

Abstract
Motivation: Homologous protein families share highly conserved sequence and structure regions that are frequent targets for comparative analysis of related proteins and families. Many protein families, such as the curated domain families in the Conserved Domain Database (CDD), exhibit similar structural cores. To improve accuracy in aligning such protein families, we propose a profile–profile method CORAL that aligns individual core regions as gap-free units.
Results: CORAL computes optimal local alignment of two profiles with heuristics to preserve continuity within core regions. We benchmarked its performance on curated domains in CDD, which have pre-defined core regions, against COMPASS, HHalign and PSI-BLAST, using structure superpositions and comprehensive curator-optimized alignments as standards of truth. CORAL improves alignment accuracy on core regions over general profile methods, returning a balanced score of 0.57 for over 80% of all domain families in CDD, compared with the highest balanced score of 0.45 from other methods. Further, CORAL provides E-values to aid in detecting homologous protein families and, by respecting block boundaries, produces alignments with improved ‘readability’ that facilitate manual refinement.
Availability: CORAL will be included in future versions of the NCBI Cn3D/CDTree software, which can be downloaded at http://www.ncbi.nlm.nih.gov/Structure/cdtree/cdtree.shtml.
Contact: fongj@ncbi.nlm.nih.gov.
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Homologous protein families contain core regions that reflect conservation in molecular evolution. Many protein family alignments in Pfam (Finn et al., 2006), SMART (Letunic et al., 2006) and SUPERFAMILY (Wilson et al., 2007) exhibit conserved regions including blocks, or ungapped regions, within an alignment. The Conserved Domain Database (CDD) (Marchler-Bauer et al., 2009) models protein domains explicitly as series of blocks. For NCBI-curated domains, the blocks represent structural core motifs based on structure superpositions as well as conserved sequence regions and motifs. Comparative analysis of proteins and protein families through sequence alignment is invaluable for grouping homologs, subdividing diverse families into sub-families, tracing evolutionary histories and identifying conserved functional sites.
In recent years, alignment methods that compare two profiles, the statistical models that represent protein families, have been shown to improve alignment quality and homolog recognition over sequence–sequence methods such as BLAST (Altschul et al., 1997) and sequence–profile methods such as PSI-BLAST (Altschul et al., 1997; Schaffer et al., 2001). Numerous profile alignment methods have been assessed in Edgar and Sjolander (2004), Heger and Holm (2001), Ohlson and Elofsson (2005), Ohlson et al. (2004), Panchenko (2003), Rychlewski et al. (2000), Soding (2005), Yona and Levitt (2002) and others. While many alignment methods focus on detecting remote homologs in order to expand coverage of functional inference, obtaining high-quality alignments remains difficult even for closely-related families. According to structure superpositions, corresponding core regions in many homologous domains differ by fewer insertions and deletions than inferred by general alignment programs, reflecting the stability of the structural core of the protein family. To better capture this property, we propose a method CORAL (CORe ALigner) to align core regions from two protein families without indels within blocks, which we will refer to as the core constraint. CORAL is implemented through a common dynamic programming engine for optimal pair-wise alignment (Needleman and Wunsch, 1970; Smith and Waterman, 1981).
Several other algorithms to align sequence or sequence profiles to core regions have been effective for detecting similarities or assigning domains. These algorithms include a profile–profile method using Gibbs sampling (Panchenko, 2003), and SALTO (Kann et al., 2005) and GLOBAL (Kann et al., 2007) which employ additional block-based constraints. SALTO aligns a consecutive sub-set of complete blocks and GLOBAL aligns a sub-set (including full or empty set) of contiguous columns within every block. All of these methods disallow indels in alignments of blocks and exclude sequence regions outside blocks. Additionally, LAMA (Pietrokovski, 1996) and CYRCA (Kunin et al., 2001) were developed to align individual blocks that represent sequence motifs (Henikoff et al., 2000). Block shift and extension operations have also proved useful to improve multiple sequence alignments (MSAs) through REFINER (Chakrabarti et al., 2006).
Here, we present the CORAL algorithm and benchmark its performance on curated domains in CDD against other widely used profile methods COMPASS (Sadreyev and Grishin, 2003), HHalign (Soding, 2005) and PSI-BLAST. Reference alignments are inferred from structure superpositions from the VAST database (Gibrat et al., 1996; Madej et al., 1995) and the SABmark benchmark set (Van Walle et al., 2005), and from a comprehensive set of expert-determined mappings, and homology is defined by CDD relationships. In particular, CORAL outperforms all other methods in the quality of alignments. We also discuss the role of profile alignment in modeling protein families.
2 METHODS
2.1 Core regions dataset
MSAs representing protein family core regions were taken from the curated domains in CDD. Sequence regions outside the cores are not aligned in CDD and are not considered in this study. Here, we use the terms domain and protein family interchangeably. NCBI-curated domains have been organized into hierarchical domain families. A superfamily, which indicates common evolutionary descent, contains one or more domain families. We define related domains with respect to CDD to be those in the same family and unrelated domains to be those in different superfamilies, in order to minimize false positives (FPs). A set of 100 domains, chosen randomly from different superfamilies, was reserved for parameter optimization (dataset ‘opt100’). Similarity between domains was estimated as the fraction identity of their consensus sequences with pair-wise sequence alignments computed by MUSCLE 3.6 (Edgar, 2004). The consensus sequences express only columns in the MSA with <50% gap content, including the most conserved columns, and hence report higher similarity values than using full length protein sequences.
2.2 Reference alignments
To test alignment accuracy, we construct three benchmark datasets. The first reference set is based on superpositions of the 3D structures that annotate curated domains in CDD v2.14. We gather structural neighbors from the VAST database that satisfy the default significance cutoff of P-value < 0.0001, such that folds are described by a continuous sequence region that overlaps the sequence fragment in the domain model by 90%. To ensure that the structure alignments involve core regions, aligned core positions are required to comprise 80% of all structurally aligned positions and 50% of the respective profiles. This procedure yields structure alignments for 2385 domain pairs within 91 CDD families.
A second set of structure alignments is taken from the superfamilies set in SABmark, that is, alignments of SCOP domains with a common evolutionary origin. CDD domains are mapped onto the SCOP domains using RPS–BLAST (Marchler-Bauer et al., 2002). Due to time of testing, a later version of CDD (v 2.16) was used for this benchmark set. SCOP folds are filtered for live sequences in Entrez and at least 50% overlap with the extent of the domain hit, resulting in structure alignments for 1627 domain pairs in 128 SCOP superfamilies. The two structural reference sets differ in coverage across and within domain families; classification by CDD versus SCOP; and curator-optimized versus RPS–BLAST-computed structure-domain alignments.
A third benchmark set provides comprehensive coverage over homologous domains in CDD. In NCBI-curated hierarchies, the MSAs of a parent domain and its sub-family contain overlapping fragments from at least one protein sequence. The shared sequence identifies aligned columns between the two MSAs and reflects the curator's assertion of how the sub-family should be mapped to its parent. Transitivity over each hierarchy extends the guide alignment to all pair-wise comparisons in multi-domain families. Guide alignments include 57 786 domain pairs over 212 CDD families.
2.3 Alignment algorithm
We describe the profile alignment algorithm with core constraint in terms of required modifications to the canonical algorithm for local alignment (Smith and Waterman, 1981). The problem is to align profiles A = a1···an and B = b1···bm with n and m columns, respectively, where each profile has been sub-divided into blocks. Let table H contain the maximum similarity score of two profile segments ending in ai and bj in entry Hi,j Scoring functions S(ai, bj) to compute the similarity between profile columns ai and bj are described in the next paragraph. To prevent gaps within blocks, the affine gap penalty is replaced with a large negative value if the last aligned column before the gap is not a block end. To ensure that the endpoints of the optimal alignment fall on the N- and C-terminal of some blocks, Hi,j may be re-initialized to S(ai, bj) (replacing initialization to 0) if ai or bj is the first column in its respective block and traceback through Hi,j is required to terminate at that position. Traceback may begin from the maximum Hi,j such that at least one of ai and bj is the end of its respective block. These changes preserve the O(nm) running time.
The optimal scores from H are normalized into Z-scores as follows. A large set of random alignments was simulated using all curated domains, each aligned with 100 domains from different superfamilies. Alignment scores were binned by the sum of lengths of the profiles. Regression curves were fitted for the means and SDs over the bins. The length-dependent values from the regressions were used to compute Z-score.
2.4 Scoring functions
Much of the previous work on profile–profile alignment algorithms sought advances through new scoring functions for comparing profile columns. Probabilistic methods are believed to be the most effective (Mittelman et al., 2003; von Ohsen et al., 2003) and are applied in state-of-the-art aligners such as prof_sim (Yona and Levitt, 2002), COMPASS and HHsearch. CORAL uses a symmetrical log-odds function similar to Picasso (Heger and Holm, 2001) and COMPASS (Sadreyev and Grishin, 2003):
To compute similarity between aligned columns a and b, Qa and Qb represent vectors of weighted observed frequencies of amino acids k in the respective columns. Likewise, R is the vector of the frequency ratios of weighted frequency for each amino acid over the background frequency of the amino acid. Q and R are defined as for PSI-BLAST (Altschul et al., 1997; Schaffer et al., 2001).
Surveys of scoring functions (Edgar and Sjolander, 2004; Mittelman et al., 2003; Panchenko, 2003) have suggested that probabilistic methods offer incremental improvements over simpler functions such as sum of pairs (Gotoh, 1993), dot product and Pearson correlation coefficient. Consequently, we also test the symmetrical dot product function:
In Section 3, the two methods will be denoted as CORAL LO and CORAL DP, respectively. The public release of CORAL will use the better performing log-odds function.
2.5 Parameter optimization
A local alignment requires that the expected column score be negative and some column score(s) be positive. To satisfy these conditions, a constant shift value is added to each column score. To initialize the search space for potential shift values, we computed the distributions of column scores for correctly aligned columns in all related domains in CDD and for all pairs of columns in a sampling of unrelated domains. A second parameter, the gap penalty, is necessary to distinguish significant alignments. Shift values between the means of each distribution and small gap weights were tested systematically over combinations of both parameters. Performance was assessed for alignment accuracy and homolog sensitivity following the testing procedures and metrics described in Section 3. Over the opt100 dataset, performance was fairly robust over a range of parameter values. We assigned shift values of −0.15 and 6.6 for the two scoring functions, respectively, and gap weights of 0.1 and 0.5, respectively.
2.6 Statistical significance
To approximate the statistical significance of each alignment, we turn to the extreme value distribution (EVD) which has been shown empirically to fit optimal ungapped alignments of random sequences (Karlin and Altschul, 1990). It is frequently used with gapped sequences and profile alignments. Supposing that the alignment scores follow an EVD, the E-value for every alignment can be computed from the alignment score z and parameters λ and μ as E=e−λ(z−μ). To determine λ and μ, normalized alignment scores from the random alignments described above were fitted to the cumulative density function,F(x)=exp(−exp(−λ(x−μ))). Parameters were computed separately for each scoring function SLO and SDP. The goodness of fit is illustrated for CORAL LO in Supplementary Figure S1.
3 RESULTS
3.1 Alignment accuracy
The quality of CORAL alignments between CDD-curated domains was evaluated against the reference alignments described in Section 2 and compared with alignments from COMPASS 3.0, HHalign 1.5.1.1 and PSI-BLAST. COMPASS is a high-performance implementation of the standard sum-of-scores optimal local alignment and its comparison with CORAL implies a lower bound in improvement that can be attributed to the core constraint. COMPASS was run with default parameters and with reduced gap penalties. To promote longer alignments, the gap open penalty was reduced arbitrarily default from 10 to 3 and the gap extension penalty default from 1 to 0.1. HHalign was run in local and global modes using one domain alignment as query and the other as template. To compute probabilities and E-values for HHalign, each HMM was calibrated against the cal.hhm database from the download site. For every pair of domains, a PSI-BLAST alignment was computed between one domain and each sequence from the MSA of the other domain, and vice versa, using the NCBI Toolkit. The sequence–profile alignment with smallest E-value was used as the PSI-BLAST alignment. CORAL and COMPASS held a speed advantage over the other methods, requiring than a 10th of a second for most inputs. HHalign required 5–10 s, largely because of the calibration step.
The following metrics are used to evaluate alignment accuracy. To measure extent of reconstructing a reference alignment, we compute Sdev, the ratio of the number of correctly aligned positions to the number of aligned columns in the reference alignment. Sdev is the same as the developer's score of (Sauder et al., 2000). To measure correctness, we compute Smod, the ratio of the number of correctly aligned positions to the number of aligned columns in the evaluated alignment where at least one of each two aligned columns is present in the reference alignment. This is analogous to the modeler's score (Sauder et al., 2000), modified to include only the profile columns that can be determined to be correct or not. The two previous measures are summarized through a balanced score, Sbalanced=(Sdev + Smod)/2. To more directly illustrate the trade-off between alignment accuracy and alignment length, we estimate the latter as Scov, the number of aligned positions divided by the length of the shorter profile. Results from multiple structure alignments for the same domain pair are averaged over the domain pair.
First, we analyze overall performance over CDD families and SCOP superfamilies, both referred to as families for brevity. An average Sbalanced for every family is taken over its domain pairs (Fig. 1). CORAL produced high-quality alignments for more families than the other methods: 44% of domain families average Sbalanced≥0.8 compared with 41% by the best non-CORAL method according to the VAST benchmark, 37% versus 28% according to the SABmark benchmark and 57% versus 45% by guide alignments. In nearly all of these families, the alignments with Sbalanced ≥0.8 were both accurate and complete. Under the three highest performing methods (CORAL LO, CORAL DP and HHalign global), over 96% of domain families with Sbalanced ≥0.8 had both Sdev ≥0.8 and Smod ≥0.8 with respect to all benchmark sets.
Fig. 1.

Distribution of balanced scores from three benchmark sets: (A) VAST structure superpositions; (B) SABmark structure alignments; and (C) curator-inferred guide alignments. SABmark alignments are grouped by SCOP superfamily and the others by CDD family. The balanced score is an average of accuracy over computed alignment and completeness in reconstructing the reference alignment.
Comparison of Sbalanced over the domain pairs present in more than one benchmark set reveals high consistency among the reference alignments. For pair-wise comparison of the reference sets, we identified domain pairs present in both benchmark sets. Sbalanced scores for the common domain pairs, averaged over domain families, were 0.026–0.032 lower according to the different alignment methods in VAST alignments than the corresponding guide alignments and 0.032–0.047 lower in SABmark alignments than the corresponding guide alignments. Approximately 2% of domain pairs had balanced score at least 0.01 higher by VAST structure alignments, 30% of domain pairs had balanced score 0.01 higher for guide alignments and the remaining two-thirds of domain pairs had negligible differences between those two references. We hypothesize that guide alignments are more accurate because they are the outcome of manual curation that reviews both structure-based alignments and patterns of sequence conservation, where the latter may overrule structure superimposition.
Nearly 19% of the domain pairs evaluated with SABmark were assigned to different CDD superfamilies (but the same SCOP superfamilies). Average Sbalanced score over these pairings was less than half that from domains in the same CDD superfamily, resulting in a larger fraction of families with low-balanced score than from the other reference alignments for all alignment methods (Fig. 1). CORAL returned highest average Sbalanced score for domains in different CDD superfamilies as well as domains from the same CDD superfamilies.
The higher Sbalanced scores for both CORAL methods over the other methods suggest that the core constraint played a significant role in improving performance for several families. For some families, including Macro and PDZ, all members benefited from the core constraint. Domain families that benefited the most and the least using CORAL are listed in Supplementary Table S1. No correlation was observed between average similarity within families and improvement, or lack thereof, from using CORAL. Better alignments generally came about because CORAL prevented spurious intra-block gaps and shifted blocks that were misaligned by COMPASS and HHalign into the right positions. The families with most negative effect from CORAL, phosphofructokinase (PFK) and Rieske, illustrate the case where long blocks must be split to enable a completely correct CORAL alignment. One example is the alignment of two Rieske domains: non-heme iron oxygenase family/nathphalene 1,2-dioxygenase sub-family (cd03535) and small sub-unit of Arsenite oxidase family (cd03476).
Families such as the kinesin/myosin motor domains contain dissimilar sub-groups such that domains within a sub-group are aligned with much higher accuracy than domains from different sub-groups. To account for varying difficulty, domain pairs were grouped by sequence identity. The distribution of sequence identity is shown in Supplementary Figure S2 with mean percent identity 29.6% and SD 10.1%. We partitioned alignments into four similarity ranges: 0–20%, 20–30%, 30–40% and ≥40%. Results from guide alignments are provided in Figure 2 and referred to in the remainder of the section; results from VAST and SABmark alignments illustrate similar trends and are provided in Supplementary Figure S3. Sdev and Smod results within each similarity range are consistent across most alignment methods (Fig. 2), pointing to the inherent ease or difficulty of aligning particular domains. CORAL has highest Sdev over all similarity ranges. Although HHalign local and COMPASS with default arguments have higher Smod at <30% identity, CORAL yields higher Sbalanced value for every similarity range. Over the entire dataset, CORAL gives an average balanced score of 0.80 and 0.77 for the log odds and dot product functions, respectively, compared with 0.74 for HHalign and 0.75 for COMPASS. The shorter alignments correlate with higher alignment accuracy (Smod), but are less informative as they exclude more homologous regions. CORAL and COMPASS parameters may be set to permit near-global alignments using a local alignment algorithm.
Fig. 2.

Alignment accuracy in terms of the (A) Sdev; (B) Smod; and (C) Scov metrics based on curator-optimized (guide) reference alignments. These metrics indicate completeness in reconstructing the reference alignment, accuracy over the computed alignment and the local–global trade-off in the resulting alignment, respectively.
PSI-BLAST performance deteriorated rapidly as sequence similarity decreases. Almost half of all domain pairs from the same family had no significant PSI-BLAST alignment. Aligning the consensus sequences by pairwise BLAST led to a similar outcome, showing that these families are not as easy to align despite the high-reported sequence identities. The default significance cut-off for PSI-BLAST is restrictive and many domain pairs may not satisfy the cut-off due to low-sequence similarity or short profile lengths. Domain pairs with no PSI-BLAST results were assigned value 0 for all metrics (following the regular definitions of Sdev and Scov, and replacing the otherwise undefined Smod term), leading to a large number of families with low Sbalanced score.
3.2 Homology recognition
Next, we evaluated the accuracy of CORAL and its E-values at detecting related domains. Although we do not propose to identify homologous protein families from core regions alone, given the evolutionary signal present in the more variable loop regions, a scoring system helps to distinguish more similar and better-aligned core regions. Related and unrelated domains are defined with respect to CDD families/superfamilies, as described in Section 2. A test set of 100 domains was taken from different superfamilies. Each domain is aligned with all domains within the same family (with a minimum of two related domains) and with 100 randomly selected unrelated domains, using the alignment methods described in the previous section. PSI-BLAST was omitted to avoid handling missing data. The distribution of sequence identity between related domains in this test set is similar to the distribution over the entire CDD (Supplementary Fig. S2).
Figure 3 shows performance measured as the fraction of true relationships (true positive, TP) that score higher than the i-th highest scoring false relationship (FP), averaged over the test set. Scores refer to the E-values for CORAL and COMPASS and probabilities for HHalign, which performed much better than its E-values. To assess sensitivity, we measure the area under curve (AUC), ROCn=1/n∑i=1…nti, for each sample domain where ti is the fraction of TPs before the i-th FP. Standard error over ROCn values is computed as 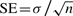 . ROC curves and AUC values reveal that the CORAL and HHalign methods detect homologs from core regions at similar rates, and better than COMPASS. Average ROC100 and SE ranges overlapped for all CORAL and HHalign methods and were: 0.962±0.009 for CORAL LO, 0.963±0.008 for CORAL DP, 0.966±0.008 for HHalign local and 0.957±0.011 for HHalign global. There was a statistically significant difference between the distribution of ROC100 values for HHalign local, the highest curve in Figure 3, from the closest methods HHalign global and CORAL according to the Wilcoxon signed-rank test (P-values 0.01–0.02), but not between the CORAL methods and HHalign global (all pair-wise P-values > 0.05).
. ROC curves and AUC values reveal that the CORAL and HHalign methods detect homologs from core regions at similar rates, and better than COMPASS. Average ROC100 and SE ranges overlapped for all CORAL and HHalign methods and were: 0.962±0.009 for CORAL LO, 0.963±0.008 for CORAL DP, 0.966±0.008 for HHalign local and 0.957±0.011 for HHalign global. There was a statistically significant difference between the distribution of ROC100 values for HHalign local, the highest curve in Figure 3, from the closest methods HHalign global and CORAL according to the Wilcoxon signed-rank test (P-values 0.01–0.02), but not between the CORAL methods and HHalign global (all pair-wise P-values > 0.05).
Fig. 3.

Recognizing homologs: ROC curve plotting percentage of TP identified before the n-th FP.
3.3 Alignment in protein family modeling
The problem of aligning conserved core regions was conceived by the need to automate domain curation and develop tools for analyzing individual families. Many domain families contain diverse members that are difficult to align. Sequence similarity, for example via characteristic motifs, can make it clear that sequence fragments are related by common descent. More powerful tools are needed to obtain an accurate alignment across the full domain model and to determine domain boundaries.
In defining diverse domain families, two important and interrelated tasks for each domain are step 1: to build a MSA and step 2: to split off sub-families when applicable for increasing functional specificity, starting with a less-diverse sub-set of sequences from the current domain. These tasks are common to many approaches to subfamily identification (see e.g. Brown et al., 2007), although, here we describe steps in the CDD curation pipeline. Typically, the higher degree of conservation in child models allows curators to extend blocks and/or define additional blocks beyond the base core structure of their parent. Aligning the child and parent domains requires the selection of a representative sequence to provide the guide alignment between a new sub-family and its parent domain. A badly aligned representative compromises the overall alignment of the child with respect to the parent, which may amplify noise present in the parent and misrepresent evolutionary distance and diversity within the superfamily. Cleaning up the child model by itself further propagates overall error, which may be difficult to detect. By iterating steps 1 and 2, the child alignment is refined, its core structure may be extended or revised, and realigning the child and parent may help to refine the core structure of the parent as well.
When subfamilies are covered by 3D structure, structure superposition helps to provide high-quality guide alignments. Profile alignments augment this information and may substitute for superpositions when structures are not known. The structure alignment may differ markedly from the guide alignment, as in the alignment of the eukaryotic translation factor 5A domain and the Hex1/S1-like RNA-binding domain (Fig. 4). In this case, CORAL validates the structural alignment and extends the aligned region. A third major step in CDD curation is annotating domain models with function and functional sites following the literature and analysis of 3D structures. The alignment of related protein families helps to confirm the locations of functional sites, which may be placed at nearby positions in parent and child domains as shown in Figure 4 for RNA-binding sites.
Fig. 4.

VAST structure, guide and CORAL alignments between the eukaryotic translation factor 5A domain (cd04468; eIF5A) and the Hex1/S1-like RNA-binding domain (cd04469; S1_Hex1) are illustrated using sequence fragments from 1X6O and 1KHI and structure superpositions. The domains share a parent (EF- and S1-like RNA-binding domain) and 28% identity. Aligned positions in the reference alignments are underlined. The structure alignment is believed to be the most accurate. Misaligned regions in the guide and CORAL alignments are colored blue. RNA-binding sites are highlighted in yellow on both sequence and structure alignments. The structure superposition is colored red for identical residues, purple for other aligned residues and grey for unaligned residues.
4 DISCUSSION
Here, we showed that profile–profile alignment with well-structured alignment constraints can achieve high-alignment accuracy and work well in detecting homologous relationships between conserved core regions of domain families. The core constraint exploits relationships between profile columns, prohibiting insertions or deletions within blocks, rather than pursuing improvements through refinement of the column scoring function. Our proposed method is a simple interpretation of a framework in which gap penalties vary according to local conservation, requiring only two different gap penalties. The core constraint may be incorporated into other alignment algorithms as well.
We benchmarked CORAL on core regions from NCBI-curated domains in CDD. Blocks in curated domains reflect sequence and structural conservation and approximate the structural core of the family. However, curators may define blocks to be longer or shorter than in structure alignments, and merge, split or delete the blocks suggested by structure alignments. They may also introduce additional blocks to record conserved features and sites outside the structural core, such as binding sites and motifs.
CORAL E-values identify 70% of all domain pairs from the same hierarchy with E-value < 0.05 compared with 3.0% of domain pairs from different superfamilies. Ranking scores from the same family, as in the homology recognition test, achieves even higher performance. In general, the CDD superfamily classification used to define homologs is comparable in specificity to SCOP superfamilies, the basis for remote homology in previous benchmark studies (Marchler-Bauer et al., 2009). Nevertheless, that curated domains in CDD are easier to classify is unsurprising, because many previous studies aligned noisier profiles constructed by PSI-BLAST and the hierarchical organization of CDD families suggests that many domains have similar conserved cores.
Constructing high-quality alignments between well-defined core regions, in contrast, benefits tremendously from the core constraint. CORAL aligns more families with high-balanced score, produces better alignments with respect to the balanced score than COMPASS or HHalign across all similarity ranges, and returns higher developer's score for almost all groups of data. Possibly even more importantly, by respecting block boundaries, it produces alignments that may be easier to revise. Automated alignments of sequences or profiles with low similarity often require manual correction to produce optimal results. Reducing error to a small number of block shifts simplifies manual analysis. Although the core constraint reduces the space of possible alignment solutions, it does not necessarily constrain the alignment to only one good solution. Our results demonstrate that weak sequence similarity between corresponding core regions increases errors in all methods. Additionally, even in the more constrained setting of global alignment, differences in profile and block lengths permit more than one possible alignment between many blocks.
The clear shortcoming of the core constraint is that at some level of divergence, core regions cannot be aligned correctly without insertions or deletions, hence methods without the core constraint are more suited to remote homolog recognition and alignment. One solution to ameliorate shift errors is to split long blocks into shorter units, randomly or by inspecting the block structure or preliminary alignments of core regions. The curated domain models already contain breaks within blocks where the sequences naturally split. In unreported experiments, we have aligned the curated domains using this alternative block definition with similar and slightly worse overall performance. Further development of this algorithm will allow for cases where additional blocks have been inserted into a sub-family model relative to its parent.
CORAL will be made available to the public as an alignment tool bundled into a future release of the NCBI Cn3D/CDTree software. This user-friendly implementation will provide fast and accurate alignment of core regions, along with access to protein family alignments from CDD. While we only tested alignments between pre-computed protein family models, core regions may be inferred from the continuous regions of any protein family alignment. However, the effective use of CORAL requires high overlap between the conserved regions of two families, for example, in the case of a common structural core, and additional processing may be needed to identify putative conserved core regions. The core constraint may also be incorporated into profile alignment algorithms with more sophisticated scoring methods to improve on both CORAL and the original method for aligning conserved cores.
ACKNOWLEDGEMENTS
We thank Anna Panchenko and John Spouge for helpful discussions.
Funding: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of Interest: none declared.
REFERENCES
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DP, et al. Automated protein subfamily identification and classification. PLoS Comput. Biol. 2007;3:e160. doi: 10.1371/journal.pcbi.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, et al. Refining multiple sequence alignments with conserved core regions. Nucleic Acids Res. 2006;34:2598–2606. doi: 10.1093/nar/gkl274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Sjolander K. A comparison of scoring functions for protein sequence profile alignment. Bioinformatics. 2004;20:1301–1308. doi: 10.1093/bioinformatics/bth090. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, et al. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Gotoh O. Optimal alignment between groups of sequences and its application to multiple sequence alignment. Comput. Appl. Biosci. 1993;9:361–370. doi: 10.1093/bioinformatics/9.3.361. [DOI] [PubMed] [Google Scholar]
- Heger A, Holm L. Picasso: generating a covering set of protein family profiles. Bioinformatics. 2001;17:272–279. doi: 10.1093/bioinformatics/17.3.272. [DOI] [PubMed] [Google Scholar]
- Henikoff JG, et al. Increased coverage of protein families with the blocks database servers. Nucleic Acids Res. 2000;28:228–230. doi: 10.1093/nar/28.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann MG, et al. A structure-based method for protein sequence alignment. Bioinformatics. 2005;21:1451–1456. doi: 10.1093/bioinformatics/bti233. [DOI] [PubMed] [Google Scholar]
- Kann MG, et al. The identification of complete domains within protein sequences using accurate E-values for semi-global alignment. Nucleic Acids Res. 2007;35:4678–4685. doi: 10.1093/nar/gkm414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Altschul SF. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl Acad. Sci. USA. 1990;87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, et al. Consistency analysis of similarity between multiple alignments: prediction of protein function and fold structure from analysis of local sequence motifs. J. Mol. Biol. 2001;307:939–949. doi: 10.1006/jmbi.2001.4466. [DOI] [PubMed] [Google Scholar]
- Letunic I, et al. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–260. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej T, et al. Threading a database of protein cores. Proteins. 1995;23:356–369. doi: 10.1002/prot.340230309. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelman D, et al. Probabilistic scoring measures for profile-profile comparison yield more accurate short seed alignments. Bioinformatics. 2003;19:1531–1539. doi: 10.1093/bioinformatics/btg185. [DOI] [PubMed] [Google Scholar]
- Murzin AG, et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Ohlson T, Elofsson A. ProfNet, a method to derive profile-profile alignment scoring functions that improves the alignments of distantly related proteins. BMC Bioinformatics. 2005;6:253. doi: 10.1186/1471-2105-6-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson T, et al. Profile-profile methods provide improved fold-recognition: a study of different profile-profile alignment methods. Proteins. 2004;57:188–197. doi: 10.1002/prot.20184. [DOI] [PubMed] [Google Scholar]
- Panchenko AR. Finding weak similarities between proteins by sequence profile comparison. Nucleic Acids Res. 2003;31:683–689. doi: 10.1093/nar/gkg154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S. Searching databases of conserved sequence regions by aligning protein multiple-alignments. Nucleic Acids Res. 1996;24:3836–3845. doi: 10.1093/nar/24.19.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlewski L, et al. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 2000;9:232–241. doi: 10.1110/ps.9.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadreyev R, Grishin N. COMPASS: a tool for comparison of multiple protein alignments with assessment of statistical significance. J. Mol. Biol. 2003;326:317–336. doi: 10.1016/s0022-2836(02)01371-2. [DOI] [PubMed] [Google Scholar]
- Sauder JM, et al. Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins. 2000;40:6–22. doi: 10.1002/(sici)1097-0134(20000701)40:1<6::aid-prot30>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Schaffer AA, et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Van Walle I, et al. SABmark–a benchmark for sequence alignment that covers the entire known fold space. Bioinformatics. 2005;21:1267–1268. doi: 10.1093/bioinformatics/bth493. [DOI] [PubMed] [Google Scholar]
- von Ohsen N, et al. Profile-profile alignment: a powerful tool for protein structure prediction. Pac. Symp. Biocomput. 2003:252–263. [PubMed] [Google Scholar]
- Wilson D, et al. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res. 2007;35:D308–D313. doi: 10.1093/nar/gkl910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona G, Levitt M. Within the twilight zone: a sensitive profile-profile comparison tool based on information theory. J. Mol. Biol. 2002;315:1257–1275. doi: 10.1006/jmbi.2001.5293. [DOI] [PubMed] [Google Scholar]