

Abstract
In the injured spinal cord, a glial scar forms and becomes a major obstacle to axonal regeneration. Formation of the glial scar involves migration of astrocytes toward the lesion. Matrix metalloproteinases (MMPs), including MMP-9 and MMP-2, govern cell migration through their ability to degrade constituents of the extracellular matrix. Although MMP-9 is expressed in reactive astrocytes, its involvement in astrocyte migration and formation of a glial scar is unknown. Here we found that spinal cord injured, wild-type mice expressing MMPs developed a more severe glial scar and enhanced expression of chondroitin sulfate proteoglycans, indicative of a more inhibitory environment for axonal regeneration/plasticity, than MMP-9 null mice. To determine whether MMP-9 mediates astrocyte migration, we conducted a scratch wound assay using astrocytes cultured from MMP-9 null, MMP-2 null, and wild-type mice. Gelatin zymography confirmed the expression of MMP-9 and MMP-2 in wild-type cultures. MMP-9 null astrocytes and wild-type astrocytes, treated with an MMP-9 inhibitor, exhibited impaired migration relative to untreated wild-type controls. MMP-9 null astrocytes showed abnormalities in the actin cytoskeletal organization and function but no detectable untoward effects on proliferation, cellular viability, or adhesion. Interestingly, MMP-2 null astrocytes showed increased migration, which could be attenuated in the presence of an MMP-9 inhibitor. Collectively, our studies provide explicit evidence that MMP-9 is integral to the formation of an inhibitory glial scar and cytoskeleton-mediated astrocyte migration. MMP-9 may thus be a promising therapeutic target to reduce glial scarring during wound healing after spinal cord injury.
Keywords: gelatinase, glial fibrillary acidic protein, chondroitin sulfate proteoglycan, extracellular matrix, actin, Rac1
Introduction
After spinal cord injury, one of the major causes leading to the regenerative failure of injured axons is the formation of a glial scar. The glial scar, consisting mainly of reactive astrocytes, not only forms a nonpermissive physical barrier to the regenerative axons but also expresses inhibitory molecules such as chondroitin sulfate proteoglycans (CSPGs), which chemically arrest the regrowth of injured axons across the lesion (Silver and Miller, 2004). Formation of the glial scar is a complex process that is primarily attributed to astrocyte migration in the vicinity of the lesion with a minor contribution resulting from astrocyte proliferation (Ridet et al., 1997; Fitch et al., 1999; McGraw et al., 2001; Matyash et al., 2002). The migratory behavior of reactive astrocytes depends on the dynamics of actin cytoskeleton mediated by the small GTPases Rho and Rac1 (Etienne-Manneville and Hall, 2002; Bourguignon et al., 2007), integrity of the intermediate filament including vimentin and glial fibrillary acidic protein (GFAP) (Menet et al., 2003), cell adhesion (Okada et al., 2006), and water fluxes across the plasma membrane (Saadoun et al., 2005).
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that can degrade a variety of protein constituents in the extracellular matrix (ECM) and thus remodel the pericellular microenvironment for cell translocation (Sternlicht and Werb, 2001). As such, MMPs modulate numerous pathological and tissue repair processes in the nervous system particularly those that require cell migration (Yong et al., 2001). The modulatory effect of MMPs in this context is largely dependent on the temporospatial expression of different MMP isoforms and their cellular sources (Yong, 2005). Various MMPs are upregulated after spinal cord injury (Duchossoy et al., 2001; Wells et al., 2003). Thus, in the injured spinal cord, it is likely that MMPs may exert both beneficial and detrimental effects, depending upon which members of the family are involved and when and where they are actively expressed.
We have previously shown that MMP-2 and MMP-9 are expressed in reactive astrocytes after spinal cord injury (Noble et al., 2002; Hsu et al., 2006), raising the possibility that these MMPs may participate in the formation of the glial scar. Consistent with that possibility, recent studies have shown that astrocyte migration is modulated by MMPs (Ogier et al., 2006; Takenaga and Kozlova, 2006). Although MMP-2 appears to promote astrocyte migration in vitro (Ogier et al., 2006), our previous study demonstrated that mice deficient in MMP-2 develop a more extensive glial scar in the injured spinal cord (Hsu et al., 2006). Interestingly, these mice also show a compensatory increase in MMP-9, raising the intriguing possibility that the more severe glial scar results from the compensatory upregulation of MMP-9. Here we test the hypothesis that MMP-9 facilitates astrocyte migration and contributes to the formation of a glial scar in the injured spinal cord. To test this hypothesis, we examined the relationship between MMP-9, glial scarring in vivo, and astrocyte migration in vitro. Our results offer new insights into the role of MMP-9 in wound healing after spinal cord injury.
Materials and Methods
Animals.
These studies were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco and in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Young breeding pairs on an FVBn background ranged in ages from 3 to 6 months. MMP-2 null, MMP-9 null, and wild-type progeny on an FVBn background were generated by breeding homozygous females and males of respective genotypes. Mice were housed in a controlled environment at 25°C with a 12 h light-and-dark cycle. Food and water were provided ad libitum. The genotypes of animals were identified by the PCR using tail tissue and specific oligonucleotide primers (Itoh et al., 1997, 1998; Vu et al., 1998; Ducharme et al., 2000) and confirmed by gelatin zymography using samples of blood.
Contusive spinal cord injury.
Male MMP-9 null and wild-type mice, weighing 30–35 g (∼4–6 months of age), were anesthetized (2.5% tribromoethanol, 0.02 ml/g body weight, i.p., T48402; Sigma) and subjected to a moderate contusion injury to the spinal cord as we described previously (Noble et al., 2002). Body temperature of the mice was maintained at 37°C with a warming pad throughout the surgery and during the recovery from anesthesia. Postoperative care included subcutaneous administration of 1 ml of 0.9% Sulfamethoxazole-Trimethoprim (GensiaSicor) and manually voiding the urinary bladder twice a day.
Tissue preparation and immunohistochemistry.
Animals were deeply anesthetized 42 d postinjury. Ice-cold normal saline was perfused transcardially, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4 for 10 min. The spinal cord was removed and immersed in the same fixative at 4°C for 4 h. Then the spinal cord was transferred into 30% sucrose in 0.1 m PBS for cryoprotection. Cryosections of the spinal cord were cut transversely at 20 μm in a cryostat, mounted onto glass slides, and stored at −20°C for further processing. Another two sets of spinal cords were obtained at 5 and 14 d postinjury, respectively, and cut horizontally.
The following primary antibodies were used for immunohistochemistry: Cy3-conjugated mouse anti-GFAP (to label astrocytes, 1:400, clone G-A-5, C9205; Sigma) and mouse anti-CSPGs (1:200, clone CS-56, C8035; Sigma). Cryosections were rinsed with 0.01 m PBS three times and blocking solution consisting of 2% normal goat serum and 0.1% bovine serum albumin in PBS at room temperature for 5 min. Incubation of the primary antibodies, diluted in above blocking solution, was performed in a humidified box at 4°C overnight. Sections immunolabeled by anti-GFAP were rinsed with PBS and coverslipped with ProLong Antifade (P-7481; Invitrogen); those immunolabeled by anti-CSPGs were further incubated with biotinylated goat anti-mouse IgM (1:200, 115–065-075; Jackson ImmunoResearch) for 30 min. The labeling of anti-CSPGs was visualized by a biotin-avidin peroxidase reaction using a Vectastain Elite ABC kit (PK-6100; Vector Laboratories) in combination with 0.05% diaminobenzidine as the chromogen and 0.001% H2O2 in 0.01 m PBS. Sections of different genotypes were grouped together and processed simultaneously in the same experimental condition to minimize staining variables for subsequent quantitative analyses. An additional group of sections was processed without using primary antibodies to serve as the negative control.
Assessment of the glial scar.
The astroglial scar was visualized by Cy3-conjugated anti-GFAP antibody. The severity of glial scarring was analyzed semiquantitatively 42 d postinjury (n = 6 per genotype) by scoring its complexity and extent as we originally described (Hsu et al., 2006). Briefly, the transverse section of the injured cord was subdivided into 12 sectors by superimposing a grid onto the digitized image of the section using Photoshop 7.0 (Adobe Systems). In each sector, the pattern of the glial scar, distribution of astrocytes, organization of astrocytic processes, astrocytic hypertrophy, and the intensity of GFAP immunoreactivity were evaluated and then a score ranging from 0 to 3 was given. A score of 0 indicates no evidence of glial scar formation, whereas a score of 1–3 represents increasing complexity of the glial scarring. The total score obtained from all 12 sectors in each transverse section reflected the severity (complexity and/or extent) of glial scar formation. In the lesion, GFAP-quiescent regions were excluded from the analysis because they were composed of nonastrocytic components such as macrophages and tissue matrix. Nine transverse sections, at intervals of 480 μm, were sampled across the entire lesion epicenter in each animal.
Assessment of CSPG immunoreactivity.
The intensity of CSPG immunoreactivity in the injured cord was quantified 42 d postinjury (n = 7 per genotype) as we previously described with some modifications (Hsu et al., 2006). Briefly, digital images of the immunostained sections were taken under consistent exposure parameters and were imported to the NIH ImageJ image processing software (1.33u). The intensity of background staining was evaluated on a reference section, which was ∼1500 μm rostral to the lesion where there was no evident tissue damage. First, the total number of pixels in the reference section was obtained from the histogram. Then a threshold value was determined so that only 1% of the darkest pixels remained in the reference section. This threshold value, which eliminated 99% of background staining, was applied to the images of 4–5 sections sampled from the lesion epicenter of the same animal to reveal the pixels that were darker than the background staining. The number of these dark pixels, which represented the intensity of CSPG immunoreactivity, was counted and the percentage of these pixels over the total pixel number of a section was calculated. All sampled sections in each animal were averaged and the mean of each genotype was reported.
Astrocyte culture.
Cortical astrocytes were cultured from the cerebral cortex of MMP-2 null, MMP-9 null, and wild-type mice at postnatal day 1 or 2, based on a previously described protocol with some modifications (Rose et al., 1993). The cerebral cortices were dissected from the brainstem, while the hippocampus, adherent meninges, and blood vessels were removed. The cerebral cortices were triturated repeatedly in 0.05% trypsin and 0.05% DNase in Ca2+ and Mg2+ free Hank's balanced salt solution. Cells were pelleted by centrifugation at 250 rpm for 5 min, resuspended, filtered through a 70 μm nylon cell strainer, and plated in a poly-l-lysine-coated flask containing Earle's MEM medium supplemented with 10% fetal bovine serum, 22 mm glucose, and 2 mm GlutaMAX-1 (35050–061; Invitrogen). After 24 h of incubation in 5% CO2 at 37°C, the flask was shaken vigorously to remove debris and unattached cells before fresh medium replacement. Shaking was repeated before changing the culture medium at 3, 5, and 7 d after astrocyte isolation and weekly thereafter. The purity of astrocyte cultures was ∼95% and was determined by the number of cells immunostained with GFAP relative to the total nuclei that had been labeled with Hoechst dye. For all in vitro studies described below, three triplicates, each of which consisted of three wells, were examined in each genotype or treatment at any given time point.
Scratch wound assay.
Cultured astrocytes were trypsinized and replated into tissue culture-treated 24-well plates (BD Falcon, BD Biosciences) at a concentration of 2 × 105 cells/well after the first passage. No ECM substrates were coated on the bottom of the culture plates. Five to seven days later when astrocytes reached confluence, a denuded area was produced by scratching the inside diameter of the well with a 10 μl pipette tip (Bourguignon et al., 2007). After 3 rinses with Earle's MEM medium, fresh culture medium containing 10 μm cytosine arabinofuranoside (C1768; Sigma) was added into each well to inhibit cell proliferation. One digital photograph was taken from each well immediately after the scratch wound was made (0 h) and at 24, 48, and 72 h later. The denuded area was gradually covered by migrating astrocytes. Using the NIH ImageJ image processing program, the size of the denuded area was determined at each time point from digital images. The astrocyte-covered area was obtained by subtracting the denuded area void of astrocytes at any given time point from the original denuded area measured at 0 h in the same well.
To determine the dependency of migration on MMPs and actin cytoskeleton, astrocytes were treated with the following inhibitors: GM6001, a general inhibitor of MMPs (364205; Calbiochem); MMP-9 inhibitor (444278; Calbiochem); cytochalasin D (20 μg/ml, C8273; Sigma); and Rac1 inhibitor (50 μm, 553530; Calbiochem). Concentrations for GM6001 and the MMP-9 inhibitor were based upon dose–response experiments, conducted in wild-type cultures, and at ranges previously shown to inhibit the migration of various cells including astrocytes in vitro (Xu et al., 2004; Mori et al., 2006; Ogier et al., 2006). GM6001 was dissolved in sterile-filtered dimethyl sulfoxide (DMSO) with final concentrations at 20 μm, 40 μm, and 80 μm. A GM6001 analog (364210; Calbiochem) was used as the negative control at a concentration of 20 μm. Likewise, MMP-9 inhibitor was dissolved in DMSO at 0.5 μm, 1.0 μm, and 2.0 μm, while the DMSO was used as the vehicle control.
Fluorescence staining of actin cytoskeleton.
To test the effects of various inhibitors on the actin cytoskeleton rearrangement, astrocytes cultured from wild-type, MMP-2 null and MMP-9 null mice were treated with 20 μg/ml cytochalasin D, 50 μm Rac1 inhibitor, or no drug for 1 h at 37°C. Subsequently, a scratch wound was made on the astrocyte monolayer using a 10 μl pipette tip as described above. After 24 h of incubation at 37°C, astrocytes were fixed with 2% paraformaldehyde, permeabilized by 90% ethanol, and incubated with Texas Red-conjugated phalloidin (Invitrogen). Another set of astrocytes without a scratch wound was stained simultaneously. The fluorescence-labeled astrocytes were examined with a confocal laser-scanning microscope.
Astrocyte proliferation assay.
Proliferating astrocytes of different genotypes were identified by 5-bromo-2′-deoxyuridine (BrdU) incorporation. After the first passage, astrocytes were transferred into a 24-well plate at 1 × 105 cells/well with a poly-l-lysine-coated coverslip on the bottom of each well. At ∼40–50% confluence, astrocytes were rinsed with Earle's MEM medium three times, incubated in nonserum culture medium for 24 h, and incubated with 30 μm BrdU (B9285; Sigma) in culture medium containing 10% fetal bovine serum for 2 h. The astrocytes were fixed with cold 70% ethanol at 4°C for 20 min, air dried, and rinsed with 2 N HCl for 20 min for DNA denaturation. After three washes with PBS, astrocytes were reacted with Alexa Fluor 594-conjugated mouse monoclonal anti-BrdU (1:100, A-21304; Invitrogen) diluted in blocking and permeablizing solution containing 2% normal goat serum, 0.2% Triton X-100, and 0.1% bovine serum albumin in PBS for 4 h at room temperature. Double labeling was performed by using rabbit polyclonal anti-GFAP (1:200, Z0334; Dako, Cytomation) diluted in above blocking and permeablizing solution, followed by fluorescein isothiocyanate-conjugated anti-rabbit secondary antibody. The nuclei of astrocytes were stained with 300 nm 4′,6-diamidino-2-phenylindole (DAPI; D3571; Invitrogen) for 3 min and the coverslip was mounted onto a slide. A proliferative astrocyte was defined by positive staining for anti-BrdU, anti-GFAP, and DAPI. The numbers of proliferative astrocytes were counted using an epifluorescent microscope with a 10× objective within six adjacent fields in the center of the coverslip. Proliferative astrocytes were expressed as a percentage of the number of BrdU positive cells relative to the total number of DAPI positive nuclei.
Astrocyte adhesion assay.
Astrocytes were replated into a poly-l-lysine-coated 12-well plate at 1 × 105 cells/well after the first passage and incubated for 24 h. The wells were shaken slightly and rinsed with Earle's MEM medium 3 times to remove unattached cells. By using an inverted phase contrast microscope with a 20× objective, the number of adherent astrocytes was determined in eight adjacent fields in the center of the well. Results of MMP-2 and MMP-9 null astrocytes were normalized with that of the wild-type astrocytes.
Astrocyte viability assay.
To assess the influence of MMP deficiency on astrocytic viability, the metabolic activity of mitochondria was examined by a quantitative colorimetric assay using [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl] tetrazolium bromide (MTT; M2128; Sigma) as previously described (Mosmann, 1983). Briefly, cultured astrocytes were transferred into a 96-well plate after the first passage at a concentration of 1 × 104 cells/100 μl medium/well and incubated for 1, 2, and 3 d. After 10 μl of 0.5% MTT was added into the well, the astrocytes were further incubated for 4 h. Then the astrocytes were dissolved with 100 μl of acidic isopropanol containing 0.04 N HCl. The optical density of the resultant formazan product was measured at 550 nm by a plate reader.
Gelatin zymography.
The gelatinase levels of MMP-2 and MMP-9 in the injured spinal cord were examined 7 d postinjury by gelatin zymography as we previously described (Noble et al., 2002; Goussev et al., 2003). Injured MMP-9 null and wild-type mice (n = 3 per genotype) were deeply anesthetized and then the spinal cord segments containing the lesion were dissected and homogenized in lysis buffer consisting of 28 mm Tris-HCl, 22 mm Tris-base, pH 8, 150 mm NaCl, 1% Nonidet P-40 (74385; Fluka BioChemika), 0.5% sodium deoxycholate, and 0.1% SDS. In another group of wild-type mice receiving only laminectomy, the matched region of the uninjured spinal cord was used as the sham control. After the protein concentration of the homogenates was determined by the bicinchoninic acid method (BCA protein assay kit, 23225; Pierce), equal amounts of protein (5 μg) were loaded on a Novex 10% zymogram gel (EC61752; Invitrogen) and separated by electrophoresis with 120 V (6 mA) at 4°C for 120 min. The gel was then incubated with renaturing buffer (LC2670; Invitrogen) at room temperature for 30 min to restore the gelatinolytic activity of the proteins. After incubation with developing buffer (LC2676; Invitrogen) at 37°C for 48 h, the gel was stained with 0.5% Coomassie Blue for 60 min and then destained with 40% methanol containing 10% acetic acid until appropriate color contrast was achieved. Clear bands on the zymogram were indicative of gelatinase activity.
Conditioned media from cultured wild-type astrocytes were collected and subjected to gelatin zymography. After astrocytes reached confluence in a 30 mm culture dish, the serum-containing medium was replaced with 1 ml of fresh serum-free medium. Then the astrocyte monolayer was treated with 6–7 scratches or transforming growth factor β (TGF-β) (10 ng/ml; 240-B; R & D Systems). After 72 h of incubation, the conditioned media were concentrated using Microcon filter (Millipore) with a 50,000 MW cutoff according to the manufacturer's instructions. Equal amounts of protein (5 μg) in the concentrated media were loaded on a gel for zymography as described above.
Statistical analysis.
Data obtained from in vitro studies of cultured astrocytes were examined by the ANOVA, followed by Bonferroni's post hoc test for multiple comparisons between the means of genotypes, treatments, or time points. Quantitative evaluation of the glial scar and CSPG immunoreactivity were performed by 2 observers who were blinded to the experimental conditions and the means obtained from MMP-9 null and wild-type mice were compared by Student's t test. Data are presented as means ± SD. A statistically significant difference is defined at p < 0.05.
Results
MMP-9 facilitates glial scarring and the expression of CSPGs in injured spinal cord
To test the hypothesis that MMP-9 contributes to glial scar formation in vivo, we examined the severity of glial scarring between wild-type mice and MMP-9 null mice by GFAP immunostaining after a contusive spinal cord injury. The difference in the pattern of glial scarring was not obvious between the wild-type mice and MMP-9 null mice at 5 d (data not shown) and 14 d postinjury (Fig. 1A,B). Nevertheless, wild-type mice had a remarkably more complicated pattern of glial scarring with prominent bundling of thick astrocytic processes than MMP-9 null mice 42 d postinjury (Fig. 1C–F). In addition, the intensity of GFAP immunoreactivity appeared higher in wild-type mice compared with MMP-9 null mice at this time point. The severity of glial scar formation was analyzed by a semiquantitative scale, based on the complexity and extent of the scarring 42 d postinjury (Fig. 1G). Wild-type mice had a significantly higher total score than MMP-9 null mice (180 ± 35 vs 136 ± 21, p < 0.05). We further analyzed whether such a higher total score in wild-type mice was a result of a more complicated (i.e., higher average score per sector) or more extensive (i.e., more sectors scored ≥1) glial scarring. We found that the average score per sector was significantly higher in the wild-type mice than in the MMP-9 null mice (2.4 ± 0.2 vs 2.2 ± 0.1, p < 0.05), although the number of sectors scored ≥1 was comparable between these two groups (74 ± 9 vs 63 ± 11). These findings indicate that wild-type mice have a more complicated pattern of glial scarring than MMP-9 null mice 42 d postinjury.
Figure 1.
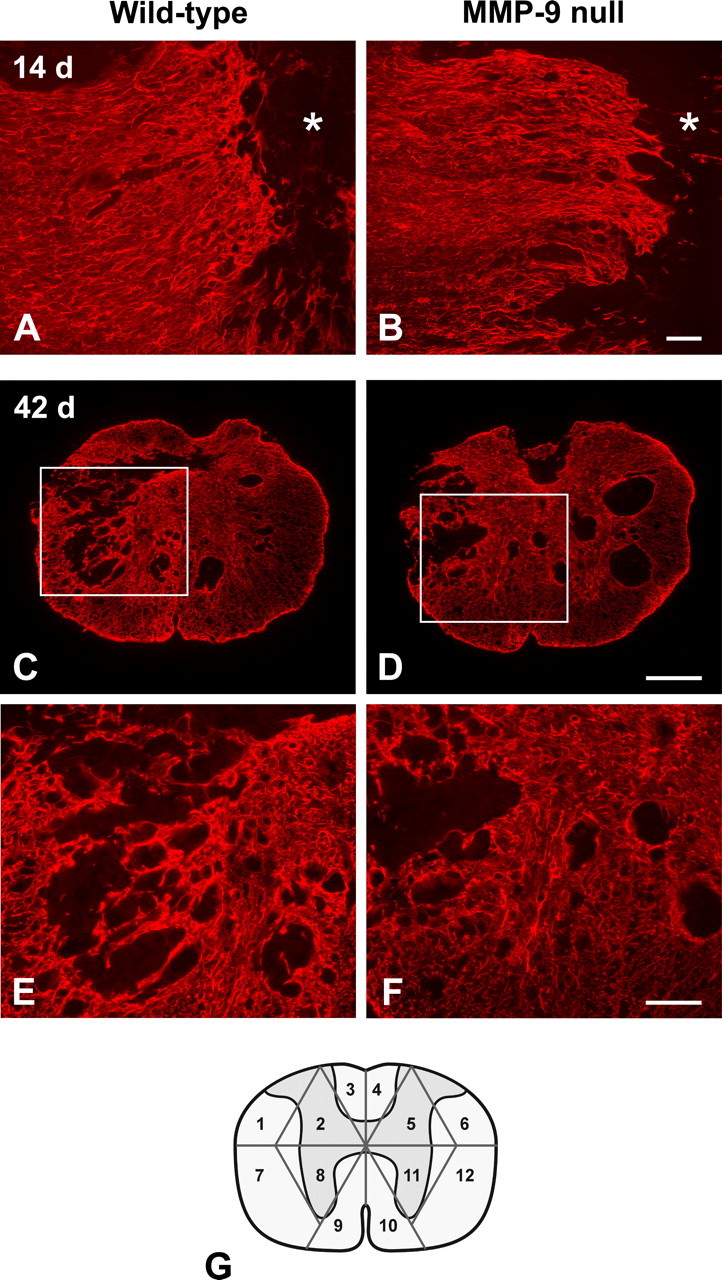
Glial scar formation visualized by GFAP-immunofluorescence in MMP-9 null and wild-type mice. A moderate reactive astroglial border is formed between the GFAP-quiescent area in the lesion epicenter (asterisks) and the intact tissue 14 d post-injury (A, B; horizontal sections). The pattern of glial scarring appears similar between the MMP-9 null and wild-type mice. At 42 d postinjury, however, the wild-type mouse exhibits a more severe glial scar formation in the lesion, evidenced by intricate tangling of astrocytic processes and intense GFAP immunoreactivity, than the MMP-9 null mouse (C, D; transverse sections). At higher magnification (E, F; taken from boxed area in C and D, respectively), entangled astrocytic processes are densely bundled to form a robust trabecular meshwork in the wild-type mouse, whereas the glial scarring is relatively modest in MMP-9 null mouse. The sampling strategy used in our semiquantitative analysis of the glial scar is illustrated in the diagram (G). Scale bars: A, B, E, F, 100 μm; C, D, 250 μm.
Expression of inhibitory molecules CSPGs by reactive astrocytes is known to cause regenerative failure of injured axons in the CNS (Davies et al., 1997; McKeon et al., 1999). Although the immunostaining of CSPGs was similar between wild-type mice and MMP-9 null mice at 5 d postinjury (data not shown), wild-type mice appeared to have higher CSPG immunoreactivity than the MMP-9 null mice at 14 d and, in particular, 42 d postinjury (Fig. 2). Quantitative assessment revealed that wild-type mice had significantly higher CSPG immunoreactivity than the MMP-9 null mice (3.8 ± 1.2% vs 2.4 ± 0.7%, p < 0.05) 42 d postinjury. In summary, these findings show that MMP-9 governs the formation of a complex and inhibitory glial scar in the injured spinal cord.
Figure 2.

Immunostaining of CSPGs (clone CS-56) in MMP-9 null and wild-type mice after spinal cord injury. At 14 d postinjury, CSPG immunoreactivity appears at the interface (arrowheads) between the lesion epicenter (asterisks) and the intact tissue in both wild-type and MMP-9 null mice (A, B; horizontal sections). The wild-type mouse displays stronger CSPG immunostaining than the MMP-9 null mouse particularly at 42 d postinjury (C, D; transverse sections). Scale bars: A, B, 100 μm; C, D, 250 μm.
MMP-9 deficiency does not alter MMP-2 expression
We previously demonstrated that MMP-2 is beneficial to wound healing because spinal cord-injured MMP-2 null mice exhibit a more complex and inhibitory glial scar, reduced white matter sparing, and impaired locomotor recovery (Hsu et al., 2006). Importantly, these MMP-2 null mice also show a compensatory increase in MMP-9. Thus, we determined whether the beneficial effect on reduced glial scarring seen in injured MMP-9 null mice is due to a compensatory upregulation of MMP-2. Gelatin zymography revealed the expression of pro- and active MMP-2 in the injured spinal cords of both MMP-9 null and wild-type mice at 7 d postinjury (Fig. 3), a time point when the expression of MMP-2 peaks in the lesion (Goussev et al., 2003). However, there were no qualitative differences in the expression of MMP-2 between these genotypes. These findings suggest a more direct involvement of MMP-9 in glial scar formation.
Figure 3.

Representative gelatin zymogram showing gelatinase levels in the injured spinal cord at 7 d postinjury. Both MMP-9 and MMP-2 are upregulated in the wild-type mouse compared with the sham-operated mouse that receives only laminectomy. The intensity of both pro- and active MMP-2 in the wild-type mouse is comparable with that of the MMP-9 null mouse, suggesting the expression of MMP-2 is unaltered in the later.
Astrocyte migration is dependent on MMP-9
To determine whether glial scar formation is a result of MMP-facilitated astrocyte migration, we first examined the expression of MMPs by cultured wild-type astrocytes treated without or with a scratch wound or exogenous TGF-β, an injury-induced cytokine known to stimulate astrocyte reactivity (Asher et al., 2000; Smith and Strunz, 2005). In agreement with previous findings (Le et al., 2003; Ogier et al., 2006; Takenaga and Kozlova, 2006), gelatin zymography showed that wild-type astrocytes expressed both MMP-2 and MMP-9 in vitro, with higher levels of both gelatinases after a scratch wound or TGF-β stimulation (Fig. 4A). We next determined whether MMPs, particularly MMP-9, are involved in astrocyte migration. When cultured wild-type astrocytes were exposed to varying concentrations of GM6001, a general inhibitor of MMPs, after a scratch wound, their migration was significantly impaired (Fig. 4B). Similarly, wild-type astrocytes treated with a more selective MMP-9 inhibitor showed significantly reduced migration at all concentrations of the inhibitor (Fig. 4C). These findings suggest that MMP-9 facilitates astrocyte migration.
Figure 4.
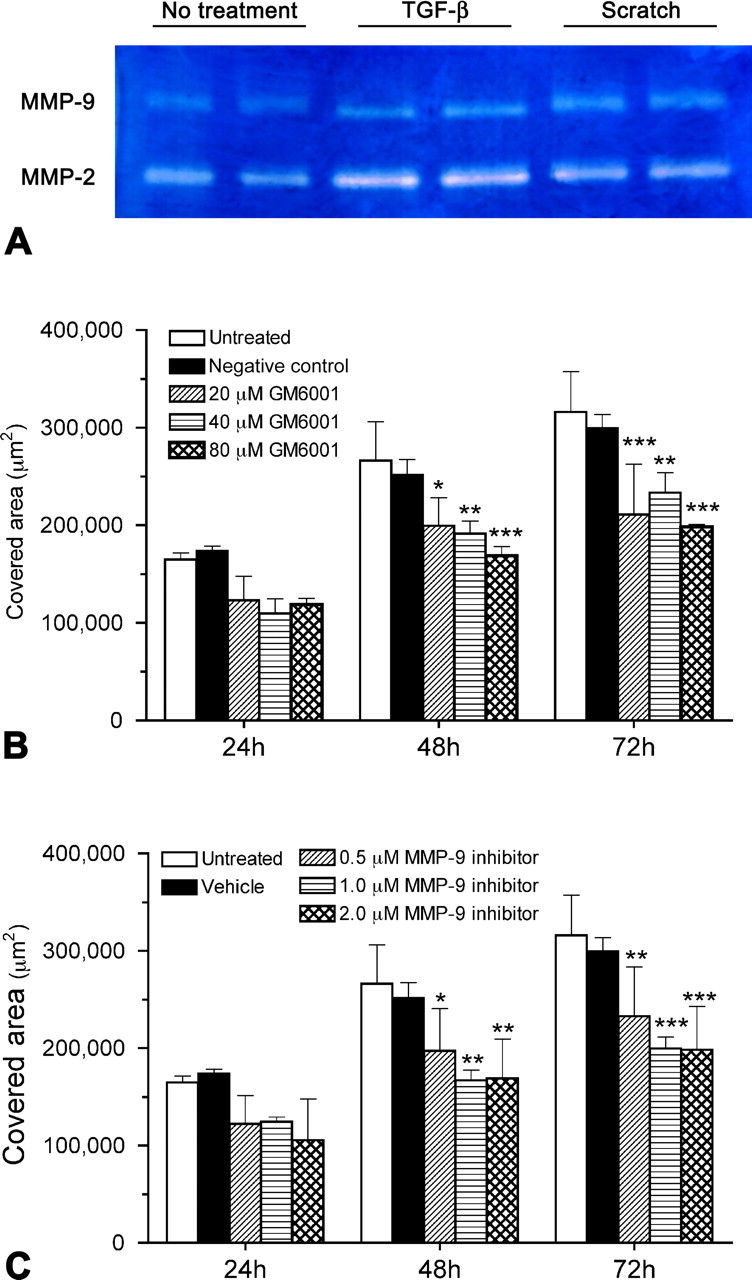
Expression of MMPs by cultured wild-type astrocytes and the effect of MMP inhibitors on astrocyte migration. A, By using conditioned media, gelatin zymography demonstrates that cultured wild-type astrocytes express both MMP-2 and MMP-9, at relatively high levels 72 h after scratches are made or TGF-β treatment. B, C, In the scratch wound assay, wild-type astrocytes are treated with various concentrations of GM6001 (B) or an MMP-9 inhibitor (C). The area covered by migrating astrocytes is measured over a period of 72 h after the scratch is made. All the concentrations examined can effectively inhibit astrocyte migration compared with the untreated group 48 and 72 h after the scratch. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the untreated group of the same time point. Error bars indicate SD.
MMP-9 modulates migration in MMP-2 null astrocytes
We have previously demonstrated that MMP-2 null mice manifest a more extensive glial scar, along with a compensatory increase in MMP-9 activity in the injured spinal cord, compared with the wild-type mice (Hsu et al., 2006). To elucidate whether this enhanced scarring results from a deficiency in MMP-2 and/or upregulation of MMP-9, we compared the migratory behavior of MMP-2 null and wild-type astrocytes in the scratch wound assay (Fig. 5). Similar to in vivo findings, migration was enhanced in MMP-2 null astrocytes. To determine the dependency of this enhanced migration on MMP-9, migration was assayed in the presence of an MMP-9 inhibitor. Migration of MMP-2 null astrocytes was significantly reduced in the presence of the MMP-9 inhibitor, which was in accordance with the attenuated migration of cultured MMP-9 null astrocytes. These findings support the hypothesis that MMP-9 plays a pivotal role in astrocyte migration in vitro.
Figure 5.
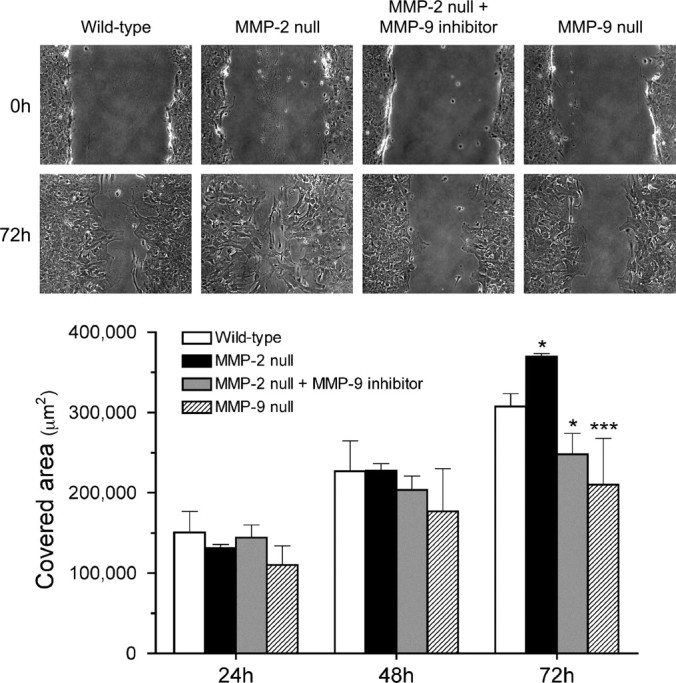
Migration of cultured astrocytes isolated from MMP-2 null, and MMP-9 null mice in the scratch assay. Representative photographs demonstrate the wound closure processes 0 and 72 h after the scratch is made. Notably, MMP-2 null astrocytes migrate faster and thus cover more denuded area than the wild-type astrocytes 72 h after the scratching. At the same time point, however, MMP-2 null astrocytes treated with 1.0 μm MMP-9 inhibitor migrate significantly slower than the wild-type astrocytes. A similar outcome of attenuated migration is also observed in MMP-9 null astrocytes. The chart summarizes the quantitative analysis of the area covered by astrocytes at various time points. The results demonstrate that MMP-9 deficiency adversely affects astrocyte migration. *p < 0.05, ***p < 0.001, compared with the wild-type at 72 h. Error bars indicate SD.
MMP-9 regulates actin-cytoskeleton organization in astrocytes
The actin cytoskeleton participates in the morphological changes and migration of astrocytes (Baorto et al., 1992; Ramakers and Moolenaar, 1998; Etienne-Manneville and Hall, 2002; Höltje et al., 2005; Bourguignon et al., 2007). In particular, Rho GTPases, such as Rac1 are known to play an important role in regulating filamentous actin (F-actin) formation and those activities required for forming astrocyte membrane protrusions and cell migration (Etienne-Manneville and Hall, 2002; Bourguignon et al., 2007). Since MMPs alter astrocyte migration, we hypothesized that these proteases would affect cytoskeletal organization.
Accordingly, we determined whether MMP-2 and MMP-9 alter the distribution of actin cytoskeleton during astrocyte migration. We observed that F-actin fibers, assessed by fluorescent phalloidin, were mostly distributed in the cell body of both wild-type (Fig. 6A-i) and MMP-2 null astrocytes (Fig. 6B-i). In contrast, only cortical actin underlying the plasma membrane and very few cell body F-actin fibers were detected in MMP-9 null astrocytes (Fig. 6C-i). These data suggest that MMP-9 regulates F-actin assembly and distribution in astrocytes. As controls, we treated astrocytes with cytochalasin D, which impairs F-actin formation, or a Rac1 inhibitor, which blocks Rac1 activation, resulting in the disassembly of both cell body F-actin fibers and cortical actin, along with dramatic changes in the cell shape in astrocytes of all 3 genotypes examined (Fig. 6A-,B-,C-ii,-iii). In the scratch wound assay, F-actin-associated membrane protrusions projecting from the wounding edges toward the migration front were evident in both wild-type (Fig. 6D-i) and MMP-2 null astrocytes (Fig. 6E-i), but negligible in MMP-9 null astrocytes (Fig. 6F-i). Treatment with either cytochalasin D or Rac1 inhibitor resulted in F-actin disarrangement, loss of scratch-induced membrane protrusions, and cell rounding in astrocytes of all 3 genotypes (Fig. 6D-,E-,F-ii,-iii) with significantly attenuated astrocyte migration (Fig. 7), suggesting a close relationship between the integrity of actin cytoskeleton, cell morphology, and astrocyte motility. Together, these findings indicate that MMP-9, but not MMP-2, is associated with F-actin assembly which contributes to the formation of membrane protrusions, migration, and wound closure by astrocytes.
Figure 6.

Fluorescence staining of F-actin in cultured astrocytes isolated from wild-type, MMP-2 null, and MMP-9 null mice using Texas Red-conjugated phalloidin. Shown here are astrocytes without cytoskeleton inhibitor (i), treated with cytochalasin D (ii), and treated with Rac1 inhibitor (iii). Before reaching a confluent monolayer (A–C), astrocytes without cytoskeleton inhibitor exhibit comparable morphology and F-actin distribution in the wild-type and MMP-2 null groups, whereas MMP-9 null astrocytes lose their stellar profile and show disintegrated F-actin distribution (C-i). After a scratch wound is made on the confluent monolayer (D–F), wild-type and MMP-2 null astrocytes, without cytoskeleton inhibitor, at the scratch border extend F-actin-containing membrane protrusions toward the denuded area (on the left). These membrane protrusions are not observed in MMP-9 null astrocytes (F-i). Regardless of having a scratch wound or not, treatments with cytochalasin D (A–F-ii) or Rac1 inhibitor (A–F-iii) cause abnormal morphology and F-actin distribution in astrocytes of all 3 genotypes. No scratch-induced membrane protrusion is found with these treatments. Scale bars: -i, 25 μm; -ii, -iii, 100 μm.
Figure 7.

The effects of cytochalasin D and Rac1 inhibitor on astrocyte migration. In the scratch wound assay, both wild-type and MMP-2 null astrocytes migrate faster and cover more denuded area than the MMP-9 null astrocytes 72 h after the scratch is made (***p < 0.001, compared with untreated wild-type). At the same time point, administration of either 20 μg/ml cytochalasin D or 50 μm Rac1 inhibitor significantly slows down astrocyte migration of all 3 genotypes (###p < 0.001, compared with respective genotypes in the untreated group). Error bars indicate SD.
Deficiency of MMP-2 or MMP-9 does not affect astrocyte proliferation, adhesion, and viability
It is possible that deficiency in either MMP-2 or MMP-9 has untoward effects on astrocyte proliferation, adhesion, and viability, which may contribute to the altered migration observed above. Proliferation, defined by BrdU incorporation was studied in cultured wild-type, MMP-2 null, and MMP-9 null astrocytes (Fig. 8A–C). Quantitative analysis showed that the number of proliferative astrocytes were comparable among the 3 genotypes (Fig. 8D). Adherence, assessed in these groups one day after the first passage, was similar among genotypes (Fig. 8E). Astrocyte viability, determined by cellular metabolic activity using the MTT assay, revealed no differences between genotypes 1, 2, and 3 d after the first passage (Fig. 8F). These findings suggest that deficiency of MMP-2 or MMP-9 does not differentially affect astrocyte proliferation, adhesion, or viability, parameters that would influence migration.
Figure 8.
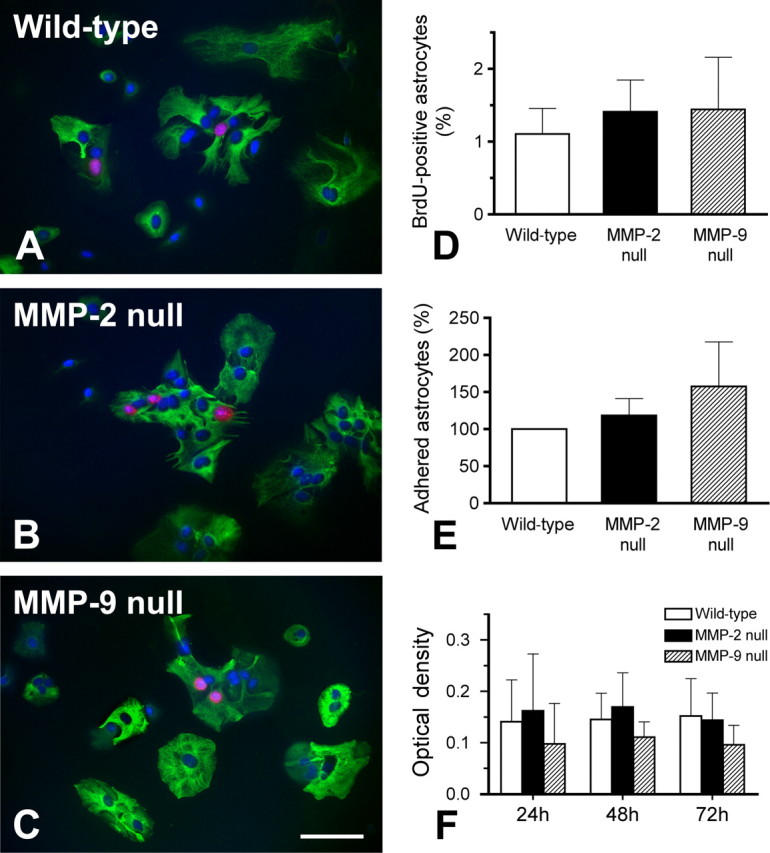
Analyses of proliferation, adhesion, and viability in cultured astrocytes isolated from wild-type, MMP-2 null, and MMP-9 null mice. Proliferative astrocytes are identified by positive immunolabeling of anti-BrdU (red) and -GFAP (green) along with DAPI nuclear stain (blue) (A– C). There is no significant difference in the number of proliferative astrocytes among the 3 groups studied (D). Astrocytes adhering to the poly-l-lysine-coated growth surface are counted 24 h after the first passage (E), while astrocyte viability is tested by colorimetric MTT assay 1, 2, and 3 d after the first passage (F). There are no significant differences among the three groups studied in all these tests. Our findings suggest that knocking out MMP-2 or MMP-9 does not affect astrocytic proliferation, adhesion, and viability. Scale bar, 100 μm. Error bars indicate SD.
Discussion
Our studies demonstrate that MMP-9 facilitates glial scar formation in the injured spinal cord and astrocyte migration in vitro. Spinal cord-injured wild-type mice developed a more severe and more inhibitory glial scar than injured MMP-9 null mice. Abrogation of glial scarring in MMP-9 null mice was independent of MMP-2 activity, which was comparable to that in the wild-type controls. Moreover, by using cultured mouse cortical astrocytes, we found that MMP-9 deficiency, as a result of either pharmacological blockade or genetic deletion, significantly attenuated astrocyte migration in a scratch wound paradigm. This defective migratory behavior was concomitant with a disarranged actin cytoskeleton. Together, these findings suggest a promotive role for MMP-9 in glial scar formation through modulation of astrocyte migration.
MMPs and glial scar formation
MMPs have diverse functions in the CNS (Yong, 2005). It has become increasingly apparent that their involvement is dictated by when and where they are activated and in the context of the injury or disease process. MMP-9 and MMP-2 are both expressed in injured spinal cord but in a temporally specific order. MMP-9 is actively expressed in the acutely injured cord (Noble et al., 2002; Wells et al., 2003), whereas MMP-2 is most prominent between 7 and 14 d after injury (Goussev et al., 2003) and is concurrent with the emergence of a glial scar. Differences between these MMPs are likely related to their distinct roles in the injured spinal cord. MMP-9, expressed in reactive astrocytes and infiltrating leukocytes, mediates early infiltration of inflammatory cells and disruption of the blood-spinal cord barrier (Noble et al., 2002). Thus, MMP-9 null mice show a significant improvement in locomotor recovery after spinal cord injury. While MMP-2 is also expressed in reactive astrocytes, it may limit the formation of an inhibitory glial scar and supports axonal regrowth as well as locomotor recovery (Hsu et al., 2006; Pastrana et al., 2006).
We provide the first evidence that MMP-9 is also involved in glial scar formation. The glial scar is reduced in MMP-9 null mice 42 d postinjury although the scarring is similar to that of the injured wild-type mice at earlier stages. Given its acute expression in reactive astrocytes, MMP-9 most likely facilitates the early migration of astrocytes to form an initially immature gliotic meshwork in the lesion, which further develops into a more complex glial scar over time during wound healing. Moreover, the presence of MMP-9 enhances the infiltration of inflammatory cells and thus increases the production of cytokines (e.g., interferon-γ and TGF-β) which are known to induce glial scar formation (Silver and Miller, 2004). Therefore, improved locomotor function in injured MMP-9 null mice may not only be due to blockade of early inflammatory cells (Noble et al., 2002) but to a reduction in the glial scar. Previously we found that MMP-2 null mice develop more extensive glial scarring than wild-type mice after spinal cord injury (Hsu et al., 2006). We also found a compensatory increase in MMP-9 in these injured MMP-2 null mice, a condition likewise occurring in experimental autoimmune encephalomyelitis (Esparza et al., 2004). Our results in this study further suggest that their extensive glial scarring and poorer recovery may not only be due to the absence of MMP-2 but to the adverse effects of increased expression of MMP-9.
Convergent evidence indicates that the glial scar, although a barrier to axonal regeneration, is also neuroprotective and beneficial to wound healing particularly at early phases after injury (Sofroniew, 2005). In response to an injury, prompt migration of reactive astrocytes seclude the lesion from uninjured tissue by compacting infiltrated inflammatory cells, resulting in more spared neuronal components and better recovery of function (Penkowa et al., 2003; Okada et al., 2006). Moreover, selective ablation of a subgroup of reactive astrocytes near a lesion in the first week after injury causes widespread inflammation, neuronal degeneration, failure of vascular barrier repair, and demyelination (Bush et al., 1999; Faulkner et al., 2004). In this context, spinal cord-injured MMP-9 null mice might be expected to display more extensive tissue damage due to compromised migration of reactive astrocytes. In fact, this does not occur in MMP-9 null mice, which exhibit restricted inflammation, preserved vascular barrier, and improved motor function relative to the wild-types in the acute stages of injury (Noble et al., 2002). Thus, the detrimental effects of MMP-9 on proteolytic tissue damage apparently outweigh its benefit of promoting astrocyte migration to restrain inflammatory responses early after the injury. In the chronic stages, conversely, MMP-9-dependent astrocyte migration culminates in an inhibitory glial scar that blocks axonal regrowth.
Reactive astrocytes in the glial scar also produce growth-inhibiting CSPGs, which restrain neurite outgrowth (McKeon et al., 1991; McKeon et al., 1999; Jones et al., 2003; Tang et al., 2003). Enzymatic treatment of the injured spinal cord with chondroitinase ABC significantly promotes axonal regeneration and recovery of motor function as a result of CSPG degradation (Bradbury et al., 2002; Chau et al., 2004; Massey et al., 2008; Tom and Houlé, 2008). Similarly, both MMP-9 and MMP-2 are capable of degrading certain CSPGs (Muir et al., 2002; Larsen et al., 2003). In degenerated peripheral nerve, MMP-9 attenuates CSPG activity with a higher proteolytic effectiveness than MMP-2 (Ferguson and Muir, 2000). In spinal cord-injured MMP-9 null mice, nevertheless, we found reduced CSPG immunoreactivity in the lesion by 42 d postinjury. This outcome was not attributable to injury-induced MMP-2 activity, which was essentially comparable between injured MMP-9 null and wild-type mice. We suggest that this finding in MMP-9 null mice reflects a reduction in the over-all complexity of the glial scar. Deficiency in MMP-9 also reduces the transmigration of hematogenous macrophages into the lesion, resulting in less CSPG deposition in the injury site because macrophages are one of the major sources of CSPG (NG2) in the subacute stage after injury (Jones et al., 2002). Interestingly, members of the CSPG family exhibit differential susceptibilities to MMP degradation (Muir et al., 2002). The anti-CSPG antibody (clone CS-56) recognizes shared epitopes on the glycosaminoglycan side chains of CSPGs. Thus we cannot rule out the possibility that some CSPGs, normally susceptible to MMP-9, are upregulated in the MMP-9 null mice, while others decrease. A decrease in the overall detectable CSPG levels would be expected if the latter dominates. As such, a less complex glial scar, concomitant with reduced expression of CSPGs, may foster a more permissive environment that would support better recovery processes seen in spinal cord-injured MMP-9 null mice (Noble et al., 2002).
MMPs and astrocyte migration
Complementary to our in vivo findings, we show that MMPs and in particular MMP-9 governs astrocyte migration in vitro. Pharmacologic blockade of MMPs, with a general inhibitor, as well as blockade of MMP-9 reduced migration. Notably, astrocyte migration was not completely blocked with either a general inhibitor or an MMP-9 inhibitor, suggesting that MMPs are not the sole modulators of astrocyte migration. Astrocytes, cultured from MMP-9 null mice, likewise showed impaired migration. Our results are consistent with previous studies showing that MMP-9 contributes to enhanced astrocyte migration (Takenaga and Kozlova, 2006; Hsieh et al., 2008). Such a promotive role in migration is further supported in other cell types where MMP-9 facilitates neuronal migration of developing granule cells in the cerebellum (Vaillant et al., 2003) and the outgrowth of oligodendrocytic processes and remyelination (Oh et al., 1999; Larsen et al., 2003) by processing the ECM molecules. Although 0 reported that astrocyte migration principally depends on MMP-2, but not MMP-9, they used a different migration assay with astrocytes embedded in agarose drops and pharmacological blocking which may not be MMP-2 specific. Our scratch assay simulates an injury and the subsequent wound closure processes, which may involve different mechanisms of cell migration from those of the agarose drop assay.
How MMP-9 modulates astrocyte migration is not well understood. Cell migration is a dynamic interplay of various processes including cell-ECM adhesion, extension of membrane projection and retraction of the trailing edge. Cell motility is also largely dependent on the organization of the actin cytoskeleton (Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996), which is regulated through multiple signaling pathways mediated by the Rho family of small GTPases, including Rho, Rac, and Cdc42 (Hall, 1998). Activation of Rho in reactive astrocytes is associated with astrocyte motility in vitro and astrogliosis after spinal cord injury (Dubreuil et al., 2003; Goldshmit et al., 2004; Höltje et al., 2005). Astrocytes are known to secrete a variety of ECM molecules such as fibronectin, laminin, and hyaluronan (Yoshida and Takeuchi, 1991; Marret et al., 1994). We previously demonstrated that Rac1 signaling is activated in astrocytes by the interaction between a transmembrane receptor CD44 and its ligand hyaluronan in the ECM, leading to increased migratory activity in vitro (Bourguignon et al., 2007). Active MMP-9 can bind to CD44, and their pericellular interactions contribute to tumor cell migration (Bourguignon et al., 1998; Yu and Stamenkovic, 1999), suggesting synergism in the extracellular regulation of cell motility. We show here that Rac1 inhibitor or cytochalasin D significantly reduced astrocyte migration regardless of the genotype in a scratch assay, providing evidence for the involvement of the actin cytoskeleton in astrocyte motility. Moreover, MMP-9 null astrocytes exhibited a disorganized actin cytoskeleton. Together, these findings suggest that MMP-9 modulates astrocyte migration likely through a CD44-dependent process, thereby stimulating intracellular signaling that affects the assembly and distribution of actin cytoskeleton.
Interaction of cell surface integrins with their ECM ligands activates the signaling pathway of the small GTPase Cdc42 and the downstream cytoskeletal dynamics, which establish cell polarity that is essential for migrating astrocytes (Etienne-Manneville and Hall, 2001). Some integrin subtypes are upregulated in reactive astrocytes (Ellison et al., 1998; Wang et al., 2005), while several MMPs bind integrins (Yong, 2005), suggesting that these MMPs may regulate astrocyte migration via integrin signaling. Although the specific role of MMP-9 in this integrin-mediated astrocyte migration remains to be determined, Ogier et al. (2006) have demonstrated a correlation between MMP-2 and β-1 integrin in migrating astrocytes in their agarose drop assay.
In conclusion, our studies demonstrate that MMP-9 facilitates glial scar formation after spinal cord injury, likely by promoting astrocyte migration as shown in vitro. Additional research with MMP conditional knock-outs and selective MMP inhibitors will better delineate how temporally specific modulation of MMP activity will affect wound healing events including the formation of the glial scar.
Footnotes
This work was supported by the National Institutes of Health, Grants R01 NS39278 (L.J.N.-H.), R01 CA66163, R01 CA78633, and P01 AR39448; a Veterans Affair Merit Review grant, and a DOD grant (L.Y.W.B.). We thank Dr. Lisa Coussens and Lidiya Korets for providing MMP-2 breeding pairs, Drs. Robert McKeon and Tiina Kauppinen for guidance in the cell culture studies, and Joel Faustino and Ying Yu for their excellent technical advices. We also thank Ms. Christina Camacho for her help in organizing graphs.
References
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in the injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baorto DM, Mellado W, Shelanski ML. Astrocyte process growth induction by actin breakdown. J Cell Biol. 1992;117:357–367. doi: 10.1083/jcb.117.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY, Gunja-Smith Z, Iida N, Zhu HB, Young LJ, Muller WJ, Cardiff RD. CD44v(3,8–10) is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–215. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Gilad E, Peyrollier K, Brightman A, Swanson RA. Hyaluronan-CD44 interaction stimulates Rac1 signaling and PKN gamma kinase activation leading to cytoskeleton function and cell migration in astrocytes. J Neurochem. 2007;101:1002–1017. doi: 10.1111/j.1471-4159.2007.04485.x. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Chau CH, Shum DK, Li H, Pei J, Lui YY, Wirthlin L, Chan YS, Xu XM. Chondroitinase ABC enhances axonal regrowth through Schwann cell-seeded guidance channels after spinal cord injury. FASEB J. 2004;18:194–196. doi: 10.1096/fj.03-0196fje. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchossoy Y, Horvat JC, Stettler O. MMP-related gelatinase activity is strongly induced in scar tissue of injured adult spinal cord and forms pathways for ingrowing neurites. Mol Cell Neurosci. 2001;17:945–956. doi: 10.1006/mcne.2001.0986. [DOI] [PubMed] [Google Scholar]
- Ellison JA, Velier JJ, Spera P, Jonak ZL, Wang X, Barone FC, Feuerstein GZ. Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke. 1998;29:1698–1706. doi: 10.1161/01.str.29.8.1698. discussion 1707. [DOI] [PubMed] [Google Scholar]
- Esparza J, Kruse M, Lee J, Michaud M, Madri JA. MMP-2 null mice exhibit an early onset and severe experimental autoimmune encephalomyelitis due to an increase in MMP-9 expression and activity. FASEB J. 2004;18:1682–1691. doi: 10.1096/fj.04-2445com. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson TA, Muir D. MMP-2 and MMP-9 increase the neurite-promoting potential of schwann cell basal laminae and are upregulated in degenerated nerve. Mol Cell Neurosci. 2000;16:157–167. doi: 10.1006/mcne.2000.0859. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–10073. doi: 10.1523/JNEUROSCI.2981-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussev S, Hsu JY, Lin Y, Tjoa T, Maida N, Werb Z, Noble-Haeusslein LJ. Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J Neurosurg Spine. 2003;99:188–197. doi: 10.3171/spi.2003.99.2.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Höltje M, Hoffmann A, Hofmann F, Mucke C, Grosse G, Van Rooijen N, Kettenmann H, Just I, Ahnert-Hilger G. Role of Rho GTPase in astrocyte morphology and migratory response during in vitro wound healing. J Neurochem. 2005;95:1237–1248. doi: 10.1111/j.1471-4159.2005.03443.x. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Wu CY, Yang CM. Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-delta-dependent ERK/Elk-1 pathway in astrocytes. Glia. 2008;56:619–632. doi: 10.1002/glia.20637. [DOI] [PubMed] [Google Scholar]
- Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, Noble-Haeusslein LJ. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci. 2006;26:9841–9850. doi: 10.1523/JNEUROSCI.1993-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998;58:1048–1051. [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci. 2002;22:2792–2803. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399–411. doi: 10.1016/s0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. J Neurosci. 2003;23:11127–11135. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, Rewcastle B, Yong VW. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci. 2003;23:4034–4043. doi: 10.1523/JNEUROSCI.23-10-04034.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marret S, Delpech B, Delpech A, Asou H, Girard N, Courel MN, Chauzy C, Maingonnat C, Fessard C. Expression and effects of hyaluronan and of the hyaluronan-binding protein hyaluronectin in newborn rat brain glial cell cultures. J Neurochem. 1994;62:1285–1295. doi: 10.1046/j.1471-4159.1994.62041285.x. [DOI] [PubMed] [Google Scholar]
- Massey JM, Amps J, Viapiano MS, Matthews RT, Wagoner MR, Whitaker CM, Alilain W, Yonkof AL, Khalyfa A, Cooper NG, Silver J, Onifer SM. Increased chondroitin sulfate proteoglycan expression in denervated brainstem targets following spinal cord injury creates a barrier to axonal regeneration overcome by chondroitinase ABC and neurotrophin-3. Exp Neurol. 2008;209:426–445. doi: 10.1016/j.expneurol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 2002;16:84–86. doi: 10.1096/fj.01-0380fje. [DOI] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Giménez y Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci USA. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mori K, Kitayama J, Shida D, Yamashita H, Watanabe T, Nagawa H. Lysophosphatidic acid-induced effects in human colon carcinoma DLD1 cells are partially dependent on transactivation of epidermal growth factor receptor. J Surg Res. 2006;132:56–61. doi: 10.1016/j.jss.2005.07.040. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Muir EM, Adcock KH, Morgenstern DA, Clayton R, von Stillfried N, Rhodes K, Ellis C, Fawcett JW, Rogers JH. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res. 2002;100:103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier C, Bernard A, Chollet AM, LE Diguardher T, Hanessian S, Charton G, Khrestchatisky M, Rivera S. Matrix metalloproteinase-2 (MMP-2) regulates astrocyte motility in connection with the actin cytoskeleton and integrins. Glia. 2006;54:272–284. doi: 10.1002/glia.20349. [DOI] [PubMed] [Google Scholar]
- Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Pastrana E, Moreno-Flores MT, Gurzov EN, Avila J, Wandosell F, Diaz-Nido J. Genes associated with adult axon regeneration promoted by olfactory ensheathing cells: a new role for matrix metalloproteinase 2. J Neurosci. 2006;26:5347–5359. doi: 10.1523/JNEUROSCI.1111-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernández J, Molinero A, Campbell IL, Hidalgo J. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp Neurol. 2003;181:130–148. doi: 10.1016/s0014-4886(02)00051-1. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ, Moolenaar WH. Regulation of astrocyte morphology by RhoA and lysophosphatidic acid. Exp Cell Res. 1998;245:252–262. doi: 10.1006/excr.1998.4224. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Frazier JM, editors. In vitro biological methods. San Diego: Academic; 1993. pp. 46–60. [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaga K, Kozlova EN. Role of intracellular S100A4 for migration of rat astrocytes. Glia. 2006;53:313–321. doi: 10.1002/glia.20284. [DOI] [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJ. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–444. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Houlé JD. Intraspinal microinjection of chondroitinase ABC following injury promotes axonal regeneration out of a peripheral nerve graft bridge. Exp Neurol. 2008;211:315–319. doi: 10.1016/j.expneurol.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant C, Meissirel C, Mutin M, Belin MF, Lund LR, Thomasset N. MMP-9 deficiency affects axonal outgrowth, migration, and apoptosis in the developing cerebellum. Mol Cell Neurosci. 2003;24:395–408. doi: 10.1016/s1044-7431(03)00196-9. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kong Q, Gonzalez FA, Sun G, Erb L, Seye C, Weisman GA. P2Y nucleotide receptor interaction with alpha integrin mediates astrocyte migration. J Neurochem. 2005;95:630–640. doi: 10.1111/j.1471-4159.2005.03408.x. [DOI] [PubMed] [Google Scholar]
- Wells JE, Rice TK, Nuttall RK, Edwards DR, Zekki H, Rivest S, Yong VW. An adverse role for matrix metalloproteinase 12 after spinal cord injury in mice. J Neurosci. 2003;23:10107–10115. doi: 10.1523/JNEUROSCI.23-31-10107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KP, Ding Y, Ling J, Dong Z, Yu FS. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:813–820. doi: 10.1167/iovs.03-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Takeuchi M. Expression of fibronectin and laminin by different types of mouse glial cells cultured in a serum-free medium. Cytotechnology. 1991;7:187–196. doi: 10.1007/BF00365930. [DOI] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]