

Abstract
Objective
Atypical teratoid/rhabdoid tumors are aggressive neoplasms of the central nervous system occurring mainly in the early childhood and rarely in adults. We described a case of this tumor in an 18-year-old male patient without previous medical history.
Material-method
The neoplasm was localized in the right fronto-temporal area of the brain and was totally excised. The specimen was fixed in formalin and embedded in paraffin. The histological and immunohistochemical features of the neoplasm were assessed, while sequencing analysis as well as interphase fluorescence in situ hybridization (FISH) were performed.
Results
Histological and immunohistochemical analysis demonstrated atypical rhabdoid cells strongly and diffusely positive for EMA and Vimentin as well as focally immunoreactive for SMA and GFAP. Additionally, though no abnormalities detected in the coding sequence of the INI1 gene, interphase FISH studies were consistent with a homozygous deletion of the INI1 gene in the majority of examined nuclei. INI1 immunostaining demonstrated diffuse loss of nuclear INI1 expression in tumor cells. Taken together, the results were consistent with a diagnosis of atypical teratoid/rhabdoid tumor (ATRT).
Conclusions
26 previous cases of ATRT have been reported in adults, thus far. To our knowledge, this is the eighth case of an ATRT reported in an adult patient having genetic confirmation and the first one in which the tumor is, partly, localized in the right temporal area of the brain. This unusual presentation underlines the necessity of considering this devastating neoplasm in the differential diagnosis of malignant brain tumors of young adults.
Keywords: Atypical, Teratoid/Rhabdoid, Tumor-malignant, neoplasm-adults
INTRODUCTION
Atypical Teratoid/Rhabdoid Tumor (ATRT), according to the World Health Organization (WHO) Classification of Tumors, is a highly malignant neoplasm (grade IV) of the Central Nervous System (CNS) that preferentially manifests in children less than three years of age [Rorke et al 1996, Judkins et al. 2007]. This tumor is mainly composed of rhabdoid cells, with the addition or not of areas demonstrating characteristics of primitive neuroectodermal tumor, epithelial tissue, neoplastic mesenchyme and neuronal or glial differentiation [Rorke and Biegel 2000, Judkins et al. 2007]. The most distinctive feature of ATRT is its association with inactivation of the hSNF5/INI1 gene, located in chromosome band 22q11.2, in the majority of cases [Versteege et al. 1998, Biegel et al. 1999].
Given that ATRT prevails in children under the age of 3, only 26 cases of this neoplasm have been reported in adult patients (18 years of age or older), so far [Horn et al. 1992, Cossu et al. 1993, Fisher et al. 1996, Ashraf et al. 1997, Byram 1999, Sugita et al. 1999, Kuge et al. 2000, Arrazola et al. 2000, Lutterbach et al. 2001, Bruch et al. 2001, Pimentel et al. 2003, Kachhara et al. 2003, Kawaguchi et al. 2004, Erickson et al. 2005, Raisanen et al. 2005, Chen et al. 2006, Rezanko et al. 2006, Chacko et al. 2007, Zarovnaya et al. 2007, Makuria et al. 2008]. In particular, only 7 of these cases had molecular genetic confirmation, based on findings by mutation analysis or fluorescence in situ hybridization (FISH) [Bruch et al. 2001, Raisanen et al. 2005, Chacko et al. 2007, Zarovnaya et al. 2007].
In this context, our aim was to present an unusual case of an ATRT in an 18-year-old male patient, in which we describe the main clinical and histological features of this particularly interesting but also devastating tumor. Moreover, we performed a short review of the relative medical literature about ATRT in adulthood. To the best of our knowledge, we present the eighth case of an ATRT with molecular genetic confirmation in an adult patient. Moreover, this is the first report of an ATRT extending to the right temporal area of the brain.
CASE HISTORY
An 18-year-old male patient, without previous medical problems, was admitted to the Department of Neurosurgery of our hospital with headache and seizures of one month duration. Neurological examination showed no major abnormalities except from mild hypoesthesia of the right face.
Magnetic Resonance Imaging (MRI) of the brain revealed a solid and partially cystic mass in the right fronto-temporal area with decreased density on T1-weighted images and intense contrast (gadolinium) enhancement [Figure 1]. Hemorrhage was mainly observed in the anterior portion of the neoplasm while extensive peritumoral edema was identified.
Figure 1.

T1-weighted Magnetic Resonance Imaging (MRI): Tumor enhancement with contrast material.
The tumor compressed the ipsilateral cerebral cortex as well as the right basal ganglia, displacing the midline and deforming the ventricles. These findings, according to the radiologist’s opinion, were compatible with that of a malignant, primary, brain tumor. Afterwards, the patient underwent right craniotomy and the neoplasm was totally excised. The patient received radiation therapy in another hospital but unfortunately succumbed to the disease 4 months later.
MATERIAL-METHODS
After total excision of the tumor, a soft red-brown mass, measuring 2.5 × 2 × 0.8 cm, was received at our pathology department. Multiple formalin-fixed paraffin-embedded tissue sections were examined with hematoxylin/eosin stain as well as with the immunohistochemical method of Envision-HRP for detection of: GFAP, MCK, EMA, Vimentin, CD56, CD57, Synaptophysin, Chromogranin-A, NSE, NFP, CD99, SMA, Desmin, AFP, hCG, Melan-A, HMB-45, c-Kit, CD34, S-100, Ki67 and p53. Immunohistochemistry for INI1 was performed as described [Judkins et al. 2004].
Genomic DNA was extracted from formalin fixed and paraffin embedded tissue using a commercially available kit (Gentra systems). Exons 2–9 of the INI1 gene were amplified and sequenced as previously described [Biegel et al. 1999]. Sections prepared from the formalin-fixed and paraffin-embedded tissue were analyzed by interphase FISH as previously described [Biegel et al. 1999].
RESULTS
Histologic examination demonstrated a highly cellular, malignant, solid, neoplasm, with extensive foci of hemorrhages and necrosis [Figure 2a, 2b]. A distinctive feature under low magnification was the sharp demarcation between neoplastic and normal tissue [Figure 2c].
Figure 2.
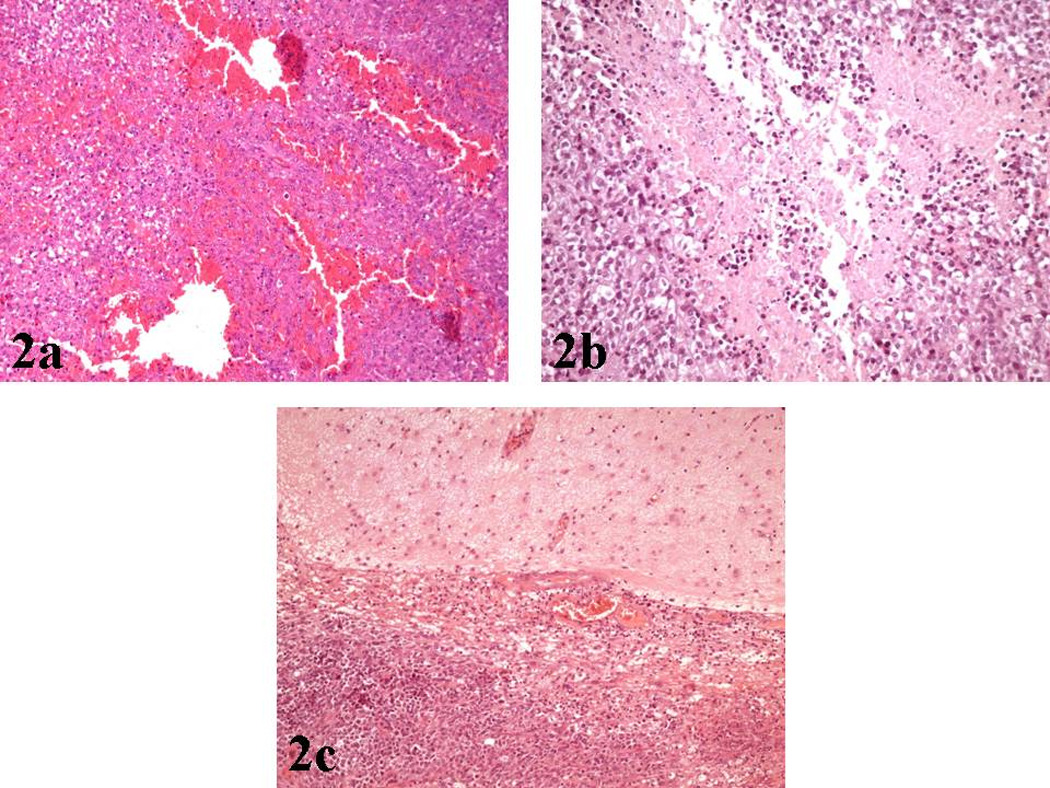
a): Hematoxylin/Eosin (X100): Area of hemorrhage within the neoplastic tissue.
b): Hematoxylin/Eosin (X200): Area of necrosis within the neoplasm.
c): Hematoxylin/Eosin (X100): Sharp demarcation of the tumor from adjacent brain parenchyma.
The majority of malignant cells were of medium size, round/oval or polygonal, with eccentric nuclei, conspicuous nucleoli and a finely granular eosinophilic or vacuolated cytoplasm [Figure 3a, 3b, 3c]. These cells with the typical ‘rhabdoid’ morphology were intimately associated with other malignant cells featured by indistinct cell borders, arranged in a more solid fashion and characterized by nuclei with a vesicular chromatin pattern [Figure 4a].
Figure 3.
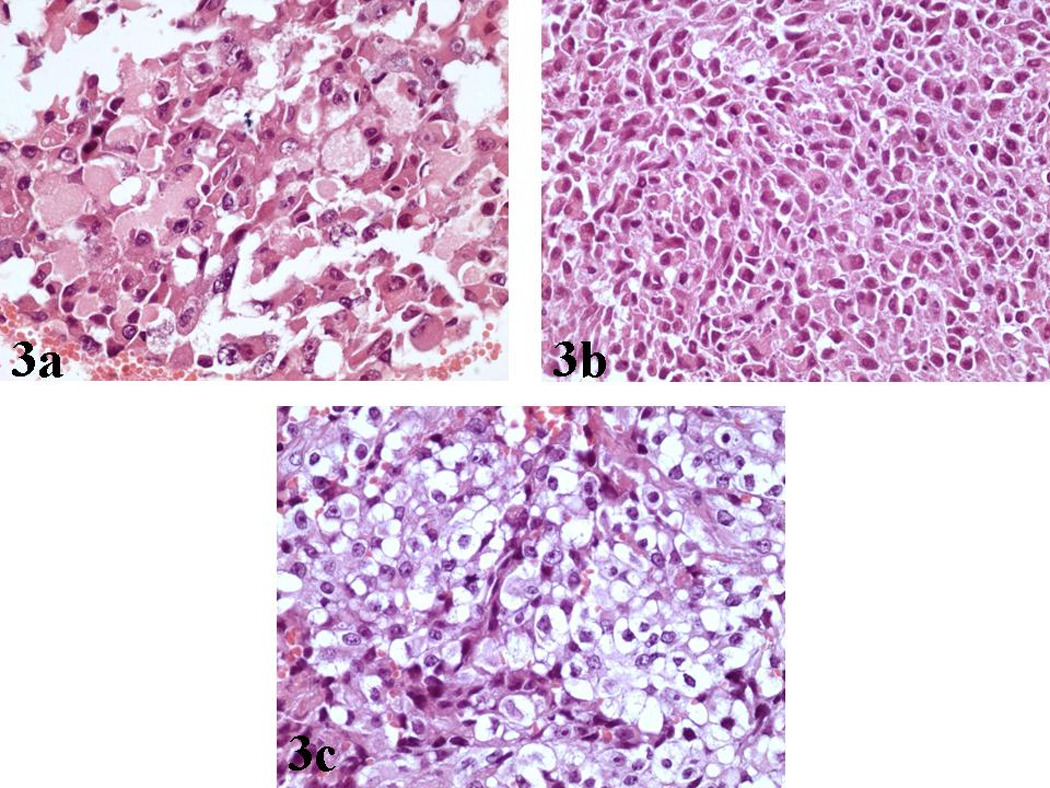
a, b): Hematoxylin/Eosin (X400): Tumor cells of medium size, round/oval or polygonal, with eccentric nuclei, conspicuous nucleoli and a finely granular eosinophilic cytoplasm.
c): Hematoxylin/Eosin (X400): Malignant cells with cytoplasmic vacuolation.
Figure 4.
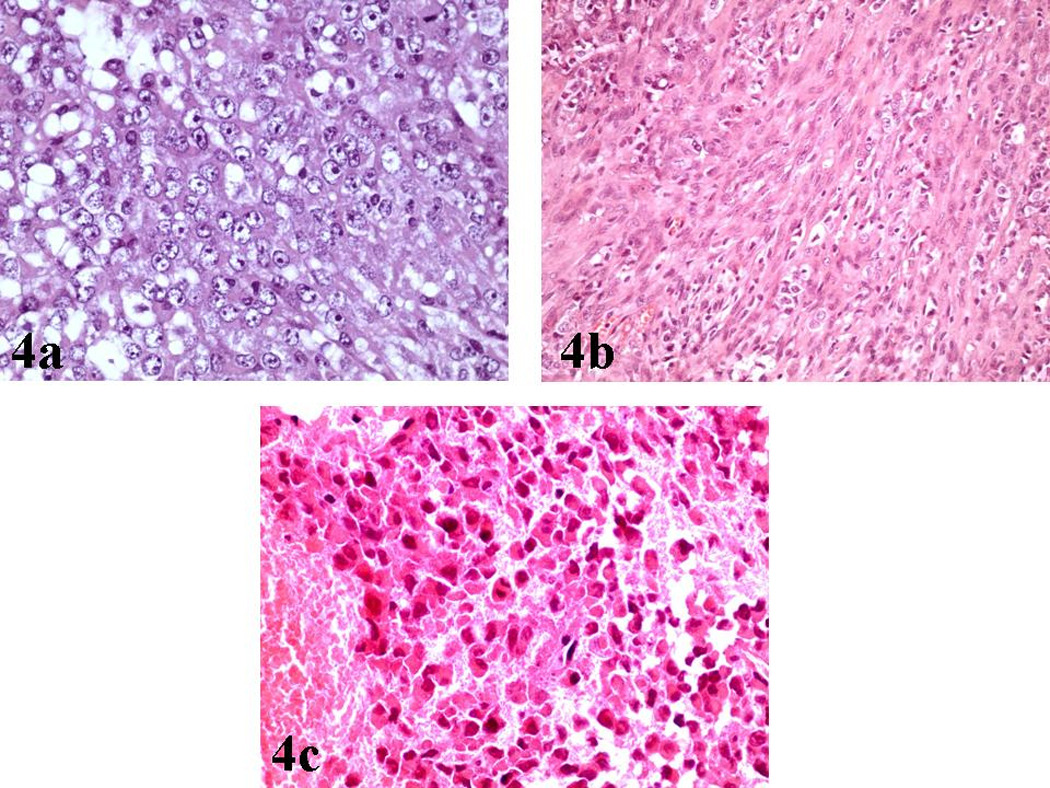
a): Hematoxylin/Eosin (X400): Nuclei of neoplastic cells with characteristic vesicular chromatin.
b): Hematoxylin/Eosin (X200): Spindle cell component of the tumor.
c): Hematoxylin/Eosin (X400): Conspicuous mitotic activity within the neoplasm.
Interestingly, a minor mesenchymal component was recognized, featured by spindle tumor cells with a sarcomatoid distribution [Figure 4b]. Finally, foci of fibrosis were identified, whereas the mitotic activity was prominent [Figure 4c].
Immunohistochemical studies showed diffuse, intense, cytoplasmic expression of Vimentin from the majority of malignant cells [Figure 5a]. Regarding EMA expression, a strong membranous and cytoplasmic expression was noted in a large fraction of the neoplastic tissue [Figure 5b]. There was intense SMA expression, though with a focal distribution [Figure 5c].
Figure 5.
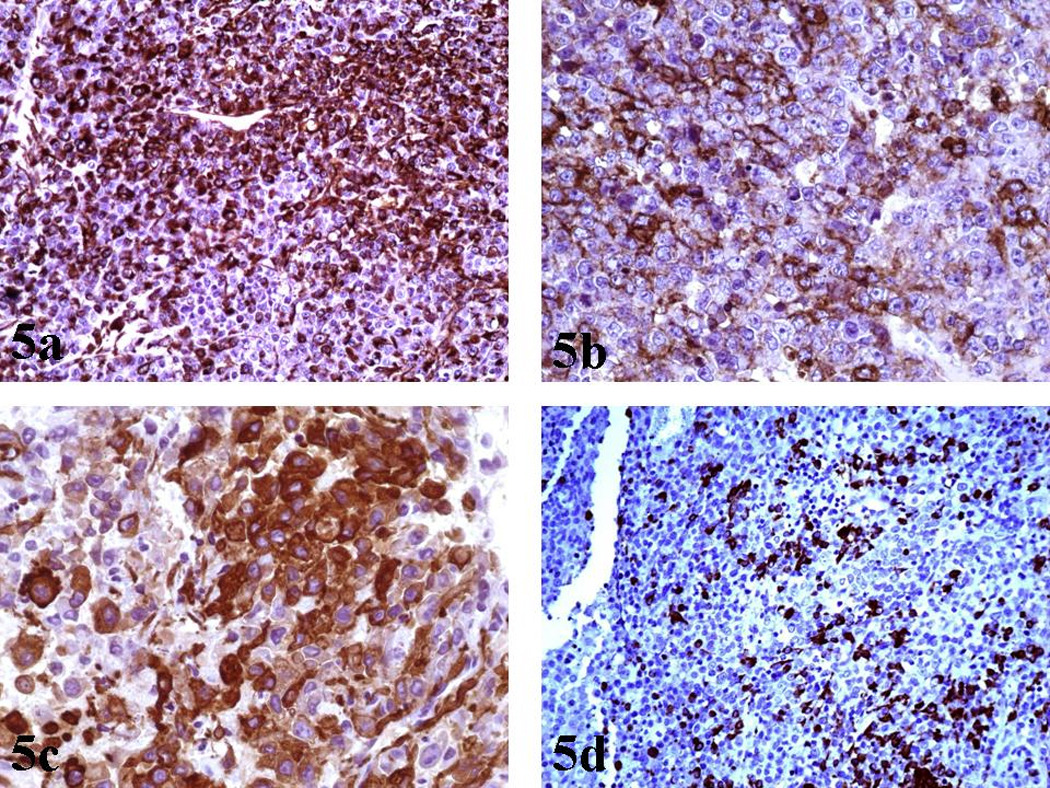
a): Vimentin (X200): Diffuse, intense, cytoplasmic expression from the majority of tumor cells.
b): EMA (X400): Strong EMA expression from many malignant cells.
c): SMA (X400): Focal, intense, SMA expression from rhabdoid cells.
d): GFAP (X200): Focal expression of GFAP.
Additionally, immunostaining for GFAP depicted positivity in some areas of rhabdoid cells [Figure 5d]. Cytokeratins (MCK) [Figure 6], Synaptophysin and NFP were focally expressed in few malignant cells. Staining for: Chromogranin-A, S-100, NSE, CD56, CD57, Desmin, AFP, hCG, Melan-A, HMB-45, c-Kit, CD99 and CD34, all showed no expression. INI1 immunostaining revealed diffuse loss of nuclear INI1 expression in tumor cells [Figure 7]. Finally, the Ki-67 proliferation index was computed at 50% [Figure 8] and the mutant p53 protein was detected in a percentage of 40%, out of the total number of malignant cells counted.
Figure 6.
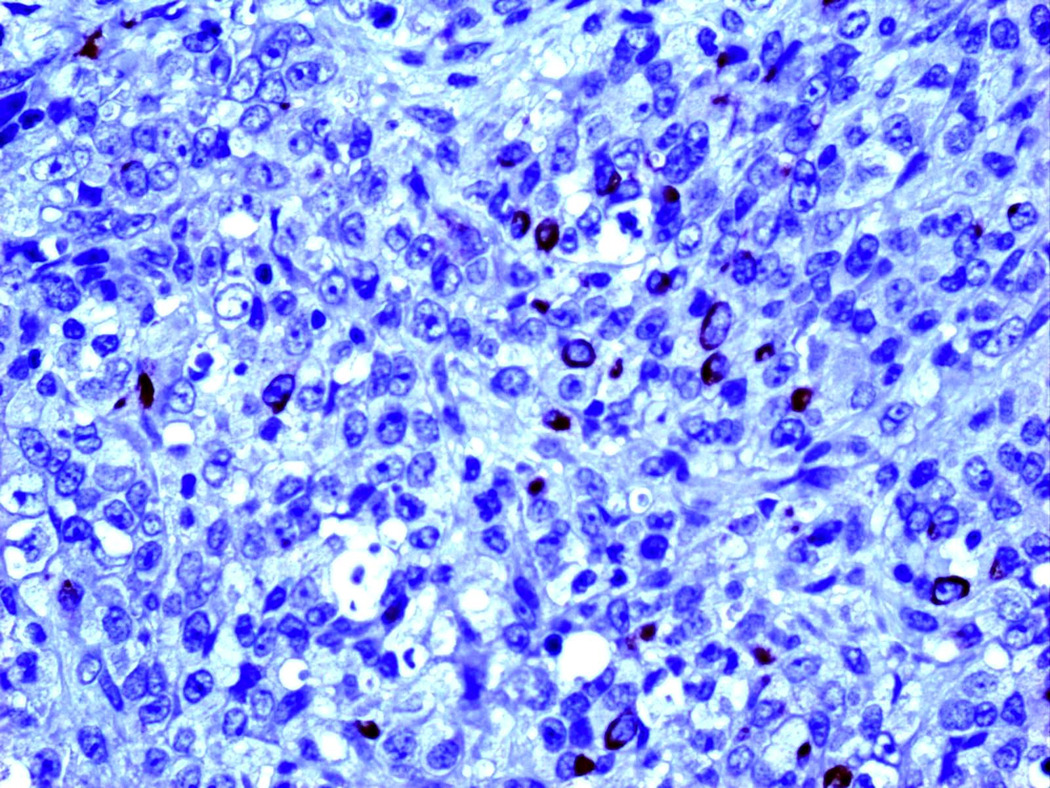
MCK (X400): Focal expression of MCK in few malignant cells.
Figure 7.
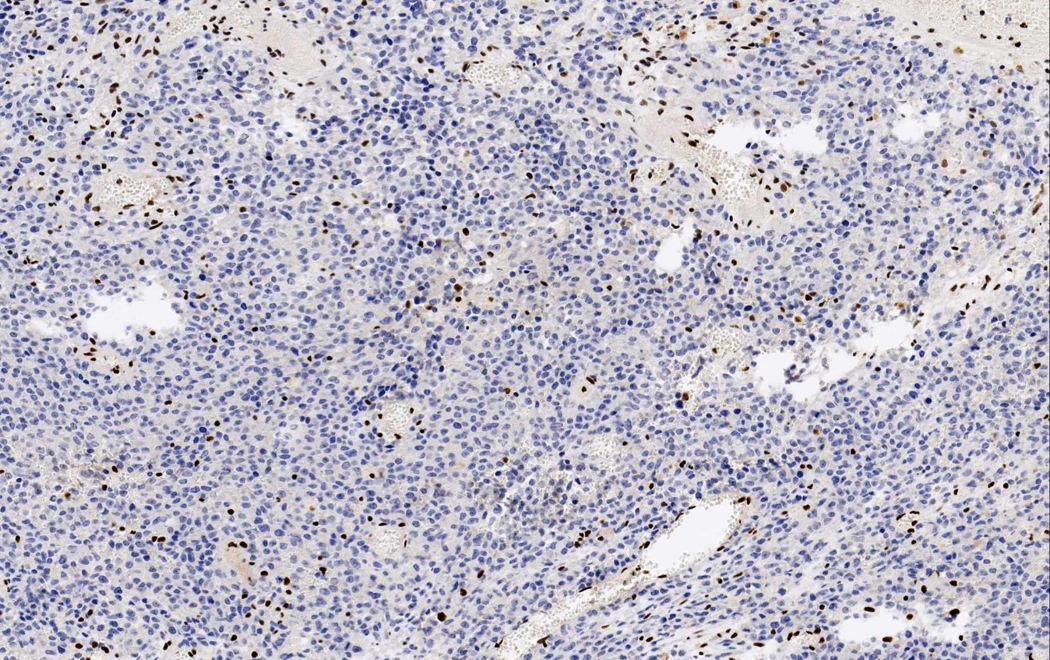
INI1 (X200): Diffuse loss of nuclear INI1 expression in tumor cells with retained expression in normal vascular structures and inflammatory cells within the tumor.
Figure 8.
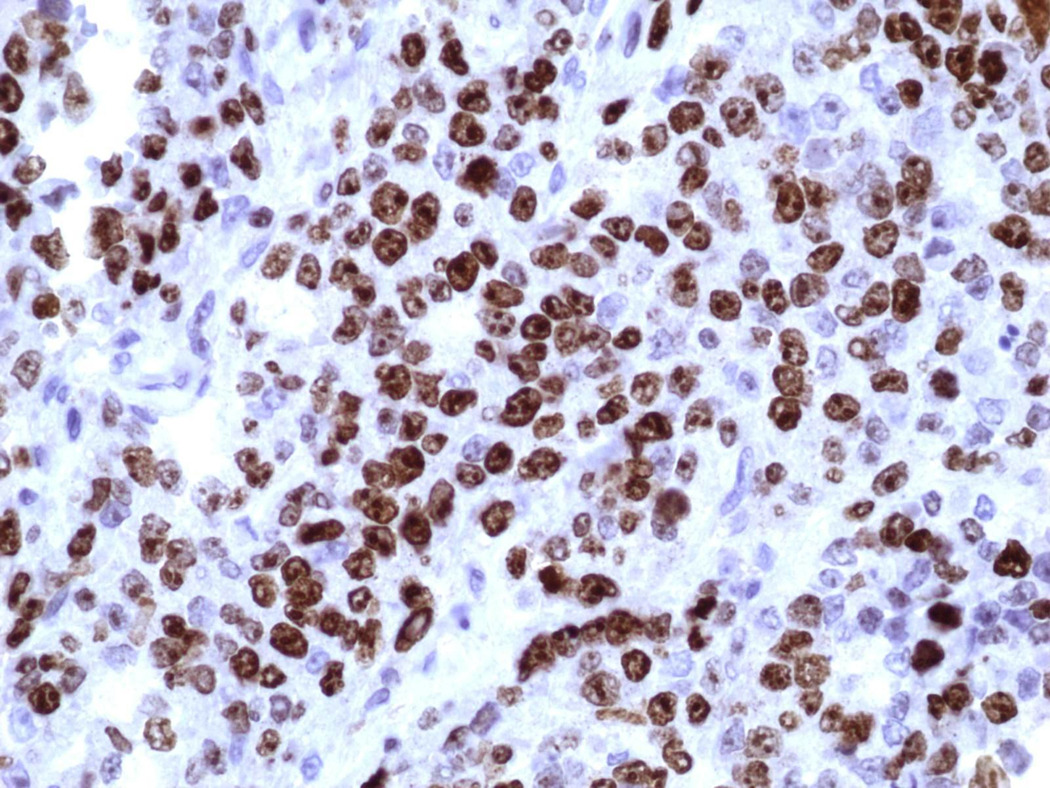
Ki67 (X400): High nuclear expression of Ki67 proliferation index.
There were no abnormalities detected in the coding sequence of the INI1 gene. Interphase FISH studies, however, demonstrated a homozygous deletion of the INI1 gene in 72/100 nuclei examined, compared to retention of both copies of the EWS 22q12 control probe.
The histopathological and immunohistochemical characteristics along with the findings of the molecular analysis were consistent with a diagnosis of ATRT .
DISCUSSION
The initial description of a rhabdoid tumor localized within the CNS, was given in 1985 [Montgomery et al. 1985]. Rorke et al. in 1996, first characterized this tumor as ‘atypical teratoid/rhabdoid tumor’, based on the disparate combination of rhabdoid, primitive neuroepithelial, epithelial and mesenchymal components [Rorke et al. 1996]. It is also of interest that this neoplasm has a male predilection and a unique similarity with the malignant rhabdoid tumor of the kidney [Rorke and Biegel 2000].
It is believed that ATRT accounts for approximately 1–2% of pediatric brain tumors [Judkins et al. 2007]. This neoplasm is extremely rare in adulthood. Based on the literature, we identified only 26 reported cases in adults (18 years of age or older) between 1992 and 2008, with a mean age at diagnosis of approximately 29 years (range: 18–45 years) [Table 1]. Among these patients, 16 were males and 10 females, with a mean age at diagnosis of approximately 29 years for both groups (range: 18–45 and 20–43 years, respectively). It should be noted, however, that only one case has been previously described affecting an 18-year-old male patient [Cossu et al. 1993].
Table 1.
Summary of cases regarding atypical teratoid/rhabdoid tumor of the CNS in adults.
| Year of publication | Age at diagnosis (years) | Gender | Tumor localization | Immunohistochemical analysis: positive markersexpression or not of INI1 protein | Molecular genetic analysis | Survival | |
|---|---|---|---|---|---|---|---|
| Horn et al. | 1992 | 21 | Male | Left temporal | Vimentin, EMA, a1-antichymotrypsin | Not performed | 72 months** |
| Cossu et al. | 1993 | 18 | Male | Left Frontal | Vimentin, EMA, Keratin | Not performed | 18 months** |
| Fisher et al. | 1996 | 32 | Male | Left frontal | Vimentin, S100, GFAP | Not performed | 1 month** |
| Ashraf et al. | 1997 | 34 | Male | Left cerebral | Vimentin | Not performed | 6 months** |
| Byram | 1999 | 35* | Male* | Left temporal | Vimentin, EMA, Keratin* | Not performed | 60 months** |
| Sugita et al. | 1999 | 27 | Male | Pineal region | Chromogranin A, synaptophysin, Vimentin, EMA, S-100, NSE, SMA, HHF-35 | Not performed | 24 months** |
| Kuge et al. | 2000 | 32 | Female | Suprasellar | Vimentin, EMA, Keratin, SMA | Not performed | No information |
| Arrazola et al. | 2000 | 20 | Male | Left parietal | EMA, Keratin,Viment, S- 100 | Not performed | Alive 24 months after the first craniotomy |
| Lutterbach et al. | 2001 | 30 | Female | Cerebellum | Vimentin, S100, NFP, Keratin, EMA, GFAP | Not performed | 11 months** |
| Bruch et al. | 2001 | 21 | Female | Spinal cord | EMA, vimentin, and SMA | Yes (22q deletion by FISH) | 6 months** |
| 34 | Female | Parietal | 6 months** | ||||
| Pimentel et al. | 2003 | 31 | Female | Right parietal | Vimentin CD68, NFP, a1-antichymotrypsin,? a1-antitrypsin, GFAP, S100, HHF35 | Not performed | 6 months** |
| Kachhara et al. | 2003 | 35 | Male | Thalamus | Vimentin | Not performed | No information |
| Kawaguchi et al. | 2004 | 22 | Male | Left cerebellar hemisphere and diffuse leptomening eal dissemination to the brain stem and the whole spinal cord | Vimentin, SMA, synaptophysin, NFP | Not performed | Alive 24 months after the diagnosis |
| Erickson et al. | 2005 | 20 | Female | Right occipital | GFAP, vimentin | Yes (FISH: no abnormalities in the INI-1 region of chromosome 22q11) | No information |
| Raisanen et al. | 2005 | 20 | Female | Sellar mass | Loss of INI1 expression | Yes (22q11.2 deletion, INI1 mutation) | Alive 28 months after the diagnosis |
| 31 | Female | Sellar mass with suprasellar extension | Loss of INI1 expression | Yes (22q11.2 deletion, INI1 mutation) | 9 months** | ||
| 45 | Male | Right cerebellar mass | Loss of INI1 expression | Yes (22q11.2 deletion, INI1 mutation) | Alive 15 months after the diagnosis | ||
| Chen et al. | 2006 | 19 | Male | Posterior fossa | No information | No information | 56.5 months** |
| Rezanko et al. | 2006 | 27 | Male | Right frontal | Vimentin, EMA, S-100 | Not performed | 4 months** |
| Chacko et al. | 2007 | 23 | Male | Right frontal | Vimentin, SMA, EMA, CD34, loss of INI1 expression | Yes (evidence of a 22q deletion by FISH, and a detectable INI1 gene mutation) | 1,5 months** |
| Zarovnaya et al. | 2007 | 43 | Female | Spinal cord | Weakly positive for EMA, loss of INI1 expression | Yes (FISH: monosomy 22) | 30 months** |
| Makuria et al. | 2008 | 23 | Male | Posterior left temporal lobe | Vimentin, Keratin, SMA, EMA, NFP, Synaptophysin, loss of INI1 expression | Not performed | In remission 30 months after the diagnosis |
| 25 | Female | Right frontal | Vimentin, SMA, Synaptophysin, CD34, NFP, loss of INI1 expression | Not performed | Alive 17 years after the diagnosis | ||
| 42 | Male | Right frontoparietal | Vimentin, EMA,Keratin, loss of INI1 expression | Not performed | Alive 18 months after the diagnosis | ||
| 37 | Male | Right frontoparietal | Vimentin, INI1 positivity | Not performed | No information | ||
information based on Lutterbach et al. 2001, Rezanko et al. 2006, Makuria et al. 2008
patient succumbed to the neoplasm
Abbreviations: EMA: epithelial membrane antigen, GFAP: glial fibrillary acidic protein, HHF35: anti-human muscle actin, NFP: neurofilament protein, NSE: neuron-specific enolase, SMA: α-smooth muscle actin.
Regarding localization, ATRTs of adults are basically located in the cerebral hemispheres [Judkins et al. 2007] [Table 1], while infratentorial neoplasms located in the cerebellum are relatively rare [Lutterbach et al. 2001, Kawaguchi et al. 2004, Raisanen et al. 2005]. It is uncommon for ATRT to be localized in the spinal cord of adults [Bruch et al. 2001, Zarovnaya et al. 2007] or children [Tanizaki et al. 2006].
Similar to the current case, a right frontal localization of the tumor was identified in 5 of the adult cases in the literature [Rezanko et al. 2004, Chacko et al. 2007, Makuria et al. 2008]. Handless frequently left temporal tumors have been reported [Horn et al. 1992, Byram 1999, Makuria et al. 2008]. However, there is no report with a neoplasm extending to the right temporal area of the brain.
With regard to imaging studies, ATRTs are usually of increased density on unenhanced Computerized Tomography (CT) images and heterogeneously enhancing with the administration of contrast material [Bambakidis et al. 2002, Judkins et al. 2007]. On MRI studies, decreased density on T1-weighted images and enhancement with gadolinium, is usually detected, as it was the case in our patient [Rorke and Biegel 2000].
Grossly, these, mainly soft and pinkish-red, neoplasms are, in part, well-demarcated from the surrounding brain structures and characteristically contain areas of hemorrhage and necrosis, similarly to our case [Rorke and Biegel 2000, Reddy 2005, Rezanko et al. 2006, Chacko et al. 2007].
As far as histopathological features are concerned, the rhabdoid cellular component is outstanding, within most ATRTs of children as well as of adults [Wick et al. 1995, Rorke et al. 1996, Arrazola et al. 2000, Judkins et al. 2007, Zarovnaya et al. 2007, Makuria et al. 2008]. Our case summarized the main features of these cells with the brisk mitotic activity and the marked proliferation rate (estimated with the Ki67 index). Furthermore, one can observe epithelial-like and mesenchymal areas [Lutterbach et al. 2001] as well as variable elements of primitive neuroectodermal tumor [Rorke and Biegel 2000, Reddy 2005]. The mesenchymal component was shown in our case, although in a lesser extent.
ATRTs are polyphenotypic tumors which characteristically show expression of EMA, vimentin and SMA, as well as various other markers, by immunohistochemical staining [Rorke and Biegel 2000, Judkins et al. 2007] [Table 1]. Additionally, expression of GFAP, Neurofilaments, S-100, Synaptophysin, NSE, CD68, α1-antitrypsin, α1-antichymotrypsin and Keratin may be detected, depending on the abovementioned different cellular composition of the neoplasm [Rorke et al. 1996, Rorke and Biegel 2000] [Table 1]. However, it should be pointed out that ATRTs typically do not express desmin or any of the markers for germ cell tumors [Rorke and Biegel 2000, Judkins et al. 2007] [Table 1]. What is more, though the INI1 nuclear protein (the product of hSNF5/INI1 gene) is typically expressed in normal brain tissue and in the majority of other neoplasms, loss of its expression has been observed in most ATRTs, including those presented in adults [Judkins et al 2004, Judkins et al. 2007] [Table 1].
In this context, it ought to be highlighted that mutations or loss of the INI1 locus at 22q11.2 represent a characteristic finding of most ATRTs, both in children and in adults [Biegel et al. 1999, Judkins et al. 2007]. In our review, we noted that in only 8/26 cases molecular genetic analysis was performed and in 7 of them a confirmation of the diagnosis was done [Table 1]. Cossu et al., in particular, described the only previous case of ATRT in an 18-year-old patient based on histological and immunohistochemical findings, without presenting molecular-genetic evidence [Cossu et al. 1993].
Regarding differential diagnosis of ATRT, variable metastatic tumors, rhabdoid meningioma, choroids plexus carcinoma and medulloblastoma/primitive neuroectodermal tumor (PNET) constitute the main entities under consideration [Rorke and Biegel 2000, Strother 2005]. The absence of any other extra-CNS primary lesion in our patient, the lack of conventional areas of meningiomas as well as the immunohistochemical and molecular features of our case, excluded these entities.
As far as histogenesis is concerned, there is a great deal of controversy. Many proposals highlight the histiocytic, mesenchymal, meningeal, neuroectodermal or even germ cell lineage of ATRT [Rorke and Biegel 2000, Judkins et al. 2007].
Finally, the prognosis of patients with ATRTs is extremely poor [Zimmerman et al. 2005, Judkins et al. 2007]. It is generally believed that most of the patients die within approximately 1 year after the initial diagnosis, probably as a result of metastatic lesions through the cerebrospinal pathway, which are common in this aggressive neoplasm [Rorke and Biegel 2000, Zimmerman et al. 2005, Strother 2005, Judkins et al. 2007]. Our analysis of adults’ cases demonstrated that the mean survival time of patients who died of the disease and whose data were available, was approximately 21 months after diagnosis (range: 1–72). Interestingly, males appear to have a more favorable outcome compared to females [mean survival: 27 months (range: 1–72) and 11,33 months (range: 6–30), respectively] [Table 1].
CONCLUSION
To summarize, we presented a case of an ATRT in an 18-year-old male patient. ATRT is a rare tumor of uncertain origin which must be considered in the differential diagnosis of intracranial neoplasms both in adult and pediatric population. Further genetic studies of the INI1 gene located in chromosome 22 may shed light upon the dismal biological behavior of this tumor.
Acknowledgments
The authors acknowledge the technical assistance of Luanne Wainwright and Laura Tooke. This study was supported in part by a grant from the NIH (CA 46274) to J.A. Biegel.
Footnotes
Conflict of Interest Statement:
We declare that we have no conflict of interest.
REFERENCES
- Arrazola J, Pedrosa I, Méndez R, Saldaña C, Scheithauer BW, Martínez A. Primary malignant rhabdoid tumour of the brain in an adult. Neuroradiology. 2000;42(5):363–367. doi: 10.1007/s002340050900. [DOI] [PubMed] [Google Scholar]
- Ashraf R, Bentley RC, Awan AN, McLendon RE, Ragozzino MW. Implantation metastasis of primary malignant rhabdoid tumor of the brain in an adult (one case report) Med Pediatr Oncol. 1997;28(3):223–227. doi: 10.1002/(sici)1096-911x(199703)28:3<223::aid-mpo14>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Bambakidis NC, Robinson S, Cohen M, Cohen AR. Atypical teratoid/rhabdoid tumors of the central nervous system: clinical, radiographic and pathologic features. Pediatr Neurosurg. 2002;37(2):64–70. doi: 10.1159/000065107. [DOI] [PubMed] [Google Scholar]
- Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A. A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Hum Pathol. 2001;32(2):156–162. doi: 10.1053/hupa.2001.21572. [DOI] [PubMed] [Google Scholar]
- Byram D, Regarding Weiss, et al. IJROBP 41:1013–1019; 1998. Int J Radiat Oncol Biol Phys. 1999;45(1):247. doi: 10.1016/s0360-3016(99)00106-6. [DOI] [PubMed] [Google Scholar]
- Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol. 2007;84(2):217–222. doi: 10.1007/s11060-007-9361-z. [DOI] [PubMed] [Google Scholar]
- Chen YW, Wong TT, Ho DM, Huang PI, Chang KP, Shiau CY, Yen SH. Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience) Int J Radiat Oncol Biol Phys. 2006;64(4):1038–1043. doi: 10.1016/j.ijrobp.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Cossu A, Massarelli G, Manetto V, Viale G, Tanda F, Bosincu L, Iuzzolino P, Cossu S, Padovani R, Eusebi V. Rhabdoid tumours of the central nervous system. Report of three cases with immunocytochemical and ultrastructural findings. Virchows Arch A Pathol Anat Histopathol. 1993;422(1):81–85. doi: 10.1007/BF01605137. [DOI] [PubMed] [Google Scholar]
- Erickson ML, Johnson R, Bannykh SI, de Lotbiniere A, Kim JH. Malignant rhabdoid tumor in a pregnant adult female: literature review of central nervous system rhabdoid tumors. J Neurooncol. 2005;74(3):311–319. doi: 10.1007/s11060-004-7560-4. [DOI] [PubMed] [Google Scholar]
- Fisher BJ, Siddiqui J, Macdonald D, Cairney AE, Ramsey D, Munoz D, Del Maestro R. Malignant rhabdoid tumor of brain: an aggressive clinical entity. Can J Neurol Sci. 1996;23(4):257–263. doi: 10.1017/s0317167100038191. [DOI] [PubMed] [Google Scholar]
- Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E. Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol. 1992;83(4):445–448. doi: 10.1007/BF00713540. [DOI] [PubMed] [Google Scholar]
- Judkins AR, Eberhart CG, Wesseling P. Atypical teratoid/rhabdoid tumour. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC Press; 2007. pp. 147–149. [Google Scholar]
- Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol. 2004;28(5):644–650. doi: 10.1097/00000478-200405000-00013. [DOI] [PubMed] [Google Scholar]
- Kachhara R, Retnam TM, Kumar S, Nair S, Bhattacharya RN, Krishnamoorthy T, Radhakrishnan VV. Rhabdoid tumor of the thalamus. Neurol India. 2003;51(2):273–274. [PubMed] [Google Scholar]
- Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science. 1994;266(5193):2002–2006. doi: 10.1126/science.7801128. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Kumabe T, Watanabe M, Tominaga T. Atypical teratoid/rhabdoid tumor with leptomeningeal dissemination in an adult. Acta Neurochir (Wien) 2004;146(9):1033–1038. doi: 10.1007/s00701-004-0313-5. [DOI] [PubMed] [Google Scholar]
- Kuge A, Kayama T, Tsuchiya D, Kawakami K, Saito S, Nakazato Y, Suzuki H. Suprasellar primary malignant rhabdoid tumor in an adult: a case report. No Shinkei Geka. 2000;28(4):351–358. [PubMed] [Google Scholar]
- Lutterbach J, Liegibel J, Koch D, Madlinger A, Frommhold H, Pagenstecher A. Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol. 2001;52(1):49–56. doi: 10.1023/a:1010683416555. [DOI] [PubMed] [Google Scholar]
- Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol. 2008;88(3):321–330. doi: 10.1007/s11060-008-9571-z. [DOI] [PubMed] [Google Scholar]
- Montgomery P, Kuhn JP, Berger PE. Rhabdoid tumor of the kidney: a case report. Urol Radiol. 1985;7(1):42–44. doi: 10.1007/BF02926849. [DOI] [PubMed] [Google Scholar]
- Pimentel J, Silva R, Pimentel T. Primary malignant rhabdoid tumors of the central nervous system: considerations about two cases of adulthood presentation. J Neurooncol. 2003;61(2):121–126. doi: 10.1023/a:1022135518846. [DOI] [PubMed] [Google Scholar]
- Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. 2005;15(1):23–28. doi: 10.1111/j.1750-3639.2005.tb00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AT. Atypical teratoid/rhabdoid tumors of the central nervous system. J Neurooncol. 2005;75(3):309–313. doi: 10.1007/s11060-005-6762-8. [DOI] [PubMed] [Google Scholar]
- Rezanko T, Tunakan M, Kahraman A, Sucu HK, Gelal F, Akkol I. Primary rhabdoid tumor of the brain in an adult. Neuropathology. 2006;26(1):57–61. doi: 10.1111/j.1440-1789.2006.00624.x. [DOI] [PubMed] [Google Scholar]
- Rorke LB, Biegel JA. Atypical teratoid/rhabdoid tumour. In: Kleihues P, Cavenee WK, editors. Pathology and Genetics Tumours of the Nervous System. Lyon: IARC Press; 2000. pp. 123–148. [Google Scholar]
- Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56–65. doi: 10.3171/jns.1996.85.1.0056. [DOI] [PubMed] [Google Scholar]
- Strother D. Atypical teratoid rhabdoid tumors of childhood: diagnosis, treatment and challenges. Expert Rev Anticancer Ther. 2005;5(5):907–915. doi: 10.1586/14737140.5.5.907. [DOI] [PubMed] [Google Scholar]
- Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M. Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int. 1999;49(12):1114–1118. doi: 10.1046/j.1440-1827.1999.00988.x. [DOI] [PubMed] [Google Scholar]
- Tanizaki Y, Oka H, Utsuki S, Shimizu S, Suzuki S, Fujii K. Atypical teratoid/rhabdoid tumor arising from the spinal cord--case report and review of the literature. Clin Neuropathol. 2006;25(2):81–85. [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol. 1995;12(3):233–248. [PubMed] [Google Scholar]
- Zarovnaya EL, Pallatroni HF, Hug EB, Ball PA, Cromwell LD, Pipas JM, Fadul CE, Meyer LP, Park JP, Biegel JA, Perry A, Rhodes CH. Atypical teratoid/rhabdoid tumor of the spine in an adult: case report and review of the literature. J Neurooncol. 2007;84(1):49–55. doi: 10.1007/s11060-007-9339-x. [DOI] [PubMed] [Google Scholar]
- Zimmerman MA, Goumnerova LC, Proctor M, Scott RM, Marcus K, Pomeroy SL, Turner CD, Chi SN, Chordas C, Kieran MW. Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol. 2005;72(1):77–84. doi: 10.1007/s11060-004-3115-y. [DOI] [PubMed] [Google Scholar]