caused or controlled by the ligands would be to say the tail wags the dog.
Considering this in analyses of K+/Na+ in the context of a toy model, the statement that unfavorable ligand-ligand interactions, ΔULL control or cause selectivity is a restatement of the definition of the selective free energy, , for a special case where ΔΔFK→Na ≈ ΔULL (which would imply , if one assumes ΔUSH − TΔS ≈ 0 (12–14)). This statement, irrespective of whether it is valid, is not a statement of causality for K+ selectivity, because the structure of the site, ρK/Na, and the resulting thermodynamic components of the selectivity, for a given ligand composition, V(R), are necessarily caused by the host, . Given Eq. 1, imposing the host, or external field, , onto the ligands of the site suggests a design principle for K+ selectivity—namely that, in the absence of other structural constraints on the binding site, it is sufficient, but not necessary, that the host enhance the number of nearest neighbors for (or overcoordinate) K+ and Na+ relative to the extent seen in bulk water. This is, obviously, not to say that one could not achieve K+ selectivity with some other choice in , which enforces some other (or additional) structural feature(s) of the complex (e.g., a more explicit penalty for radially accommodating Na+).
Conclusion
We have probed K+ selectivity (i.e., the condition, ΔΔFK→Na > 0) in naive toy binding site models, and in toy models that capture the qualitative features of the eightfold coordinated construct as observed in MD simulations (12,14,24,25) of KcsA. Our results illustrate what can be deduced from statistical thermodynamic considerations—that the extent of K+ selectivity in a given toy can be changed by altering its ligand composition. However, from a formal perspective, a given ligand composition cannot be considered to cause the observed selectivity. Accordingly, for all examined toys, the difference in dipolar strength between the carbonyl moieties that make up the toy and the water molecules that make up bulk aqueous solution does not cause K+ selectivity. Rather, selectivity is due to the fact that the toy exerts an appropriately designed external field, Utoy, on the ligands available to the ion. This field causes the coordinative ensemble experienced by the ion to be different from that experienced in the bulk aqueous environment. There is a multitude of ways this ensemble may be controlled by Utoy to yield K+ selectivity. In principle, Utoy may also be designed to select Na+ (with either water or carbonyls). However, if Utoy, in the absence of other constraints, serves to provide more than six nearest (water or carbonyl) neighbors for both K+ and Na+, the result is a K+-selective environment that either meets or exceeds what can be expected of K+ channels (Table S1).
We believe these findings show, with the toy model, what can be supported by various other means (1–5,18,25,26)—namely, that topological forces arising from the system remainder, or host, are the primary factor in determining the selectivity and structure of a given binding site. Obviously this is not to say that carbonyl moieties and water molecules are equivalent. Nor is it to say that differences in ligand composition, given a particular configurational ensemble for an ion, will yield the same extent (or sign in) selective free energy. Such non sequitur would be formally incorrect. However, our analysis suggests that the intimate connection between the host (i.e., its external field), the coordinative ensemble (structure) it provides, and the free energy of a bound ion ultimately provides a basis for using simplified models to derive design principles for ion-selective binding sites.
Acknowledgments
We thank P. Fowler and M. S. P. Sansom for generously providing cation-oxygen structural analyses from their MD simulations of site S2 in KcsA (25).
This work is supported by the National Science Foundation (0434578, PHYS0216576, and MCB-0413858), and the National Institutes of Health (RR06009).
Supporting Material
References
- 1.Bostick D.L., Brooks C.L., III Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proc. Natl. Acad. Sci. USA. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bostick D.L., Brooks C.L., III The statistical determinants of selective ionic complexation: ions in solvent, transport proteins, and other hosts. Biophys. J. 2008;96 doi: 10.1016/j.bpj.2009.03.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varma S., Sabo D., Rempe S.B. K+/Na+ selectivity in K channels and valinomycin: over-coordination versus cavity-size constraints. J. Mol. Biol. 2008;376:13–22. doi: 10.1016/j.jmb.2007.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varma S., Rempe S.B. Tuning ion coordination architectures to enable selective partitioning. Biophys. J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varma S., Rempe S.B. Structural transitions in ion coordination driven by changes in competition for ligand binding. J. Am. Chem. Soc. 2008;130:15405–15419. doi: 10.1021/ja803575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reference deleted in proof.
- 7.Evans R. The nature of the liquid-vapor interface and other topics in the statistical mechanics of non-uniform, classical fluids. Adv. Phys. 1979;28:143–200. [Google Scholar]
- 8.Henderson J.R., Schofield P. Statistical mechanics of a fluid drop. Proc. R. Soc. Lond. A. 1982;380:211–227. [Google Scholar]
- 9.Hansen J.P., McDonald I.R. Academic Press; San Diego, CA: 1986. Theory of Simple Liquids. 556 p. [Google Scholar]
- 10.Reference deleted in proof.
- 11.Bostick D.L., Brooks C.L., III On the equivalence point for ammonium (De)protonation during its transport through the AmtB channel. Biophys. J. 2007;92:L103–L105. doi: 10.1529/biophysj.107.109165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noskov S.Y., Bernèche S., Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 13.Noskov S.Y., Roux B. Ion selectivity in potassium channels. Biophys. Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 14.Noskov S.Y., Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J. Gen. Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenman G. Cation selective electrodes and their mode of operation. Biophys. J. 1962;2:259–323. doi: 10.1016/s0006-3495(62)86959-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenman G., Horn R. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. J. Membr. Biol. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- 17.Åqvist J., Alvarez O., Eisenman G. Ion-selective properties of a small ionophore in methanol studied by free energy perturbation simulations. J. Phys. Chem. 1992;96:10019–10025. [Google Scholar]
- 18.Thomas M., Jayatilaka D., Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys. J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory J.K., Clary D.C., Liu K., Brown M.G., Saykally R.J. The water dipole moment in water clusters. Science. 1997;275:814–817. doi: 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]
- 20.Guillot B. A reappraisal of what we have learnt during three decades of computer simulations on water. J. Mol. Liq. 2002;101:219–260. [Google Scholar]
- 21.Sharma M., Resta R., Car R. Dipolar correlations and the dielectric permittivity of water. Phys. Rev. Lett. 2007;98:247401. doi: 10.1103/PhysRevLett.98.247401. [DOI] [PubMed] [Google Scholar]
- 22.Badyal Y.S., Saboungi M.-L., Price D.L., Shastri S.D., Haeffner D.R. Electron distribution in water. J. Chem. Phys. 2000;112:9206–9208. [Google Scholar]
- 23.Ren P., Ponder J.W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B. 2003;107:5933–5947. [Google Scholar]
- 24.Asthagiri D., Pratt L.R. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J. Chem. Phys. 2006;125 doi: 10.1063/1.2205853. 024701. [DOI] [PubMed] [Google Scholar]
- 25.Fowler P.W., Tai K., Sansom M.S.P. The selectivity of K+ ion channels: testing the hypotheses. Biophys. J. 2008;95:5062–5072. doi: 10.1529/biophysj.108.132035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miloshevsky G.V., Jordan P.C. Conformational changes in the selectivity filter of the open-state KcsA channel: an energy minimization study. Biophys. J. 2008;95:3239–3251. doi: 10.1529/biophysj.108.136556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huetz P., Boiteux C., Compoint M., Ramseyer C., Girardet C. Incidence of partial charges on ion selectivity in potassium channels. J. Chem. Phys. 2006;124 doi: 10.1063/1.2159483. 044703. [DOI] [PubMed] [Google Scholar]
- 28.Warren G.L., Patel S. Hydration free energies of monovalent ions in transferable intermolecular potential four point fluctuating charge water: an assessment of simulation methodology and force field performance and transferability. J. Chem. Phys. 2007;127 doi: 10.1063/1.2771550. 064509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.