

Abstract
The purpose of this study was to investigate whether topically administered P-glycoprotein (P-gp) substrates/modulators can alter vitreal kinetics of intravitreally administered quinidine. Male New Zealand rabbits were used under anesthesia. Vitreal kinetics of intravitreally administered quinidine (0.75-μg dose) was determined alone and in the presence of verapamil (coadministered topically/intravitreally) or prednisolone hemisuccinate sodium (PHS) (coadministered topically). In the presence of topically instilled verapamil (1% w/v), elimination half-life (t1/2) (176 ± 7 min), apparent elimination rate constant (λz) (0.0039 ± 0.0001 min–1), and mean retention time (MRT) (143 ± 30 min) of intravitreally administered quinidine were significantly different from those of the control (105 ± 11 min, 0.0066 ± 0.0007 min–1, and 83 ± 13 min, respectively). A 2-fold increase in the t1/2 with a corresponding decrease in λz and a 1.5-fold increase in the MRT of quinidine were observed in the presence of topically coadministered 2% w/v PHS. Intravitreal coadministration of quinidine and verapamil resulted in a significant increase in t1/2 (159 ± 9 min) and a decrease in λz (0.0043 ± 0.0002 min–1) of quinidine. The vitreal pharmacokinetic parameters of sodium fluorescein, alone or in the presence of topically instilled verapamil, did not show any statistically significant difference, indicating that ocular barrier integrity was not affected by topical verapamil administration. Results from this study suggest that topically applied P-gp substrates/modulators can alter vitreal pharmacokinetics of intravitreally administered P-gp substrates, possibly through the inhibition of P-gp expressed on the basolateral membrane of the retinal pigmented epithelium.
The retina is the primary target for most posterior segment ocular disorders such as age-related macular degeneration, diabetic macular edema, retinitis pigmentosa, endophthalmitis, and proliferative vitreoretinopathy (Kim et al., 2007). However, drug delivery to the posterior chamber ocular tissues is challenged by various physiological barriers such as the cornea, conjunctiva, sclera, and the blood-ocular barriers (Dey et al., 2003; Duvvuri et al., 2003b; Majumdar et al., 2003a,b; Cunha-Vaz, 2004; Yasukawa et al., 2004). The retinal pigmented epithelium (RPE), which forms the outer blood-retinal barrier, limits vitreal penetration of drugs administered by the systemic and transscleral routes (Duvvuri et al., 2003a; Ghate and Edelhauser, 2006; Janoria et al., 2007). P-glycoprotein (P-gp), a 170-kDa ATP-dependent membrane-bound efflux protein, expressed on the RPE plays a major role in restricting diffusion of P-gp substrates from the choroidal stroma into the neural retina across the RPE (Kennedy and Mangini, 2002; Steuer et al., 2005).
P-gp displays broad specificity, accepting many structurally, functionally, and mechanistically unrelated compounds (Ambudkar et al., 2003), and its role in limiting drug penetration across biological barriers is well established. P-gp-mediated drug efflux at the blood-brain barrier is a major factor behind poor penetration of chemotherapeutic agents that are P-gp substrates into the brain after systemic administration (Golden and Pollack, 2003; Kemper et al., 2004). A number of reports also illustrate the role of intestinal P-gp in limiting systemic bioavailability of orally administered agents. Moreover, up-regulation of P-gp expressed by tumor cells is considered to be a major mechanism behind multidrug resistance (Matheny et al., 2001; Fromm, 2003, 2004; Kunta and Sinko, 2004). In addition, it has also been demonstrated that P-gp expressed on the canalicular membrane of the hepatocytes and the luminal surface of the proximal kidney tubule cells, including nephrons, expedites hepatic and renal elimination of substrates.
In general, P-gp is expressed on the apical membrane of epithelial cells, preventing drug transport from the lumen into the systemic circulation (e.g., intestinal epithelium) or from the systemic circulation into the brain (endothelial cells of the blood-brain barrier) (Matheny et al., 2001). However, an earlier report suggests that P-gp is expressed on both the apical as well as basal membranes of the RPE cells (Kennedy and Mangini, 2002). P-gp on the RPE cells may thus affect permeation of substrates from the vitreous humor into the systemic circulation and vice versa (Dey et al., 2003; Duvvuri et al., 2003b) and could be a major factor behind the inability of systemic, periocular, and trans-scleral routes of administration to generate and maintain therapeutic concentrations of P-gp substrates in the retina. Thus, factors/agents that can modulate the efflux activity of RPE P-gp could probably alter ocular pharmacokinetics of P-gp substrates.
In the past, a number of strategies attempting to modulate the activity or expression of efflux proteins on various mammalian tissues have been investigated. These include the use of chemosensitizers, prodrugs, polymers, nanoparticles, transcriptional regulators, and monoclonal antibodies (Jain et al., 2004; Katragadda et al., 2005; Nobili et al., 2006). We were surprised to find that there are only three studies, to our knowledge, in which drug-drug interaction at the level of the RPE P-gp and its effect on ocular drug pharmacokinetics in vivo have been investigated. These recent reports evaluated the effect of systemic/systemic, systemic/intravitreal, or intravitreal/intravitreal coadministration of substrates or inhibitors on ocular pharmacokinetics (Duvvuri et al., 2003a; Senthilkumari et al., 2008a,b). However, so far, the effect of topically administered P-gp substrates/inhibitors on the functional activity of P-gp expressed on the RPE has not been reported.
Topical eye drops containing antimicrobial and anti-inflammatory agents, steroids, and other therapeutic compounds are routinely administered to treat various ocular infections and disorders. Many of these agents are P-gp substrates/inhibitors and can diffuse into the RPE. The objective of this study was to determine whether topically administered P-gp substrates could modulate the functional activity of RPE P-gp and alter the vitreal pharmacokinetics of another P-gp substrate, quinidine, administered intravitreally. Erythromycin (Matheny et al., 2001), prednisolone (P-gp substrates commonly applied topically) (Karssen et al., 2002), and verapamil (a P-gp inhibitor used in earlier reports investigating inhibition of RPE P-gp) were administered topically. Quinidine, used in an earlier study to evaluate functional activity of RPE P-gp (Duvvuri et al., 2003a), was used as a model P-gp substrate in this study and its pharmacokinetic parameters were evaluated.
Materials and Methods
Animals. New Zealand male White rabbits were procured from Myrtle's Rabbitry (Thompson Station, TN). Experiments conformed to the tenets of the Association for Research in Vision and Ophthalmology statement on the Use of Animals in Ophthalmic and Vision Research and followed the University of Mississippi Institutional Animal Care and Use Committee approved protocols.
Materials. Microdialysis probes (CMA/20; 20,000 Da molecular mass weight and 10 mm shaft) were obtained from CMA/Microdialysis Inc. (North Chelmsford, MA). Erythromycin, prednisolone hemisuccinate sodium (PHS), verapamil hydrochloride, fluorescein sodium, and quinidine hydrochloride were purchased form Sigma-Aldrich (St. Louis, MO). Ketamine hydrochloride and xylazine were procured from Fort Dodge Animal Health (Fort Dodge, IA) and Lloyd Laboratories (Shenandoah, IA), respectively. Pentobarbital was obtained from Virbac AH, Inc. (Fort Worth, TX). Solvents used were purchased from Thermo Fisher Scientific (Waltham, MA).
In Vitro Probe Recovery. Probe recovery was determined by placing the probe in an isotonic phosphate-buffered saline (IPBS) solution (pH 7.4) at 37°C, containing a known concentration of quinidine (equivalent to an intravitreal dose of 0.75 μg) alone or in the presence of verapamil or PHS. The probe was perfused with sterile IPBS (with or without verapamil) at a flow rate of 2 μl/min, and the dialysate was collected every 20 min. Relative recovery was calculated using eq. 1:
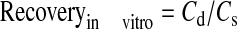 |
(1) |
where Cd is the dialysate quinidine concentration and Cs is the quinidine concentration in IPBS. The concentration of quinidine in the vitreous humor samples was calculated by dividing the dialysate concentration by the in vitro recovery factor obtained as described above.
Probe Implantation. Rabbits (weighing 2–2.5 kg) were anesthetized using ketamine (35 mg/kg)/xylazine (3.5 mg/kg) administered intramuscularly and were maintained under anesthesia throughout the duration of the experiment (ketamine/xylazine administered intramuscularly every 40 min). Before probe implantation, 1% tropicamide was applied topically to dilate the pupil. A 22-guage needle was then inserted into the posterior chamber of the eye. The point of insertion was approximately 3 mm below the corneal-scleral limbus. The needle was withdrawn, and the vitreal probe was implanted immediately. The position of the probe was adjusted so that the semipermeable membrane was in the mid-vitreous section. The probes were continuously perfused with sterile IPBS (pH 7.4) at a flow rate of 2 μl/min using a CMA/100 microinjection pump (CMA/Microdialysis Inc.). After probe implantation, animals were allowed to stabilize for a period of 2 h before drug administration. Vitreal samples were collected every 20 min for a period of 9 h. Samples were collected in microcentrifuge tubes and stored at –20°C until further analysis. At the end of the study, animals were euthanized, under deep anesthesia, with an overdose of sodium pentobarbital administered through the marginal ear vein.
Drug Administration. Quinidine was administered intravitreally (0.75-μg dose in 50 μl of IPBS). Studies were performed with quinidine administered alone (control) or in the presence of topically coadministered erythromycin (0.2% w/v, pH 7.4), verapamil (0.5% w/v and 1% w/v, pH 6.0), and PHS (1% w/v and 2% w/v, pH 7.4). One hundred microliters of the inhibitor solution was instilled in the conjunctival sac. In the preliminary studies, erythromycin (0.2% w/v) was applied topically at 0, 2, and 4 h after intravitreal quinidine injection. Subsequently, the topical P-gp substrate/inhibitor administration time was modified to 2, 4, and 6 h after quinidine administration to prolong the residence of the topically applied agent in the RPE tissue. Further studies with verapamil and PHS were carried out with topical instillation at 2, 4, and 6 h after intravitreal administration. All solutions were prepared in sterile IPBS.
Vitreal pharmacokinetics of quinidine (0.75 μg) was also studied in the presence of intravitreally administered verapamil (100 μg). In this study, verapamil was coadministered intravitreally with quinidine (injection volume 50 μl; codissolved) after the probe stabilization period. In addition, IPBS (pH 7.4) containing verapamil (1 mg/ml) was continuously perfused through the concentric probes to maintain high verapamil levels in the vitreous humor throughout the duration of the experiment.
Fluorescein Kinetics. Vitreal kinetics of intravitreally administered fluorescein (dose 10 μg, injection volume 50 μl), alone or in the presence of topically coadministered verapamil (1% w/v, IPBS, pH 6.0), was studied to ensure preservation of the barrier properties of the RPE in the presence of topical verapamil (1% w/v). In these experiments, 100 μl of 1% w/v verapamil solution was applied at 2, 4, and 6 h after intravitreal fluorescein administration.
Distribution of PHS and Verapamil in Ocular Tissues After Topical or Intravitreal Application. In a separate set of studies, verapamil (1% w/v, pH 6.0) or PHS (2% w/v, pH 7.4) was applied topically in the cul-de-sac of the rabbit's eye at 2, 4, and 6 h after probe stabilization. At the end of 7 h (for the verapamil studies) and 9 h (for the PHS studies), rabbits were euthanized, eyes were enucleated, and ocular tissues were collected and analyzed for drug content using a high-performance liquid chromatography (HPLC) system. In addition, ocular tissue concentrations of verapamil at the end of 7 h after intravitreal administration, as described earlier, were also determined.
Bioreversion of PHS to Prednisolone. PHS, a hemisuccinate ester prodrug of prednisolone (Augustijns et al., 1998), requires hydrolysis (chemical or enzymatic) of the ester bond to generate free prednisolone. The presence of esterase activity in rabbit ocular tissues has been demonstrated and well documented with ester prodrugs of pilocarpine, dipivefrin, ganciclovir, and acyclovir (Tsuji et al., 1987; Majumdar et al., 2006, 2009). Bioreversion of PHS was studied in vitreous humor and in the ocular tissues such as the cornea, iris-ciliary body, and RPE/choroid tissue as described previously (Majumdar et al., 2009). Vitreous humor was centrifuged, and the supernatant was used. All of the other ocular tissues were homogenized in 5 ml of chilled IPBS with a tissue homogenizer (Tissuemiser; Thermo Fisher Scientific) for periods of 30 s, with 1-min intervals, in an ice bath. The homogenates were centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatants were used for the PHS enzymatic hydrolysis studies. Protein content in the supernatant was measured using the Bradford Protein estimation kit (Sigma-Aldrich), and the final protein content was adjusted to 1 mg/ml with IPBS.
Hydrolysis studies were carried out in triplicate at 37°C in a shaking water bath (75 reciprocations/min). One hundred microliters of PHS stock solution was added to the required volume of the tissue homogenates and to vitreous humor to obtain a final PHS concentration of 10 μg/ml. At predetermined time points, 100-μl samples were withdrawn, and an equal volume of ice-cold methanol was immediately added to the sample to arrest the enzymatic degradation process. The stability of PHS (10 μg/ml) in IPBS was also studied as a control.
Analytical Procedures. Sample preparation. For studies involving distribution of PHS and verapamil in ocular tissues after topical or intravitreal application, enucleated eyes were rinsed with ice-cold IPBS to remove any traces of blood and blotted dry using Kimwipes. Aqueous and vitreous humor samples were collected using a 27-gauge needle attached to a 1-ml tuberculin syringe. Eyes were then dissected and iris-ciliary bodies, lenses, and RPE/choroid tissues were isolated and weighed. Tissues were homogenized in ice-cold IPBS using a Tissuemiser. Homogenates were diluted with an equal volume of ice-cold acetonitrile-methanol (50:50) mixture, and centrifuged at 13,000 rpm for 20 min at 4°C. The supernatant was analyzed for drug content. Aqueous and vitreous humor samples were used as such or diluted with IPBS and taken for analysis. Extraction efficiency of PHS and verapamil from the ocular tissues was almost 100%.
Chromatography. Quinidine and verapamil were analyzed using an HPLC system comprising a Waters 717 Plus autosampler, Waters 2475 multi λ fluorescence detector, Waters 600 controller pump, and Agilent 3395 integrator. A Symmetry C18 (4.6 × 250 mm) column was used, and the flow rate was set at 1 ml/min for both the compounds. Quinidine analysis was performed using 20 mM phosphate buffer (pH 2.5) with 20% acetonitrile as the mobile phase, at an excitation wavelength of 250 nm and emission wavelength of 440 nm. Verapamil quantification was performed at an excitation wavelength of 280 nm and emission wavelength of 320 nm using acetonitrile and 0.07% v/v o-phosphoric acid in deionized water (33:67) as the mobile phase. Fluorescein and prednisolone analyses were performed using reversed-phase HPLC procedures as described previously (Macha and Mitra, 2001; Cho, 2003).
Data analysis. Vitreal pharmacokinetic parameters of quinidine were determined by noncompartmental analysis using WinNonlin (version 5.2; Pharsight, Mountain View, CA). Terminal slopes of the vitreous concentration-time profile were estimated by log-linear regression, and the apparent elimination rate constant (λz) was derived from the slope. Elimination half-life (t1/2) was calculated from the equation t1/2 = 0.693/λz. The area under the vitreal concentration-time curve from time 0 to time t and from time 300 to time 540 was calculated by the linear trapezoidal method and extrapolated to infinity according to eq. 2:
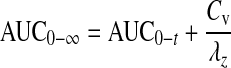 |
(2) |
The area under the statistical moment curve (AUMC0–∞) was calculated using eq. 3:
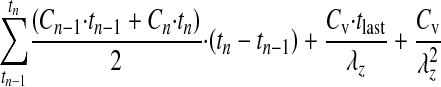 |
(3) |
where Cv is the quinidine concentration at the last time point (540 min). The mean retention time (MRT) was calculated using the equation: MRT = AUMC0–∞/AUC0–∞. The total clearance was calculated as CL = Dose/AUC0–∞. CL300–540 = Dose/AUC300–540. The apparent volume of distribution at steady state, Vss = (Dose × AUMC0–∞)/(AUC0–∞)2. The time course of fluorescein after a single intravitreal bolus dose was described by a biexponential profile (Macha and Mitra, 2001a) as expressed in eq. 4:
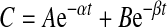 |
(4) |
in which A and B are zero-time concentration coefficients, t is the time (minutes), and α and β are the disposition rate constants of the initial and terminal phases, respectively. Vitreous humor was considered as part of the apparent central compartment and all other exchanging compartments including the anterior chamber were considered as part of the apparent peripheral compartment. Elimination was assumed to take place through plasma from the apparent central compartment. This model is illustrated as follows:
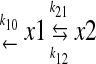 |
Quinidine is administered into the vitreous humor (X1), and the variables X1 and X2 represent the amount of the drug in the vitreous humor and other exchanging compartments, respectively. A similar model has been used to describe the kinetics of intravitreally administered fluorescein and other drugs (Macha and Mitra, 2001a,b, 2002). In the open two-compartment model represented by eq. 4, the rate constant of drug transfer from the apparent peripheral to the apparent central compartment (k21) was calculated according to eq. 5:
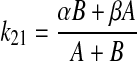 |
(5) |
The elimination rate constant of fluorescein from the apparent central compartment (k10) was determined using eq. 6:
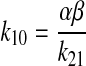 |
(6) |
The rate constant of fluorescein transfer from the apparent central compartment to the apparent peripheral compartment (k12) was calculated using eq. 7:
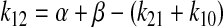 |
(7) |
Data obtained were subjected to statistical analysis using Student's t test. p ≤ 0.05 was considered to denote a statistically significant difference.
Results
Effect of Topically Applied Erythromycin on Vitreal Kinetics of Intravitreally Administered Quinidine. Vitreal kinetics of intravitreally administered quinidine (0.75-μg dose, 50-μl injection volume) was studied either alone or in the presence of topically coadministered erythromycin (100 μl of a 0.2% w/v solution in IPBS, pH 7.4). The erythromycin solution was administered either at 0, 2, and 4 h or at 2, 4, and 6 h after intravitreal quinidine administration. When erythromycin was applied at 0, 2, and 4 h, vitreal pharmacokinetic parameters, i.e., t1/2 (104 ± 7 min), CL (0.0086 ± 0.0030 ml/min), and λz (0.0066 ± 0.0004 min–1), were not significantly different from the control values (105 ± 11 min, 0.0048 ± 0.0012 ml/min, and 0.0066 ± 0.0007 min–1, respectively). A change in the topical erythromycin dosing times to 2, 4, and 6 h after intravitreal quinidine administration also did not produce a significant difference in the pharmacokinetic parameters. Higher doses of erythromycin, in a solution form, could not be administered because of the limited aqueous solubility (2 mg/ml) of erythromycin.
Effect of Topically Coadministered Verapamil on Intravitreal Kinetics of Quinidine. The effect of topically coadministered verapamil (100 μl applied at 2, 4, and 6 h) on the intravitreal kinetics of quinidine (0.75 μg) was examined at two different verapamil concentrations (0.5% and 1% w/v). Verapamil had limited solubility at pH 7.4 (1 mg/ml) (Duvvuri et al., 2003a). Thus, verapamil solutions used in this study were prepared in IPBS (pH 6.0 ± 0.1). The concentration-time profiles of quinidine after intravitreal administration, alone or in the presence of topically coadministered verapamil (0.5% and 1% w/v), are illustrated in Fig. 1, A and B. The vitreal pharmacokinetic parameters are provided in Table 1. At a concentration of 0.5% w/v, verapamil did not produce any significant change in the vitreal kinetics of quinidine. However, at 1% w/v, topical verapamil produced a 1.7-fold decrease inλz (from 0.0066 ± 0.0007 to 0.0039 ± 0.0001 min–1), a 1.7-fold increase in t1/2, and a 1.7-fold increase in the MRT. Statistically significant differences between the mean vitreal quinidine concentrations of the 1% verapamil-treated group and the control group were observed from the 360-min time point onward (Fig. 1B). Significant differences in AUC0–∞ and CL from those of the control were not observed. However, when partial areas were taken into account, a 1.7-fold increase in AUC300–540 and a 1.7-fold decrease in CL300–540 were observed.
Fig. 1.

A, vitreal concentration-time profile of quinidine (0.75 μg) alone (control) or in the presence of topically coadministered verapamil 0.5% w/v (100 μl administered at 2, 4, and 6 h after quinidine administration). Data points represent mean ± S.D. of four determinations. B, vitreal concentration-time profile of quinidine (0.75 μg) alone (control) or in the presence of topically coadministered verapamil 1% w/v (100 μl administered at 2, 4, and 6 h after quinidine administration). Data points represent mean ± S.D. of four determinations.
TABLE 1.
Vitreal pharmacokinetic parameters of intravitreally administered quinidine (0.75-μg dose) alone or in the presence of topically coadministered verapamil (0.5% w/v or 1% w/v)
Verapamil was administered at 2, 4, and 6 h after intravitreal quinidine administration. Values represent the mean ± S.D. (n = 4).
| Kinetic Parameters | Quinidine | Quinidine + Verapamil (0.5% w/v) | Quinidine + Verapamil (1% w/v) |
|---|---|---|---|
| λz (min-1) | 0.0066 ± 0.0007 | 0.0066 ± 0.0010 | 0.0039 ± 0.0001*** |
| AUC0-∞ (μg × min/ml) | 168 ± 56 | 180 ± 41 | 158 ± 37 |
| CL (ml/min) | 0.0048 ± 0.0012 | 0.0043 ± 0.0008 | 0.0049 ± 0.0012 |
| Vss (ml) | 0.41 ± 0.14 | 0.37 ± 0.12 | 0.72 ± 0.30 |
| MRT∞ (min) | 83 ± 13 | 84 ± 13 | 143 ± 30** |
| AUC300-540 (μg × min/ml) | 7.73 ± 1.32 | 9.05 ± 0.38 | 12.86 ± 1.25* |
| CL300-540 (ml/min) | 0.099 ± 0.017 | 0.080 ± 0.004 | 0.060 ± 0.013* |
P < 0.001.
P < 0.01.
P < 0.05.
Vitreal Kinetics of Quinidine in the Presence of Intravitreally Administered Verapamil. The effect of intravitreal coadministration of verapamil (100 μg) on the vitreal kinetics of quinidine was also examined. In these studies the microdialysis probe perfusion solution contained verapamil (1 mg/ml) to maintain a significantly higher verapamil/quinidine ratio in the vitreous humor. Figure 2 represents the vitreous concentration-time profile of quinidine in the presence of intravitreally coadministered verapamil. A 1.6-fold increase in the MRT (from 83 ± 13 to 131 ± 16 min), a 1.5-fold decrease in λz (from 0.0066 ± 0.0007 to 0.0043 ± 0.0002 min–1), and a corresponding 1.5-fold increase in t1/2 (from 105 ± 11 to 159 ± 9 min) of quinidine were observed in the presence of intravitreally coadministered verapamil (Table 2). Statistically significant differences between the mean vitreal quinidine concentrations of the treated and control groups were observed from the 100-min time point onward (Fig. 2).
Fig. 2.

Vitreal concentration-time profile of quinidine (0.75 μg) alone (control) or in the presence of intravitreally coadministered verapamil (100 μg, administered along with quinidine). Data points represent mean ± S.D. of four determinations.
TABLE 2.
Vitreal kinetic parameters of quinidine (0.75-μg dose) alone or in the presence of intravitreally coadministered verapamil (100 μg)
Verapamil solution (1 mg/ml in IPBS) was used as the perfusate. Values represent the mean ± S.D. (n = 4).
| Kinetic Parameters | Quinidine | Quinidine + Intravitreal Verapamil |
|---|---|---|
| λz (min-1) | 0.0066 ± 0.0007 | 0.0043 ± 0.0002*** |
| AUC0-∞ (μg × min/ml) | 168 ± 56 | 180 ± 72 |
| CL (ml/min) | 0.0048 ± 0.0012 | 0.0046 ± 0.0014 |
| Vss (ml) | 0.41 ± 0.14 | 0.63 ± 0.27 |
| MRT∞ (min) | 83 ± 13 | 131 ± 16** |
| AUC100-540 (μg × min/ml) | 40 ± 0.03 | 56 ± 11* |
| AUC300-540 (μg × min/ml) | 7.73 ± 1.32 | 13.31 ± 2.28** |
| CL300-540 (ml/min) | 0.099 ± 0.017 | 0.057 ± 0.009** |
P < 0.001.
P < 0.01.
P < 0.05.
Effect of Topical PHS on Vitreal Kinetics of Quinidine. Prednisolone is practically insoluble in water (Karssen et al., 2002), and therefore its water-soluble derivative PHS was used in this study. The effect of topically administered PHS (1% and 2% w/v) on the vitreal kinetics of intravitreally administered quinidine was studied (Table 3; Fig. 3, A and B). One hundred microliters of a 1 or 2% w/v PHS solution was instilled at 2, 4, and 6 h after quinidine administration. Topical coadministration of 1% w/v PHS resulted in a 1.4-fold increase in the t1/2 and a 1.4-fold decrease in the λz of quinidine. However, statistically significant changes in AUC300–540 and CL300–540 were not observed at this dose. Coadministration of 2% w/v PHS produced a 2.0-fold increase in the t1/2 and a 2-fold decrease in the λz of quinidine. A 1.4-fold increase in CL300–540 and AUC300–540 was also observed. Moreover, a 1.6-fold increase in MRT of quinidine was observed compared with that of the control. Statistically significant differences between the mean vitreal quinidine concentrations of the 2% PHS-treated group and the control group were noted from the 360-min time point onward (Fig. 3B).
TABLE 3.
Vitreal pharmacokinetic parameters of quinidine (0.75-μg dose) after intravitreal administration in the presence and absence of topically coadministered PHS
Values represent the mean ± S.D. (n = 4).
| Kinetic Parameters | Quinidine | Quinidine + 1% PHS (w/v) | Quinidine + 2% PHS (w/v) |
|---|---|---|---|
| λz (min-1) | 0.0066 ± 0.0007 | 0.0048 ± 0.0004** | 0.0033 ± 0.0004***,† |
| AUC0-∞ (μg × min/ml) | 168 ± 56 | 191 ± 42 | 151 ± 21.6 |
| CL (ml/min) | 0.0048 ± 0.0012 | 0.0040 ± 0.0009 | 0.0050 ± 0.0007 |
| Vss (ml) | 0.41 ± 0.14 | 0.37 ± 0.14 | 0.65 ± 0.20 |
| MRT∞ (min) | 83 ± 13 | 87 ± 16 | 129 ± 21**,† |
| AUC300-540 (μg × min/ml) | 7.73 ± 1.32 | 8.70 ± 0.79 | 11.76 ± 0.40*,† |
| CL300-540 (ml/min) | 0.099 ± 0.017 | 0.087 ± 0.008 | 0.063 ± 0.002*,† |
Statistically significant difference between quinidine (control) and treated groups (Quinidine + 2% PHS and Quinidine + 1% PHS)
P < 0.05
P < 0.01
P < 0.001.
Statistical significant difference between Quinidine + 2% PHS and Quinidine + 1% PHS: P < 0.05.
Fig. 3.

A, vitreal concentration-time profile of quinidine (0.75 μg) alone (control) or in the presence of topically coadministered prednisolone hemisuccinate sodium (1% w/v, 100 μl administered at 2, 4, and 6 h after quinidine administration). Data points represent mean ± S.D. of four determinations. B, vitreal concentration-time profile of quinidine (0.75 μg) alone (control) or in the presence of topically coadministered prednisolone hemisuccinate sodium (2% w/v, 100 μl administered at 2, 4, and 6 h after quinidine administration). Data points represent mean ± S.D. of four determinations.
Ocular Tissue Distribution of Verapamil and PHS. Table 4 presents ocular tissue concentrations of verapamil (after topical and intravitreal administration) and PHS (after topical administration). Topical and intravitreal routes of administration generated similar verapamil concentrations in the RPE/choroid tissue at the end of 7 h. The 7-h time point was selected for verapamil because the effect of topical verapamil on the vitreal quinidine kinetics became evident at approximately this time (significant change in the mean vitreal concentrations between the treated and control groups) in the pharmacokinetic profile (Fig. 1A). Very low/insignificant verapamil concentrations were observed in the vitreous humor after topical verapamil administration, indicating that the 2-h stabilization period was sufficient to seal the scleral port created during probe implantation. PHS tissue concentrations were evaluated at the end of the experiment (9 h). It is interesting to note that after topical administration, the fraction of PHS sodium appearing in the vitreous humor (concentration in the vitreous humor as a percentage of the topically administered dose) was significantly higher than that observed with verapamil.
TABLE 4.
Ocular distribution of verapamil and PHS
Verapamil tissue concentrations were determined 7 h after topical (100 μl of a 1% w/v solution applied 2, 4, and 6 h after intravitreal quinidine administration) and intravitreal administration (dose: 100 μg). Ocular distribution of topically applied PHS (100 μl of a 2% w/v solution applied at 2, 4, and 6 h after intravitreal quinidine administration) was determined 9 h after intravitreal quinidine administration, and the values are reported for both intact PHS and free prednisolone concentrations observed. Values represent the mean ± S.D. (n = 4).
|
Tissues
|
Verapamil Concentration
|
PHS Topical Application (2% w/v)
|
||
|---|---|---|---|---|
| Topical Application | Intravitreal Administration | Intact PHS Concentration | Concentration of Free Prednisolonea | |
| Aqueous humor (μg/ml) | 7.9 ± 3.2 | 1.5 ± 0.3 | 10.2 ± 1.1 | 5.0 ± 1.4 |
| Iris-ciliary body (μg/g) | 4.4 ± 1.6 | 27.0 ± 10.0 | 7.0 ± 1.5 | 13.3 ± 4.3 |
| Lens (μg/g) | 6.3 ± 1.2 | 45.1 ± 4.2 | ||
| Vitreous humor (μg/ml) | 0.086 ± 0.003 | 31.5 ± 1.4 | 0.64 ± 0.21 | 0.49 ± 0.29 |
| Retina-choroid (μg/g) | 47.0 ± 8.1 | 52.0 ± 12.2 | 8.8 ± 1.2 | 16.7 ± 1.6 |
Generated as a result of bioreversion of PHS in the ocular tissues.
Bioreversion of PHS. Table 5 depicts the apparent pseudo-first-order degradation rate constants and half-lives of PHS in ocular tissue homogenates (1 mg/ml protein content), including vitreous humor. Tissue homogenates were prepared in IPBS (pH 7.4). PHS was hydrolyzed to the parent drug, prednisolone, suggesting the role of esterases in the bioreversion of PHS. Degradation rate constants were obtained from log concentration of PHS remaining versus time plots. Hydrolysis rate constants obtained from the control (PHS in IPBS) were subtracted from the overall observed rate constants to estimate rate constants for the enzyme-mediated hydrolytic process.
TABLE 5.
Apparent first-order degradation rate constants and half-lives of PHS in ocular tissue homogenates (1 mg/ml protein content)
Values represent the mean ± S.D. (n = 4).
| Drug/Kinetic Parameters for PHS | Control | Cornea | Vitreous Humor | Iris-Ciliary Body | RPE/Choroid Tissue |
|---|---|---|---|---|---|
| k (×103 min-1 | 0.33 ± 0.05 | 0.81 ± 0.07 | 3.06 ± 0.34 | 2.00 ± 0.06 | 1.20 ± 0.05 |
| t1/2 (min) | 2118 ± 333 | 852 ± 74 | 228 ± 28 | 347 ± 10 | 578 ± 27 |
Intravitreal Kinetics of Fluorescein in the Presence of Topically Applied Verapamil. Figure 4 illustrates the vitreous concentration-time profile of fluorescein, alone or in the presence of topically coadministered verapamil. Vitreal fluorescein concentration-time data could be best fitted to a two-compartment open model. One hundred microliters of verapamil (1% w/v, pH 6.0) was applied topically at 2, 4, and 6 h after fluorescein administration to evaluate the effect of topical verapamil administration on the barrier properties of the RPE. The vitreal pharmacokinetic parameters of fluorescein, such as elimination half-life, CL, AUC, steady-state volume of distribution, and apparent elimination rate constant, remained unchanged in the presence of topically coadministered verapamil (Table 6).
Fig. 4.

Vitreal concentration-time profile of fluorescein (10.0 μg) alone or in the presence of topically coadministered verapamil (1% w/v, 100 μl administered at 2, 4, and 6 h after intravitreal fluorescein administration). Data points represent mean ± S.D. of four determinations.
TABLE 6.
Vitreal kinetics of intravitreally administered fluorescein (10 μg)
Values represent the mean ± S.D. (n = 4).
| Kinetic Parameters | Fluorescein |
|---|---|
| k10 (min-1) | 0.021 ± 0.01 |
| k12 (min-1) | 0.02 ± 0.01 |
| k21 (min-1) | 0.0135 ± 0.004 |
| AUC (μg × min/ml) | 3272 ± 632 |
| K10t1/2 (min) | 38.6 ± 20.3 |
| CL (ml/min) | 0.003 ± 0.0005 |
| MRT∞ (min) | 124 ± 49 |
| Vss (ml) | 0.37 ± 0.1 |
| β (min-1) | 0.0049 ± 0.0009 |
| β t1/2 (min) | 122 ± 39 |
Discussion
The goal of this novel study was to evaluate whether a drug-drug interaction could occur between intravitreally and topically coadministered P-gp substrates and to determine its effect on the ocular pharmacokinetics of the intravitreally administered compound.
Duvvuri et al. (2003a) demonstrated that intravitreal coadministration of verapamil and quinidine resulted in increased vitreal elimination of quinidine. Moreover, when quinidine was administered systemically and verapamil was coadministered intravitreally, the vitreal AUC of quinidine increased significantly. In another in vivo study, Senthilkumari et al. (2008a) reported a significant increase in the ocular tissue concentrations of intravitreally administered rhodamine-123 in the presence of a P-gp inhibitor applied intravenously. The authors hypothesized that the increased vitreal rhodamine-123 concentrations were probably the result of inhibition of efflux mediated by P-gp expressed on the ocular tissues. In a subsequent study, the investigators studied systemic coadministration of both compounds (rhodamine-123 and the inhibitor) but did not observe an increase in vitreal rhodamine-123 concentrations, probably because of inadequate inhibitor concentrations at the target site (Senthilkumari et al., 2008b).
The above three studies, which to our knowledge are the only published reports investigating in vivo RPE P-gp-mediated efflux, evaluated the effect of either intravitreal or systemically coadministered inhibitors on vitreal kinetics of P-gp substrates. From a therapeutic point of view, with respect to delivery of P-gp substrates to the posterior chamber ocular tissues, the use of an intravitreal inhibitor is not feasible considering that high intravitreal levels of the inhibitor can only be maintained through multiple intravitreal injections. On the other hand, the use of systemic inhibitors is not attractive because of nonspecific systemic exposure to the inhibitor and the limited, clinically relevant, inhibitor dose that can be administered.
A hitherto uninvestigated and novel alternative approach that could be therapeutically effective and minimize systemic exposure is modulation of efflux mediated by P-gp expressed on the RPE through topical substrate/inhibitor application. As discussed earlier, several therapeutic agents that are P-gp substrates are currently administered topically. The literature also suggests that a fraction of topically administered agents may reach the RPE (Salminen and Urtti, 1984; Oztürk et al., 1999, 2000; Acheampong et al., 2002; Tan et al., 2002). Taking both factors into consideration, we undertook this study to evaluate the feasibility of modulating ocular kinetics of intravitreally administered substrates through local application of P-gp substrates/inhibitors.
Erythromycin, at a concentration of 0.2% w/v, applied at 0, 2, and 4 h or at 2, 4, and 6 h after intravitreal quinidine administration, did not significantly affect the pharmacokinetic parameters of quinidine. Higher doses of erythromycin were not tested because of limited aqueous solubility. Topical verapamil, at a concentration of 0.5% w/v, also did not produce any significant change in the vitreal quinidine kinetics. The inability of 0.5% w/v verapamil and 0.2% w/v erythromycin to affect the pharmacokinetic parameters of quinidine could be caused by insufficient inhibitor concentrations at the RPE at these doses. Coadministration of topical 1% w/v verapamil resulted in a significant decrease in the apparent elimination rate constant and an increase in the vitreal half-life and mean retention time of quinidine in the posterior chamber (Table 1). Significant differences in AUC0–∞ and CL from those of the control were not observed, possibly because the inhibitory effect of the topically administered agents becomes significant only when vitreal quinidine concentrations are reduced to the low levels observed at the midpoint of the study or because adequate inhibitor concentrations are achieved at the RPE at that point. Consistent with the findings with verapamil and erythromycin, PHS demonstrated a dose-dependent effect on the vitreal pharmacokinetic parameters of quinidine (Table 3). At a concentration of 2% w/v, PHS produced a much more significant change than 1% w/v, probably because of higher concentrations of prednisolone generated at the target site, the RPE.
In the studies involving topical application of 1% verapamil (Fig. 1B) and 2% PHS (Fig. 2B), statistically significant differences between the mean vitreal quinidine concentrations of the control and the treated groups were observed from the 360-min time point. With 1% PHS a statistically significant difference between the means of the control and treated groups was observed only after 460 min. When partial areas were taken into account, significant differences in AUC300–540 and CL300–540 were observed in the 1% w/v verapamil- and 2% w/v PHS-treated groups from those of the control, demonstrating an interaction of topically administered P-gp substrates with RPE P-gp. These results illustrate the importance of the permeability kinetics of the topically applied agents.
Fluorescein has been used as a marker compound to monitor the integrity and tightness of the blood-retinal barriers. The vitreal pharmacokinetic parameters of fluorescein were not affected by topical coadministration of 1% w/v verapamil (Table 6), and the values obtained in this study were consistent with values published previously (Macha and Mitra, 2001a). The results thus strongly suggest that the observed effect of topical verapamil on the vitreal kinetics of quinidine is a result of verapamil interacting with RPE P-gp.
Because prednisolone rather than PHS is known to interact with P-gp, bioreversion of PHS to prednisolone is necessary. In vitro metabolism studies confirmed bioreversion of PHS, to generate free prednisolone, in the ocular tissues (Table 5). At higher topical doses of PHS, greater quantities of PHS would be reaching the RPE, and thus greater concentrations of prednisolone would be generated. It is interesting to note that the ocular tissue distribution studies, after topical administration of verapamil and PHS, revealed that significantly higher fractions of the topically administered PHS dose reached the vitreous humor (almost 5-fold higher) compared with those for verapamil. Cheruvu and Kompella (2006) indicated that compounds with high logP values demonstrate lower trans-scleral permeability, possibly because of an interaction with the proteins expressed on Bruch's membrane. The higher vitreal PHS concentrations observed could be a result of the greater aqueous solubility of PHS at physiological pH ranges, favoring greater diffusion across Bruch's membrane and/or changes in the binding affinity with Bruch's membrane proteins. The ocular tissue concentration data (Table 4) also suggest that verapamil and PHS migrate laterally along the cornea/aqueous humor route and possibly also across the conjunctiva into the sclera. The results further demonstrate that after topical administration, sufficient verapamil and PHS concentrations can accumulate in the RPE/choroid tissue and inhibit P-gp on the basolateral membrane of RPE. In fact, the concentration of verapamil in the RPE/choroid tissue 7 h after topical verapamil instillation was similar to that obtained after intravitreal administration. However, insignificant vitreal concentrations suggest that very little verapamil could traverse across the RPE into the neural retina.
The elimination rate of quinidine from the vitreous humor was observed to decrease in the presence of intravitreally administered verapamil (Table 2), suggesting functional involvement of P-gp expressed on the basolateral membrane of the RPE or apical membrane of the retinal endothelial cells. These results are contrary to those of Duvvuri et al. (2003a), who observed that quinidine elimination increased in the presence of intravitreal verapamil. The authors had suggested that P-gp expressed on the neural retina, facing the vitreous, probably influences elimination of P-gp substrates from the vitreous humor. However, there are no other reports corroborating P-gp expression on the neural retina. Besides P-gp expression on the apical membrane of the retinal endothelial cells, most in vitro and ex vivo studies suggested that functional activity of P-gp is localized on the basolateral membrane of the RPE. A study by Steuer et al. (2005) demonstrated higher permeability of verapamil (2.6-fold) and rhodamine (3.5-fold) across isolated RPE in the neural retina to choroid direction compared with the choroid to neural retina direction, demonstrating the significance of P-gp localized on the basolateral membrane (choroidal side) of the RPE. The results by Senthilkumari et al. (2008a) also suggested functional expression of P-gp on the basolateral membrane of the RPE. The biochemical and functional study performed by Kennedy and Mangini (2002) is the only study, to our knowledge, suggesting expression of P-gp on the apical membrane of the RPE. However, the authors postulated that P-gp localized on the apical RPE probably serves additional purposes, such as modulation of volume-sensitive chloride efflux or functioning as a lipid translocase. However, because the intravitreal verapamil dose administered was not mentioned by the authors (Duvvuri et al., 2003a), there is a possibility that the verapamil dose administered in the earlier study was sufficient to inhibit P-gp on the neural retina but not to interact with P-gp expressed on the basolateral membrane of the RPE.
Taken together, the above results demonstrate that topically administered P-gp substrates can migrate along the corneal-scleral pathway and possibly across the conjunctiva into the sclera and significantly alter elimination profiles of intravitreally administered P-gp substrates. This interaction can be used to modulate drug elimination from the posterior chamber of the eye. The affect of this strategy might be significantly more marked with topical and systemic coadministration (currently under investigation), considering the low plasma concentrations of the P-gp substrates generated compared with the vitreous humor concentrations obtained after intravitreal administration. This technique may also help modulate efflux activity of other transporters, such as the multidrug-resistant proteins, expressed on the RPE. Further investigation of this novel approach is warranted.
Acknowledgments
We appreciate the support and technical help extended by the animal facility staff, in particular Dr. Harry Fyke (university veterinarian) and Penni Bolton (animal care supervisor). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This work was supported by the National Institutes of Health National Eye Institute [Grant EY018426-02].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.108.026450.
ABBREVIATIONS: RPE, retinal pigmented epithelium; P-gp, P-glycoprotein; PHS, prednisolone hemisuccinate sodium; IPBS, isotonic phosphate buffer saline; HPLC, high-performance liquid chromatography; AUMC, area under the statistical moment curve; MRT, mean retention time; AUC, area under the curve.
References
- Acheampong AA, Shackleton M, John B, Burke J, Wheeler L, and Tang-Liu D (2002) Distribution of brimonidine into anterior and posterior tissues of monkey, rabbit, and rat eyes. Drug Metab Dispos 30 421–429. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, and Gottesman MM (2003) P-glycoprotein: from genomics to mechanism. Oncogene 22 7468–7485. [DOI] [PubMed] [Google Scholar]
- Augustijns P, Annaert P, Heylen P, Van den Mooter G, and Kinget R (1998) Drug absorption studies of prodrug esters using the Caco-2 model: evaluation of ester hydrolysis and transepithelial transport. Int J Pharm 166 45–53. [Google Scholar]
- Cheruvu NP and Kompella UB (2006) Bovine and porcine transscleral solute transport: influence of lipophilicity and the Choroid-Bruch's layer. Invest Ophthalmol Vis Sci 47 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CY, Shin BS, and Yoo SD (2003) Sensitive analysis of prednisolone and prednisone in human plasma by reverse phase high-performance liquid chromatography with ultraviolet detection. Analytical Letters 36 1573–1585. [Google Scholar]
- Cunha-Vaz JG (2004) The blood-retinal barriers system: basic concepts and clinical evaluation. Exp Eye Res 78 715–721. [DOI] [PubMed] [Google Scholar]
- Dey S, Anand BS, Patel J, and Mitra AK (2003) Transporters/receptors in the anterior chamber: pathways to explore ocular drug delivery strategies. Expert Opin Biol Ther 3 23–44. [DOI] [PubMed] [Google Scholar]
- Duvvuri S, Gandhi MD, and Mitra AK (2003a) Effect of P-glycoprotein on the ocular disposition of a model substrate, quinidine. Curr Eye Res 27 345–353. [DOI] [PubMed] [Google Scholar]
- Duvvuri S, Majumdar S, and Mitra AK (2003b) Drug delivery to the retina: challenges and opportunities. Expert Opin Biol Ther 3 45–56. [DOI] [PubMed] [Google Scholar]
- Fromm MF (2003) Importance of P-glycoprotein for drug disposition in humans. Eur J Clin Invest 33 (Suppl 2): 6–9. [DOI] [PubMed] [Google Scholar]
- Fromm MF (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25 423–429. [DOI] [PubMed] [Google Scholar]
- Ghate D and Edelhauser HF (2006) Ocular drug delivery. Expert Opin Drug Deliv 3 275–287. [DOI] [PubMed] [Google Scholar]
- Golden PL and Pollack GM (2003) Blood-brain barrier efflux transport. J Pharm Sci 92 1739–1753. [DOI] [PubMed] [Google Scholar]
- Jain R, Majumdar S, Nashed Y, Pal D, and Mitra AK (2004) Circumventing P-glycoproteinmediated cellular efflux of quinidine by prodrug derivatization. Mol Pharm 1 290–299. [DOI] [PubMed] [Google Scholar]
- Janoria KG, Gunda S, Boddu SH, and Mitra AK (2007) Novel approaches to retinal drug delivery. Expert Opin Drug Deliv 4 371–388. [DOI] [PubMed] [Google Scholar]
- Karssen AM, Meijer OC, van der Sandt IC, De Boer AG, De Lange EC, and De Kloet ER (2002) The role of the efflux transporter P-glycoprotein in brain penetration of prednisolone. J Endocrinol 175 251–260. [DOI] [PubMed] [Google Scholar]
- Katragadda S, Budda B, Anand BS, and Mitra AK (2005) Role of efflux pumps and metabolising enzymes in drug delivery. Expert Opin Drug Deliv 2 683–705. [DOI] [PubMed] [Google Scholar]
- Kemper EM, Boogerd W, Thuis I, Beijnen JH, and van Tellingen O (2004) Modulation of the blood-brain barrier in oncology: therapeutic opportunities for the treatment of brain tumours? Cancer Treat Rev 30 415–423. [DOI] [PubMed] [Google Scholar]
- Kennedy BG and Mangini NJ (2002) P-glycoprotein expression in human retinal pigment epithelium. Mol Vis 8 422–430. [PubMed] [Google Scholar]
- Kim SH, Lutz RJ, Wang NS, and Robinson MR (2007) Transport barriers in transscleral drug delivery for retinal diseases. Ophthalmic Res 39 244–254. [DOI] [PubMed] [Google Scholar]
- Kunta JR and Sinko PJ (2004) Intestinal drug transporters: in vivo function and clinical importance. Curr Drug Metab 5 109–124. [DOI] [PubMed] [Google Scholar]
- Macha S and Mitra AK (2001a) Ocular pharmacokinetics in rabbits using a novel dual probe microdialysis technique. Exp Eye Res 72 289–299. [DOI] [PubMed] [Google Scholar]
- Macha S and Mitra AK (2001b) Ocular pharmacokinetics of cephalosporins using microdialysis. J Ocul Pharmacol Ther 17 485–498. [DOI] [PubMed] [Google Scholar]
- Macha S and Mitra AK (2002) Ocular disposition of ganciclovir and its monoester prodrugs following intravitreal administration using microdialysis. Drug Metab Dispos 30 670–675. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Gunda S, and Mitra A (2003a) Functional expression of a sodium dependent nucleoside transporter on rabbit cornea: role in corneal permeation of acyclovir and idoxuridine. Curr Eye Res 26 175–183. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Hingorani T, Srirangam R, Gadepalli RS, Rimoldi JM, and Repka MA (2009) Transcorneal permeation of l- and d-aspartate prodrugs ester of acyclovir: delineation of passive diffusion versus transporter involvement. Pharm Res 26 1261–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Kansara V, and Mitra AK (2006) Vitreal pharmacokinetics of dipeptide monoester prodrugs of ganciclovir. J Ocul Pharmacol Ther 22 231–241. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Tirucherai GS, Pal D, and Mitra AK (2003b) Functional differences in nucleoside and nucleobase transporters expressed on the rabbit corneal epithelial cell line (SIRC) and isolated rabbit cornea. AAPS PharmSci 5 E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheny CJ, Lamb MW, Brouwer KR, and Pollack GM (2001) Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Pharmacotherapy 21 778–796. [DOI] [PubMed] [Google Scholar]
- Nobili S, Landini I, Giglioni B, and Mini E (2006) Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets 7 861–879. [DOI] [PubMed] [Google Scholar]
- Oztürk F, Kortunay S, Kurt E, Inan UU, Ilker SS, Basci N, and Bozkurt A (1999) The effect of long-term use and inflammation on the ocular penetration of topical ofloxacin. Curr Eye Res 19 461–464. [DOI] [PubMed] [Google Scholar]
- Oztürk F, Kurt E, Inan UU, Kortunay MC, Ilker SS, Ba°ci NE, and Bozkurt A (2000) Penetration of topical and oral ofloxacin into the aqueous and vitreous humor of inflamed rabbit eyes. Int J Pharm 204 91–95. [DOI] [PubMed] [Google Scholar]
- Salminen L and Urtti A (1984) Disposition of ophthalmic timolol in treated and untreated rabbit eyes. A multiple and single dose study. Exp Eye Res 38 203–206. [DOI] [PubMed] [Google Scholar]
- Senthilkumari S, Velpandian T, Biswas NR, Saxena R, and Ghose S (2008a) Evaluation of the modulation of P-glycoprotein (P-gp) on the intraocular disposition of its substrate in rabbits. Curr Eye Res 33 333–343. [DOI] [PubMed] [Google Scholar]
- Senthilkumari S, Velpandian T, Biswas NR, Sonali N, and Ghose S (2008b) Evaluation of the impact of P-glycoprotein (P-gp) drug efflux transporter blockade on the systemic and ocular disposition of P-gp substrate. J Ocul Pharmacol Ther 24 290–300. [DOI] [PubMed] [Google Scholar]
- Steuer H, Jaworski A, Elger B, Kaussmann M, Keldenich J, Schneider H, Stoll D, and Schlosshauer B (2005) Functional characterization and comparison of the outer blood-retina barrier and the blood-brain barrier. Invest Ophthalmol Vis Sci 46 1047–1053. [DOI] [PubMed] [Google Scholar]
- Tan AY, LeVatte TL, Archibald ML, Tremblay F, Kelly ME, and Chauhan BC (2002) Timolol concentrations in rat ocular tissues and plasma after topical and intraperitoneal dosing. J Glaucoma 11 134–142. [DOI] [PubMed] [Google Scholar]
- Tsuji A, Tamai I, and Sasaki K (1987) Hydrolysis of prednisolone succinate by esterase in rabbit ocular tissue. Ophthalmic Res 19 322–329. [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Ogura Y, Tabata Y, Kimura H, Wiedemann P, and Honda Y (2004) Drug delivery systems for vitreoretinal diseases. Prog Retin Eye Res 23 253–281. [DOI] [PubMed] [Google Scholar]