

Abstract
Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) plays an important role in immune surveillance and preferentially induces apoptosis in cancer cells over normal cells, suggesting its potential in cancer therapy. However, the molecular basis for its selective killing of cancer cells is not well understood. Recent studies have identified the CCN family of integrin-binding matricellular proteins as important regulators of cell behavior, including cell adhesion, proliferation, migration, differentiation, and survival. We show here that CCN1 (CYR61) supports the adhesion of prostatic carcinoma cells as an adhesion substrate through integrins and heparan sulfate proteoglycans (HSPGs). Knockdown of CCN1 expression in PC-3 and DU-145 androgen-independent prostate cancer cells strongly inhibited their proliferation without causing apoptosis, indicating that CCN1 promotes their growth. However, CCN1 also significantly enhances TRAIL-induced apoptosis through interaction with integrins αvβ3, α6β4, and the cell surface HSPG syndecan-4, acting through a PKCα-dependent mechanism without requiring de novo protein synthesis. Knockdown of CCN1 expression in PC-3, DU-145, and LNCaP cells severely blunted their sensitivity to TRAIL, an effect that was reversed by exogenously added CCN1 protein. These findings reveal a functional dichotomy for CCN1 in prostate carcinoma cells since it contributes to both cell proliferation and TRAIL-induced cell death, and suggest that CCN1 expression status may be an important parameter in assessing the efficacy of TRAIL-dependent cancer therapy.
Introduction
Prostate cancer is the most commonly diagnosed malignancy in men and the second leading cause of cancer-related deaths in the United States (1;2). In early stages of the disease, prostate cancer cells are dependent on androgens that increase proliferation and inhibit apoptosis. While androgen ablation therapy often results in cancer regression, this process also selects for androgen-independent cancer cells, which can develop into later stage cancers that are metastatic, resistant to apoptosis, and recalcitrant to therapy (1). TRAIL (tumor necrosis factor-related apoptosis inducing ligand) is a type II transmembrane protein of the tumor necrosis factor superfamily that binds five known receptors. Whereas DcR1, DcR2, and OPG are thought to be decoy receptors, ligation of TRAIL to DR4 or DR5 triggers receptor trimerization, recruitment of FADD to the receptor via the receptor death domain, and activation of the apoptotic initiators, caspases-8 and -10 (3). TRAIL is expressed mainly in immune cells and plays a critical role in immune surveillance by natural killer (NK) cells (4), and TRAIL-deficient mice are more susceptible to autoimmune diseases and more prone to tumor metastasis (5;6). TRAIL preferentially induces apoptosis in a variety of cancer cells but exhibits limited cytotoxicity in normal cells. Thus, using TRAIL to induce apoptosis has emerged as a promising cancer therapeutic approach, especially as an adjuvant therapy for advanced prostate cancer in combination with irradiation or chemotherapy (7-9). However, the molecular basis for the susceptibility of prostate cancer cells to TRAIL-induced apoptosis is not well understood, and may involve multiple contributing factors that function in a cell type-dependent manner (10;11).
CCN1 (CYR61) is a secreted, integrin-binding protein that regulates multiple cellular activities, including cell adhesion, migration, proliferation, survival, and apoptosis (12), and is considered a matricellular protein given its predominantly regulatory rather than structural roles (13). Whereas CCN1 is a potent angiogenic inducer and is essential for cardiovascular development during embryogenesis, its expression in the adult is associated with pathological conditions in which angiogenesis and inflammation play important roles, such as wound healing, restenosis, atherosclerosis, and tumorigenesis (12;14-16). Consistent with its angiogenic activity, overexpression of CCN1 in cancer cells promotes tumor growth and vascular density in immunodeficient mice (14), and overexpression of CCN1 has been associated with human breast cancers, gliomas, gastric adenocarcinomas, and melanomas (17;18). In the prostate, CCN1 is overexpressed in benign prostatic hyperplasia (BPH) and promotes prostate epithelial and stromal cell proliferation (19). Down-regulation of CCN1 by staurosporine in prostate cancer cells is associated with neuronal differentiation and suppression of malignancy (20), implicating CCN1 in prostate cancer. Furthermore, stromal expression of CCN2, a close homolog of CCN1, has been shown to promote prostate cancer angiogenesis and tumorigenesis (21).
Here we show that CCN1 expression in prostate carcinoma cells is a double-edged sword: it enhances both cell proliferation and TRAIL-induced cytotoxicity. Knockdown of constitutive CCN1 expression in the androgen-independent prostate carcinoma cell lines PC-3 and DU145 severely inhibits cell proliferation and curtails TRAIL-dependent apoptosis. Mechanistically, CCN1 cooperates with TRAIL in a PKCα-dependent manner, triggered by its interaction with integrins αvβ3, α6β4, and syndecan-4. These findings indicate that prostate carcinoma cell growth and apoptosis are regulated by the extracellular matrix microenvironment through integrin-mediated signaling, and point to CCN1 expression as a critical factor in both prostatic cell proliferation and sensitivity to TRAIL.
Materials and Methods
Cell culture
Normal human PrECs were obtained from Cambrex, along with the basal medium (BM) and growth medium (GM) supplements. PC-3, DU145 and LNCaP cells were obtained from the American Type Culture Collection (ATCC) and grown in RPMI 1640 (Gibco) with 10% FBS (Intergen). All cells were grown at 37°C with 5% CO2.
Proteins, reagents & antibodies
Wild-type CCN1, CCN2, and mutant CCN1 proteins were purified from a baculovirus expression system as described (14;22). Mouse mAb against PKCα (clone 3), fibronectin (FN), and laminin (LN) were from BD Biosciences. Heparin (sodium salt), 4′,6′-diamidino-2-phenylindole (DAPI), human vitronectin (VN), TPA/PMA (phorbol 12-myristate 13-acetate), NAC (N-acetyl-cysteine), BHA (butylated hydroxyanisole), MTT, BSA, heparinase, chondroitinase, and anti-β-actin mAb (AC-15) were from Sigma. Epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) were supplied by Collaborative Biomedical Products. Transforming growth factor-β (TGF-β) was from R&D Systems. Recombinant soluble human TRAIL was from Axxora. Synthetic peptides GRGDSPK and GRGESPK were purchased from American Peptide Company. Function-blocking mAbs against integrins, including GoH3 (anti-α6), anti-β4 (ASC-3), JB1A and P5D2 (anti-β1), P1D6 (anti-α5), and PiF6 (anti-αvβ5) were purchased from Chemicon. Function-blocking anti-αvβ3 antibody (anti-VNR-1) was a generous gift from Dr. Stephen Lam (Univ. Illinois). Polyclonal phospho-Ser PKC substrate antibodies were from Cell Signaling. Rabbit polyclonal Syndecan-4 antibodies were from Santa Cruz. DCF-DA was obtained from Molecular Probes. Anti-mouse and anti-rabbit secondary antibodies were purchased from Amersham. Rabbit polyclonal anti-CCN1 antibodies were raised in rabbits as previously described (14). Caspase-3 inhibitor Z-DEVD-fmk, caspase-8 inhibitor Z-IETD-fmk, caspase-9 inhibitor Z-LEHD-fmk, caspase10 inhibitor Z-AEVD-fmk, cycloheximide, Jnk inhibitor I (cell permeable inhibitory peptide from the JNK binding domain of JIP), Jnk inhibitor II (SP600125), p38 inhibitor (SB202190), and PKC inhibitors (Gö6976, bisindolylmaleimide I (BisI)), and Chelerythrine chloride (C.C.)) were from Calbiochem. Rabbit polyclonal antibodies against GAPDH, agonistic mAbs against DR4 (DR-4-02) and DR5 (79103) were from Abcam and R & D Systems, respectively.
Northern Hybridization
Cell were grown to sub-confluency in RPMI 1640 medium without phenol red with 5% delipidated FBS, and starved in serum-free RPMI 1640 24 hrs prior to treatment with TPA, bFGF, EGF, or TGF-β for 1 hour at 37°C, 5% CO2. Total RNA was isolated and electrophoresed on 1% agarose gel, followed by RNA blotting and hybridization with radioactively labeled human CCN1 and GAPDH cDNA probes using standard protocols.
Adhesion Assays
Adhesion assays were done as previously described (23). Briefly, CCN1, fibronectin (FN), vitronectin (VN), laminin (LN), or BSA were coated on 96-well plate (50 μl protein per well in 1 × PBS) and kept at 4°C overnight. The wells were then blocked for 1 hr with 1% BSA at room temperature. Cells were brought to suspension in 1 × PBS, 2.5 mM EDTA, 0.1% glucose. Following two washing in PBS, cells were suspended in serum-free media and allowed to adhere to the protein-coated wells at 37°C for 10-15 min. Where indicated, antibodies, heparin, or heparinase were added to the cell suspension followed by a 30 min. gentle agitation at room temp. prior to plating. Adherent cells were fixed overnight with formalin, washed and stained with 1% methylene blue in sodium borate buffer (10 mM, pH6.0) for 30 min. at room temperature. The cells were then washed with boric acid buffer, and the dye was extracted with 50% ethanol/0.1N HCl and quantified by absorbance at 620 nm.
Apoptosis assays
For DAPI staining, cells were plated in 24- or 48-well plates (50,000 or 25,000 cells/well, respectively) in culture media and allowed to attach overnight. Cells were rinsed with PBS and incubated at 37°C with serum-free media with test molecules for 6 hrs. Cells were fixed with 4% formaldehyde overnight, and then stained with DAPI at 1μg/ml in PBS for 5 min. Using a fluorescent microscope, 3 randomly selected fields (∼150 cells per field) per well were counted. For terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) assay, PC-3 cells were plated on cover slips (in a 24-well plate, 50,000 cells/well). Apoptosis was carried out as described above, and fixed with 1% paraformaldehyde for 10 min. at room temperature. TUNEL assay was performed according to manufacturers instructions (ApopTag, Red; Chemicon, Inc.). Samples were counter-stained with DAPI before mounting.
Apoptosis in LNCaP cells were scored by the M30 assay, which detects the caspase-3-cleaved form of keratin XVIII (24;25). The M30 Apoptosense ELISA was performed according to manufacturers instruction (DiaPharma). Absorbance was measured at 450 nm. Cell viability was determined with the MTT assay for metabolic activity, carried out as described (26).
Oligonucleotide transfection
PC-3 and DU145 cells were plated in 24-well plates (20,000 cells/well) overnight and transfected with phosphorothiolated oligonucleotides (Sigma Genosys) using lipofectamine reagent (Invitrogen) according to manufacturer's instructions. Briefly, 0.4 μg oligonucleotide was complexed with 0.5μl lipofectamine in the presence of 4 μl Plus reagent (Invitrogen) in a total volume of 250 μl serum- and antibiotic-free RPMI 1640. Transfection was carried out for 3 hrs, at which time medium was added to make a final volume of 500 μl RPMI 1640 with 10% FBS. Cells were washed with PBS 24 hrs post-transfection, and normal growth media were added. After 24 or 48 hrs, cell lysates were collected for immunoblotting. The CCN1 anti-sense sequence was 5′-GCAGGCACGGGGCAGGTGG-3′ and the sense sequence was 5′-CCACCTGCCCGCTGCCTGC-3′ (19). The antisense sequence for PKCα was 5′-CAGCCATGGTTCCCCCCAAC-3′ and the sense sequence was 5′-CCAGTCACTCGCACCATCGC-3′ (27).
Growth curves
PC-3 and DU145 cells were plated in 6-well plates, transfected with oligonucleotides as described above, and growth was assessed at 24, 48, and 72 hours. At each time point, cells were collected by trypsin and the number of cells was counted using a hemocytometer. Cell death was measured in parallel at each time point with DAPI staining.
Immunoblotting
Immunoblotting was done according to standard protocols. Samples represent an equivalent amount of protein from 250,000 cells and were resolved on 10% SDS-PAGE prior to transfer to nitrocellulose membranes.
Measurement of ROS
ROS generation was measured as previously described {Chen, 2007 8802 /id}. Briefly, intracellular peroxide was measured using the cell-permeable dye, H2DCF-DA, which becomes fluorescent upon oxidation by intracellular peroxide/hydroperoxides. PC-3 cells plated (50,000 cells/well) on glass coverslips were serum starved in phenol-red free media for 30 minutes prior to treatment. Media were then replaced after 1 hour after treatment with PBS containing 5μM H2DCF-DA and incubated for 10 minutes at 37°C. Coverslips were then mounted on slides and imaged by fluoresce microscopy. The integrated densities were measured using NIH ImageJ software.
Results
CCN1 supports adhesion and promotes cell proliferation of prostate cancer cells
To investigate the potential role of CCN1 in prostate carcinoma cells, we examined its expression. CCN1 is expressed in the malignant, androgen-independent PC-3 and DU145 prostate carcinoma cells, and its expression is induced to a higher level by TPA and EGF in DU145 cells (Fig. 1A). By contrast, CCN1 mRNA is undetectable in the androgen-dependent LNCaP cells, but is induced upon by stimulation of cells with TPA or EGF (Fig. 1A). Since CCN1 is a cell-adhesive ECM protein that induces adhesive signaling in various cell types in endothelial cells through integrin αvβ3 and fibroblasts through α6β1 and heparan sulfate proteoglycans (HSPGs)(12), we tested the ability of prostate carcinoma cells to adhere to CCN1-coated surfaces. Indeed, CCN1 supported the adhesion of PC-3 and DU-145 cells with similar efficiency compared to other ECM proteins, including fibronectin (FN), vitronectin (VN), or laminin (LN)(Fig. 1B-C). Furthermore, inhibitory monoclonal antibodies (mAbs) against integrins α6 (GoH3), β1 (JB1A), or β4 (ASC-3), but not mAb against integrin αvβ3, blocked the adhesion of PC-3 cells to CCN1 (Fig. 1D). Similar results were observed for DU145 cells (data not shown). Since CCN1 supports cell adhesion through integrin α6β1 and HSPGs as coreceptors in several cell types including fibroblasts and smooth muscle cells (12), we tested the requirement of HSPGs. Treatment of cells with heparinase, but not chondroitinase, abolished cell adhesion to CCN1 (Fig. 1E). To corroborate these results, we tested the functions of the CCN1 mutants TM and D125A, which contain missense mutations targeting specific receptor binding sites and are unable to bind α6β1-HSPG or αvβ3, respectively (22;28). Whereas TM was completely unable to support cell adhesion, D125A was as active as wild type CCN1 (Fig. 1F). These results show that CCN1 supports PC-3 prostate carcinoma cell adhesion through integrin α6β1, α6β4, and HSPGs.
Figure 1. CCN1 is expressed in prostate cells and supports prostatic cell adhesion through integrins.
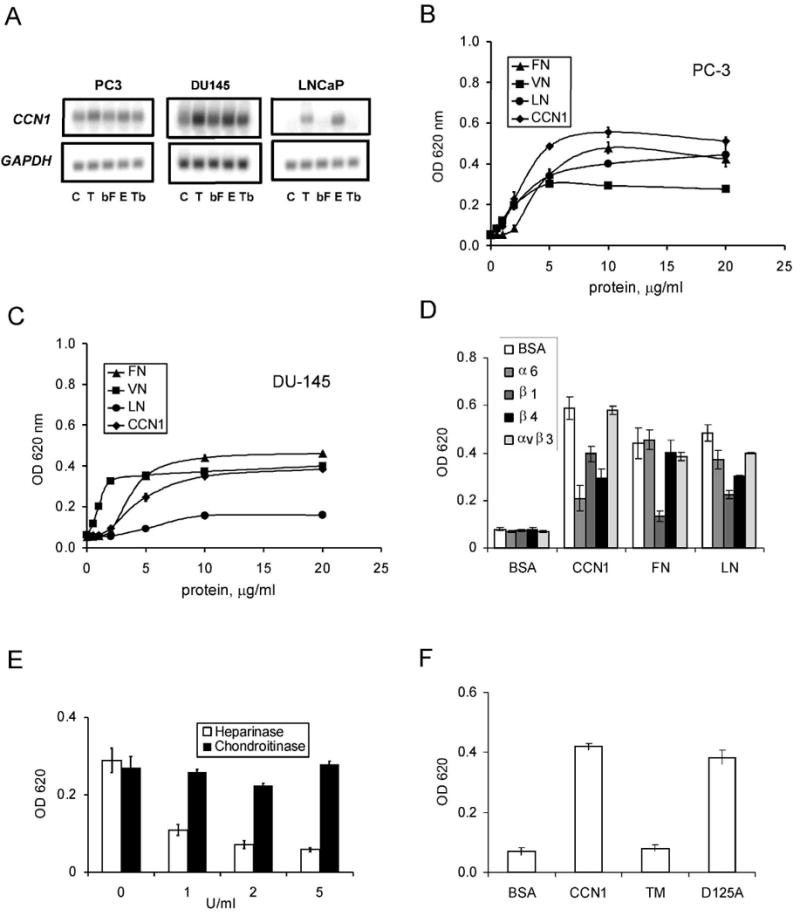
A. RNA blot of total RNA from PC-3, DU145, and LNCaP cells treated with TPA (T; 10 nM), bFGF (bF; 10 ng/ml), EGF (E; 3 ng/ml), or TGF-β (Tb; 10 ng/ml) for 1 hr, hybridized to cDNA probes for CCN1 and GAPDH. B. PC-3 cells adhered on plates coated with various concentrations of CCN1, FN, VN, and LN for 30 min. Adherent cells were stained with methylene blue and extracted dye was quantified by absorbance at 620 nm. C. Adhesion of DU145 cells on various substrates as described above. D. PC-3 cells were pre-treated with antibodies against α6, β1, β4, or αvβ3, integrins before adhering to plates coated with indicated substrates and cell adhesion measured. E. PC3 cells were treated with 1-5 u/ml of heparinase or chondroitinase and their adhesion to CCN1-coated plates measured. F. PC-3 cell adhesion to plates coated with BSA, CCN1, or the CCN1 mutants TM (α6β1-HSPG binding-defective) and D125A (αv binding-defective) measured by methylene blue staining.
CCN1 is known to enhance growth factor-induced DNA synthesis in some cell types, and to stimulate the proliferation of prostate epithelial and stromal cells (19;29). The basal expression of CCN1 in PC-3 and DU145 cells suggests that CCN1 expression may promote cell proliferation in these aggressive prostate carcinoma cells. To test this possibility, we knocked down CCN1 expression in PC-3 and DU145 cells using antisense oligonucleotide, and counted the total number of cells at 24, 48, and 72 hrs post-transfection (Fig. 2). Transfection of antisense oligonucleotide, but not the sense oligonucleotide, significantly reduced CCN1 expression in PC-3 and DU145 cells (Fig. 2A,D). Knockdown of CCN1 in both PC-3 and DU-145 cells resulted in substantially slower rates of growth compared to control cells transfected with the sense oligonucleotide (Fig. 2B,E). Cell division was essentially negligible within the first 48 hrs in CCN1-depleted cells, whereas control cells proliferated substantially. No apoptosis was observed in these cells, and thus the inhibition of cell proliferation by CCN1 knockdown was not due to apoptotic cell death (Fig. 2C,F). Together, these results show that as expected, endogenous CCN1 expression promotes cell proliferation and confers a substantial growth advantage to prostate cancer cells in culture.
Figure 2. CCN1 is critical for proliferation of prostate carcinoma cells.
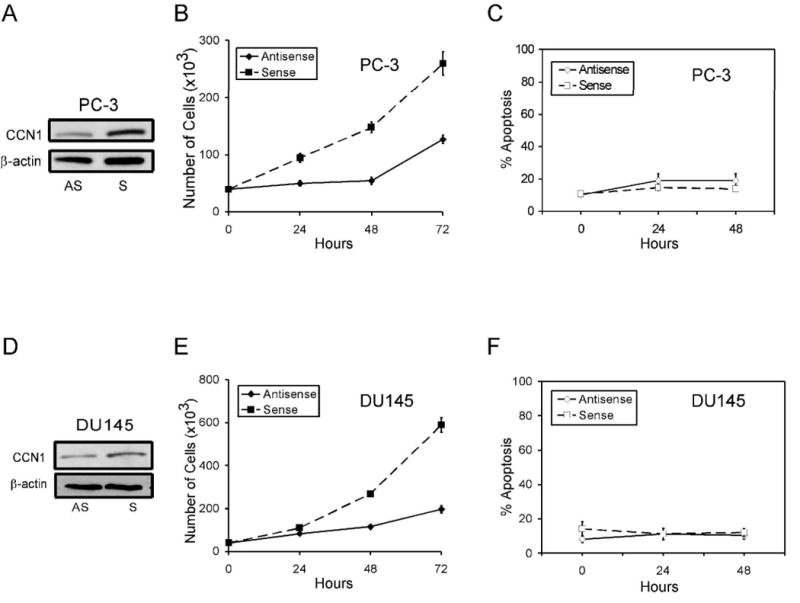
PC-3 (A) and DU145 (D) cells were treated with anti-sense (AS) or sense (S) CCN1 oligonucleotides. To show CCN1 knockdown, cell lysates were collected 24 hrs after transfection and electrophorsed on 10% SDS-PAGE, followed by immunoblotting with antibodies against CCN1 and β-actin. Transfected PC-3 (B) and DU-145 (E) cells were cultured in growth media, and total cell numbers counted 24, 48, and 72 hrs later. Parallel plates of PC-3 (C) and DU-145 (F) cells were scored for apoptosis at each time point.
CCN1 cooperates with TRAIL to induce apoptosis in prostate cells
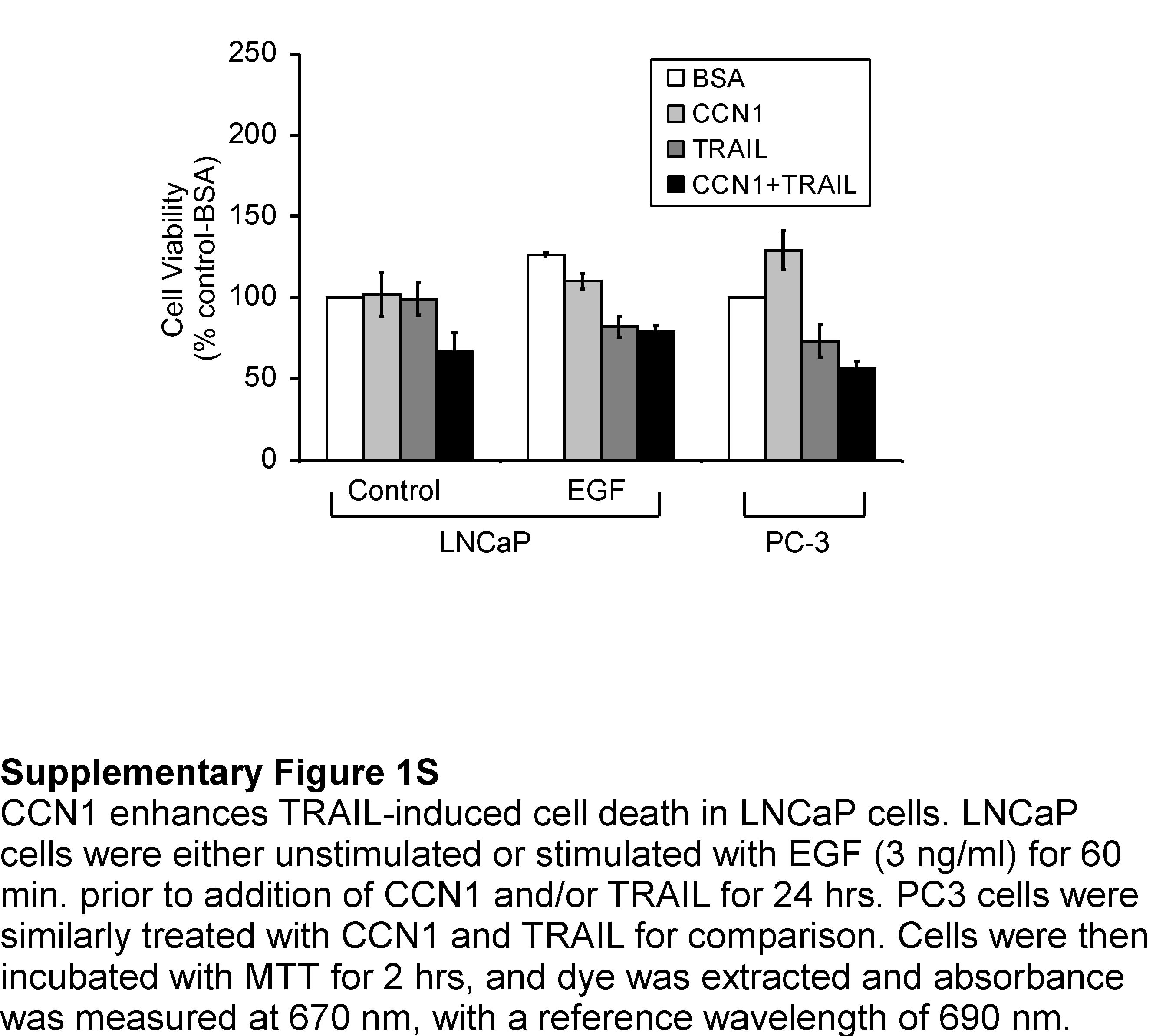
As a matricellular protein, CCN1 regulates cell survival and cell death through integrin-mediated signaling (29;30). Recently, we found that CCN1 allows TNFα to induce apoptosis in human skin fibroblasts by overriding the antioxidant effect of NFκB without requiring the inhibition of protein synthesis or NFκB signaling (31). Given the potential application of the TNF family member TRAIL as a cancer therapeutic, we tested the possibility that CCN1 might regulate the susceptibility of prostatic cancer cells to TRAIL-induced apoptosis. Whereas CCN1 (even as high as 10 μg/ml) or a low dose of TRAIL (5 ng/ml) alone did not induce appreciable cell death in normal prostate epithelial cells (PrECs) or in DU145 and PC-3 prostate carcinoma cells, the combination of both proteins triggered ∼30-50% cell death within 6 hrs (Fig. 3A,3B). Thus, even though TRAIL alone can also induce apoptosis in DU145 and PC-3 cells when used at high concentrations (>10 ng/ml; data not shown)(32), CCN1 strongly enhances TRAIL-induced prostatic cell death and does so in a dose-dependent manner (Fig. 3A and data not shown). Likewise, exogenously added CCN1 cooperated with TRAIL to induce apoptosis in the androgen-dependent LNCaP cells as judged by the M30 assay for caspase-3 activity (Fig. 3C) and the MTT assay for cell viability (Supplementary Fig. 1S). Since CCN1 expression in LNCaP cells is inducible by TPA or EGF (Fig. 1A), pre-treatment with EGF to induce CCN1 expression enhanced TRAIL-induced apoptosis (Fig. 3C), consistent with CCN1 cooperation with TRAIL.
Figure 3. CCN1 cooperates with TRAIL to induce apoptosis in prostate cells.
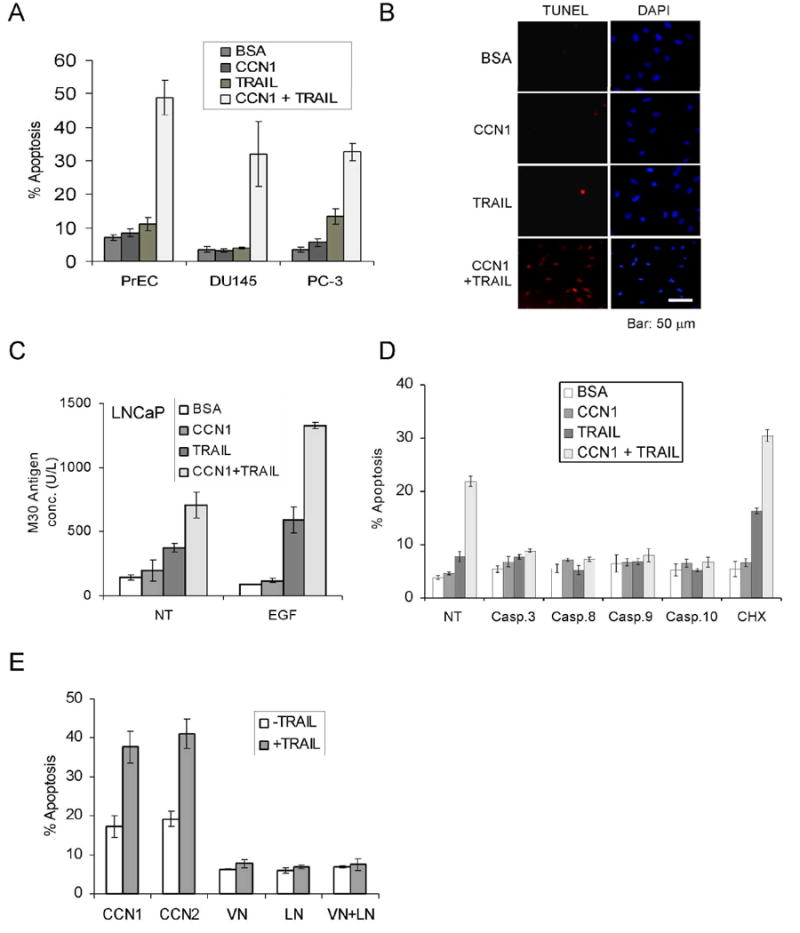
A. PrEC, DU145, and PC-3 cells were treated with CCN1 (10 μg/ml) and/or TRAIL (5 ng/ml) for 6 hrs and scored for apoptosis. B. PC-3 cells were treated with CCN1 and/or TRAIL as above, followed by TUNEL assay and counterstaining with DAPI. C. LNCaP cells were either untreated (NT) or treated with EGF for 60 min. prior to stimulation with CCN1 and/or TRAIL for 6 hrs, and subjected to M30 apoptosis assay. D. Cells were pre-treated with cycloheximide (5 μM) or caspase inhibitors for 30 min., including 50 μM Z-DEVD (caspase-3), Z-IETD (caspase-8), Z-LEHD (caspase-9), or Z-AEVD (caspase-10), then treated with CCN1 and/or TRAIL for 6 hrs as above and scored for apoptosis. E. PC-3 cells were adhered to plates coated with CCN1, CCN2, VN, LN (10 μg/ml each), or VN and LN and cultured in serum-free medium for 6 hrs with or without TRAIL (5 ng/ml). Cells were then fixed and scored for apoptosis.
Inhibitors against caspases-3, -8, -9, and -10 each abrogated CCN1/TRAIL-mediated cell death (Fig. 3D). These results, together with TUNEL-positive staining (Fig. 3B), indicate that CCN1/TRAIL-induced cell death is apoptotic in nature. Furthermore, the caspase requirements suggest that apoptotic signals initiated through the TRAIL receptors (caspase-8 and -10) are amplified through the mitochondria, resulting in the activation of caspase-9 (33). CCN1 also cooperates with agonistic antibodies against DR4 and DR5 to induce cell death, indicating that CCN1 can cooperate with apoptotic signaling through either death receptor of TRAIL (Supplementary Fig. 2S). Treatment of cells with cycloheximide enhanced TRAIL-induced apoptosis as expected (34), and did not prevent CCN1/TRAIL cooperation (Fig. 3D). Thus, de novo protein synthesis is not required for CCN1/TRAIL-dependent cell death.
Since CCN1 is an ECM-associated cell adhesive molecule, we tested its ability to promote TRAIL-induced apoptosis as a cell adhesion substrate. PC-3 cells were adhered to plates coated with CCN1, the closely related family member CCN2 (connective tissue growth factor, CTGF)(23), or ECM proteins such as VN and LN. Cells adhered to CCN1- or CCN2-coated surfaces were highly apoptotic in the presence of TRAIL, whereas cells adhered to VN or LN did not undergo apoptosis (Fig. 3E). Furthermore, a combination of VN and LN, which can bind HSPGs and several integrins including α6β4 and αvβ3, the CCN1 receptors required for cooperation with TRAIL (see below), was unable to promote TRAIL-mediated apoptosis (Fig. 3E). These results indicate that CCN proteins appear unique among ECM cell adhesive molecules in being able to cooperate with TRAIL.
Endogenous CCN1 expression drives TRAIL sensitivity
Since exogenously added CCN1 protein can enhance TRAIL-induced apoptosis, we postulated that endogenously expressed CCN1 in PC-3 and DU145 cells may sensitize these cells to TRAIL-induced apoptosis. To test this hypothesis, we knocked down the expression of CCN1 in these cells as described above, and then treated them with various concentrations of TRAIL in the presence or absence of exogenously added CCN1 24 hrs post-transfection. Remarkably, knockdown of CCN1 by antisense oligonucleotide greatly diminished the sensitivity of cells to TRAIL-induced apoptosis. When CCN1 was depleted, there was essentially no response to TRAIL at lower doses, and it was not until TRAIL concentrations reached 50 ng/ml that appreciable levels of apoptosis were observed (Fig. 4A,B). By contrast, control cells transfected with sense oligonucleotide were still responsive to TRAIL. Exogenously added CCN1 reversed the effect of antisense oligonucleotides, restoring the apoptotic response to a level similar to that in control cells transfected with sense oligonucleotides. CCN1 protein added to control cells further enhanced apoptosis in the presence of TRAIL. These results show that endogenously expressed CCN1 is critical for the susceptibility of PC-3 and DU145 cells to TRAIL-induced apoptosis. Similarly, antisense oligonucleotide eliminated EGF-induced CCN1 expression in LNCaP cells, and concomitantly, the EGF enhancement of TRAIL-dependent apoptosis was abolished (Fig. 4C). Thus, endogenous expression of CCN1 drives TRAIL sensitivity, and CCN1 knockdown in PC-3, DU145, and LNCaP prostate carcinoma cells significantly reduced TRAIL-induced apoptosis.
Figure 4. Downregulation of CCN1 in prostate carcinoma cells inhibits TRAIL-induced apoptosis.
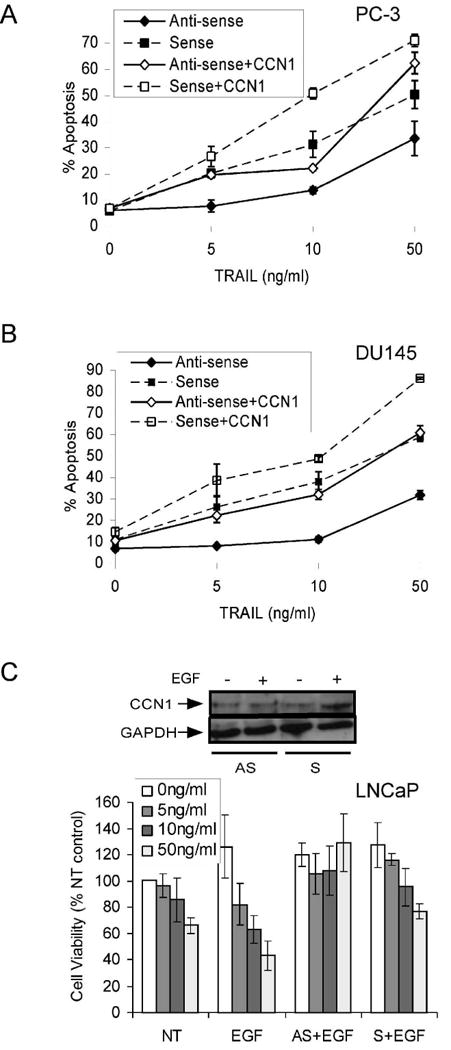
PC-3 (A) and DU145 (B) cells were transfected with sense or antisense CCN1 oligonucleotides as described in Figure 2, and treated with various concentrations of TRAIL (0-50 ng/ml) as indicted, either in the presence or absence of soluble CCN1 for 6 hrs prior to scoring for apoptosis by DAPI staining. C. LNCaP cells were transfected with sense or antisense CCN1 oligonucleotides and treated with EGF (3 ng/ml) where indicated for 1 hr before exposure to various concentrations of TRAIL and CCN1 (10 μg/ml) for 6 hrs. Cell viability was measured by the MTT assay. To show EGF induction of CCN1 and antisense knockdown, cell lysates were electrophorsed on 10% SDS-PAGE, followed by immunoblotting with antibodies against CCN1 and GAPDH (upper panel).
CCN1 cooperates with TRAIL through interaction with integrins and HSPGs
To analyze the mechanism of CCN1/TRAIL cooperation, we have focused on PC-3 cells in subsequent studies. Since PC-3 cells adhered to CCN1 through α6β1, α6β4 and HSPGs, we investigated whether these adhesive receptors for CCN1 also play a role in CCN1/TRAIL cooperation. Pretreatment of cells with mAbs against integrins α6 (GoH3) or β4 (ASC-3) subunits blocked CCN1/TRAIL-induced apoptosis, whereas mAbs against β1 (P1D6) and α5 (JB1A) had no effect (Fig. 5A). Since α6 heterodimerizes with β1 or β4 chains, these results indicate that although both α6β1 and α6β4 are involved in mediating adhesion to CCN1, only α6β4, but not α6β1, is critical for mediating CCN1 enhancement of TRAIL activity in PC-3 cells. Furthermore, pretreatment of PC-3 cells with function-blocking anti-αvβ3 mAb abrogated apoptosis, even though αvβ3 is not required for adhesion to CCN1. Function-blocking anti-α5β1 or anti-αvβ5 mAbs had no effect on CCN1 enhancement of TRAIL activity (Fig. 5B). Consistently, the peptide GRGDSP, which inhibits ligand binding to αv integrins, α5β1, and α8β1, obliterated CCN1/TRAIL-induced apoptosis in PC-3 cells, whereas the control peptide GRGESP had no effect (Fig. 5B). These results indicate that αvβ3 and α6β4 play a critical role in CCN1/TRAIL cooperation, whereas αvβ5, α5β1 and α6β1 are not important for this process.
Figure 5. CCN1/TRAIL cooperation is mediated through αvβ3, α6β4, and syndecan-4.
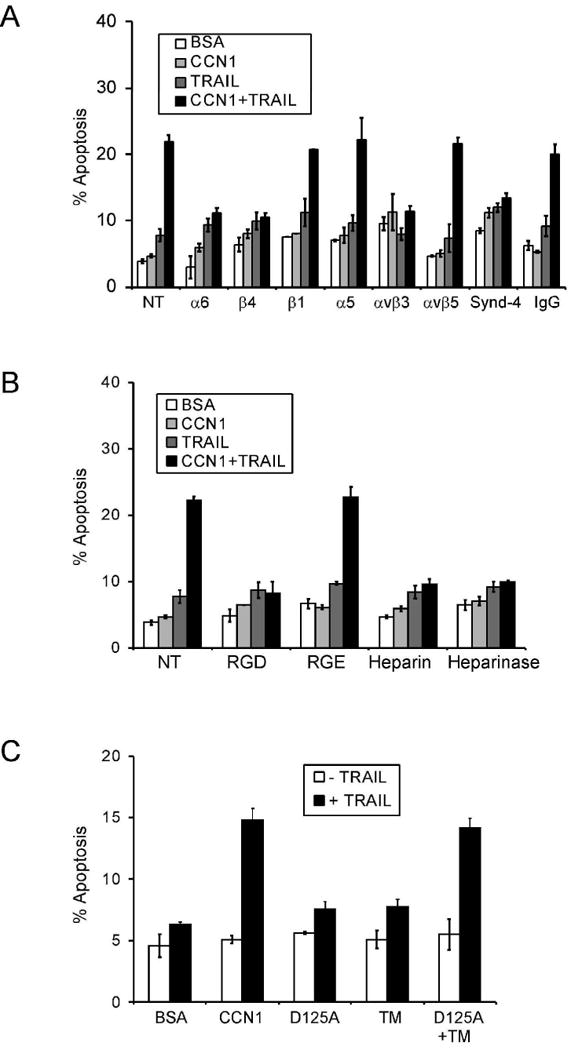
PC-3 cells were scored for apoptosis following indicated treatments. A. Cells were pre-treated with mAbs (50 μg/ml each) against α6 (GoH3), β4 (ASC-3), β1 (JB1A), α5β1 (JB-55), αvβ5 (P1F6), αvβ3 (anti-VNR-1), or syndecan-4 or mouse IgG for 1 hr prior to stimulation with CCN1 (10 μg/ml) and/or TRAIL (5 ng/ml). B. Cells were pre-treated for 30 min. with GRGDSP or GRGESP peptides (0.2 mM each), soluble heparin (1 mg/ml), or with heparinase (20 U/ml) for 24 hrs before addition of CCN1 and/or TRAIL. C. Cells were treated with WT CCN1, D125A (αv binding-defective mutant), TM (α6β1-HSPG binding-defective mutants), or D125A and TM with or without TRAIL and scored for apoptosis.
To evaluate the potential role of cell surface HSPGs, we added soluble heparin in the culture medium or treated the cells with heparinase prior to apoptosis assay. Either treatment blocked CCN1 enhancement of TRAIL-induced cell death, indicating that cell surface HSPGs are involved (Fig. 5B). Antibodies against syndecan-4 abrogated CCN1/TRAIL cooperation, whereas control IgG had no effect (Fig. 5A), in agreement with the role of syndecan-4 as the HSPG receptor for CCN1 (30;31). Together, these results indicate that CCN1 sensitizes PC-3 cells to TRAIL-induced apoptosis through interaction with integrins αvβ3, α6β4, and syndecan-4. Consistent with this interpretation, CCN1 mutants that are either defective for binding α6 integrin and HSPGs (TM) or αvβ3 (D125A) are incapable of apoptotic cooperation with TRAIL (Fig. 5C). Interestingly, the combination of D125A and TM reconstituted wild type activity, indicating that the mutant proteins are biologically active, and that interaction of CCN1 with αvβ3 and α6-HSGPs need not occur through the same CCN1 molecule. However, a combination of VN and LN, which can bind HSPGs and several integrins including α6β4 and αvβ3, the receptors for CCN1 cooperation with TRAIL, was unable to enhance TRAIL activity (Fig. 3E). These results indicate that CCN proteins are unique among ECM molecules in being able to cooperate with TRAIL.
CCN1 cooperates with TRAIL in a PKCα-dependent manner
Published studies have implicated the roles of reactive oxygen species (ROS) and the stress-induced kinases, including JNK and p38 MAPK, in TRAIL-mediated apoptosis (26;35-37). Recently, we have found that CCN1 is a potent inducer of reactive oxygen species (ROS) in fibroblasts (31), and we have confirmed that CCN1 induces ROS accumulation in PC-3 cells (Supplementary Fig. 3S). However, the presence of the ROS scavengers N-acetyl cysteine (NAC) or butylated hydroxyanisole (BHA) had no effect on CCN1/TRAIL-induced apoptosis in PC-3 cells (Supplementary Fig. 3S). Further, neither a JNK inhibitory peptide, nor a chemical inhibitor of JNK (SP600125), nor the p38 MAPK inhibitor SB202190 had any effect on CCN1/TRAIL-mediated apoptosis in PC-3 cells (Supplementary Fig. 4S). Therefore, ROS, JNK, and p38 do not appear to be involved in mediating CCN1/TRAIL apoptotic cooperation.
Since TRAIL-induced apoptosis is dependent on protein kinase C (PKC) activity in LNCaP cells (36), we tested whether PKC plays a role in CCN1/TRAIL cooperation. We found that each of three different inhibitors of classical PKCs, Gö6976, BisI, and Chelerythrine chloride, blocked CCN1-enhanced apoptosis in PC-3 cells (Fig. 6A). Treatment of cells with CCN1 and TRAIL resulted in enhanced phosphorylation of PKC substrates compared to stimulation with either CCN1 or TRAIL alone, indicating that CCN1 and TRAIL cooperate to induce PKC activity (Fig. 6B). This phosphorylation event was blocked by BisI, indicating its dependence on PKC. Among the various PKC isoforms, PKCα is uniquely activated by syndecan-4 (38;39), a coreceptor required for CCN1/TRAIL cooperation (Fig. 5A). Thus, we investigated the role of PKCα by using a well-characterized antisense oligonucleotide to downregulate its expression (27). Transfection of PKCα antisense oligonucleotide, but not the sense oligonucleotide, reduced the level of PKCα in PC-3 cells (Fig. 6C). Remarkably, knockdown of PKCα completely abrogated CCN1 enhancement of TRAIL-induced apoptosis (Fig. 6C). These results indicate that CCN1-TRAIL cooperation proceeds through a PKCα-dependent mechanism.
Figure 6. PKCα is required for CCN1/TRAIL cooperation.
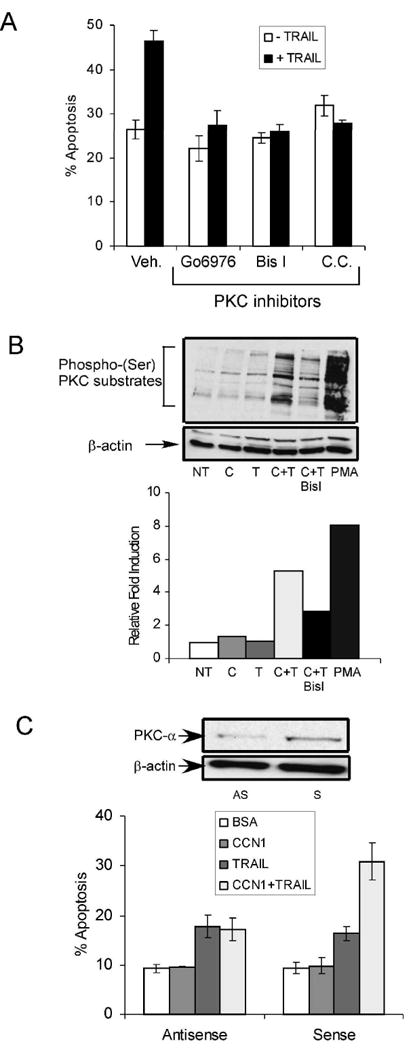
A. PC-3 cells were pre-treated with Gö6976 (1 μM), BisI (1 μM), or chelerythrine chloride (1 μM), or a vehicle control (Veh) and adhered to surfaces coated with CCN1 (10 μg/ml). TRAIL was then added and incubated for 6 hrs and scored for apoptosis. B. Adherent PC-3 cells were treated with CCN1 (C), TRAIL (T), CCN1 And TRAIL (C+T) or PMA for 30 min., with precincubation with BisI for 30 min where indicated. Cell lysates were resolved on 10% SDS-PAGE and immunoblotted with antibodies against phospho-(Ser)PKC substrates and β-actin. Graph shows quantification of signals using ImageJ software from NIH. C. PC-3 cells were transfected with anti-sense (AS) or sense (S) oligonucleotides against PKCα, treated with CCN1 and/or TRAIL for 6 hrs and scored for apoptosis. To analyze the oligonucleotide knockdown, cell lysates were collected 48 hrs after transfection and resolved on 10% SDS-PAGE, followed by immunoblotting with antibodies against PKCα or β-actin (upper panel).
Discussion
The present study reveals a surprising functional dichotomy for the matricellular protein CCN1 in prostatic cells. Whereas expression of CCN1 promotes prostatic cell proliferation, it also enhances the cytotoxicity of TRAIL. Thus, expression of CCN1 in prostate carcinoma cells appears to be a double-edged sword, conferring a growth advantage on the one hand, while sensitizing them as targets of TRAIL-induced apoptosis on the other (Fig. 7). These findings indicate that the ECM microenvironment may play critical roles in prostate carcinoma cell growth and survival, and implicate CCN1 expression as a potentially important parameter in assessing the efficacy of TRAIL-dependent cancer therapy.
Figure 7. A model of CCN1 functions in prostate carcinoma cells.
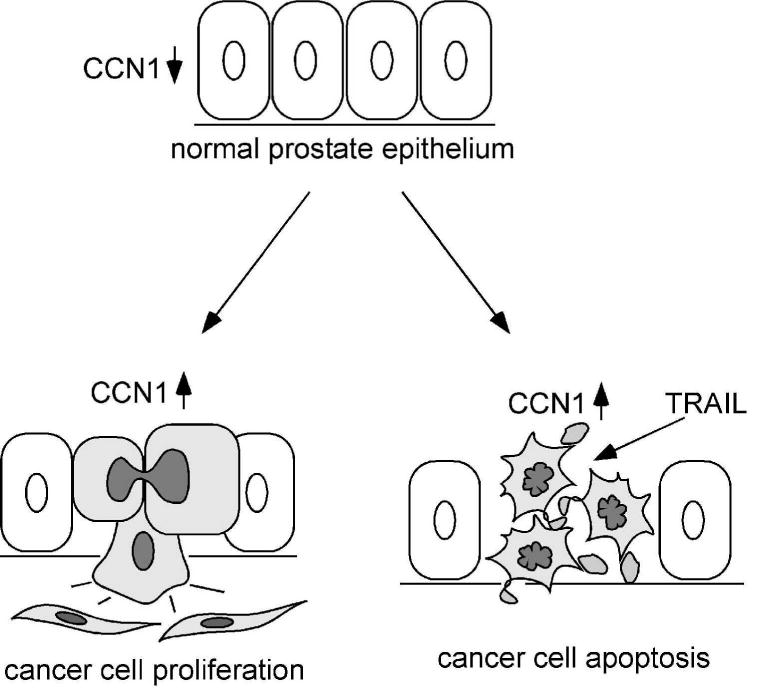
In the normal prostate epithelium, CCN1 expression is low. Elevated CCN1 expression (cells in grey) promotes prostate cancer cell proliferation, thus enhancing tumor growth. CCN1 is also a known angiogenic inducer and can upregulate the expression of MMPs (14;47), attributes that can promote tumor growth invasion. However, the expression of CCN1 makes the prostate cancer cells hypersensitive to TRAIL-induced apoptosis.
CCN1 has been shown to promote DNA synthesis and proliferation of various cell types, including prostate epithelial and stromal cells (19). Thus, it is not surprising that endogenous expression of CCN1 elevates cell proliferation in PC-3 and DU-145 cells (Fig. 2). While CCN1 supports endothelial cell survival through interaction with integrin αvβ3, it can also induce apoptosis in some cell types, including fibroblasts, through an α6β1-HSPG-dependent pathway (29;30). Although CCN1 does not induce cell death in prostatic cells on its own, it cooperates with TRAIL to trigger apoptosis through interaction with integrins αvβ3, α6β4 and syndecan-4 (Fig. 5). The ability of CCN1 to promote TRAIL-induced cell death in both p53-null (PC-3) and p53 mutant (DU145) cells indicates that it functions through a p53-independent pathway, and suggests that it may be involved in apoptosis of other cancer cells, many of which have lost p53 expression or function. The requirement of multiple CCN1 receptors for cooperation with TRAIL may serve to specify the target cells, ensuring that only those expressing the correct receptors are eliminated. It is interesting to note that a combination of VN and LN, which together can bind α6β4, αvβ3, and HSPGs, is unable to enhance TRAIL cytotoxicity (Fig. 3E), suggesting that unique functions of CCN1 are required. It is possible that CCN1 might engage an additional receptor, or that ligation to CCN1 may trigger a unique assembly of receptors that is necessary for crosstalk with the TRAIL-induced signaling pathway. Aside from CCN1, the related family member CCN2 is also able to enhance the cytotoxicity of TRAIL (Fig. 3E), suggesting that this activity may be common among multiple CCN family members. Since CCN2 is also expressed in PC3 and DU145 cells (data not shown), it is likely that knockdown of both CCN1 and CCN2 may produce a greater cytoprotective effect against TRAIL-induced apoptosis than depletion of CCN1 alone.
TRAIL is structurally related to TNFα, a pro-inflammatory cytokine and a potent activator of NFκB. TNFα also induces apoptotic signals that are normally suppressed by NFκB activity, leading to cell death only when NFκB signaling is blocked (40). Recently, we have found that CCN1 is able to unmask the cytotoxicity of TNFα, converting it from a cytokine that normally promotes cell proliferation in fibroblasts into one that induces rapid apoptosis without perturbation of NFκB signaling (31). Although TNFα and TRAIL are related cytokines, they act through distinct receptors and signaling mechanisms. CCN1 enables the cytotoxicity of TNFα in fibroblasts by inducing a high level of ROS, thereby overriding the anti-apoptotic effects of NFκB to achieve a prolonged activation of JNK that is necessary for TNFα-induced apoptosis (31). By contrast, we show that CCN1/TRAIL cooperation does not require ROS or JNK activation, but is instead dependent on the activation of PKCα (Fig. 6 and Supplementary Figs. 3,4).
The requirement of PKCα for CCN1 to enhance TRAIL activity is somewhat unexpected, since PKCα signaling is generally associated with cell survival (41). Syndecan-4 can directly activate PKCα (38), suggesting that CCN1 may activate PKCα through its interaction with syndecan-4. PKCα is pro-mitogenic in many cancer cells, and anti-sense approaches targeting PKCα are undergoing clinical trial as cancer therapy (42). Nevertheless, PKCα has also been shown to mediate apoptosis in some cell types. For example, in LNCaP prostate carcinoma cells, phorbol ester-induced apoptosis proceeds through a PKCα-dependent pathway through a mechanism requiring p38 MAPK (36). By contrast, the PKCα-dependent apoptotic cooperation of CCN1 and TRAIL does not require activation of p38 MAPK or JNK. Future studies will be required to elucidate precisely how PKCα signaling mediates apoptosis in this context.
The expression of CCN1 has been detected in stromal cells of prostate carcinomas (43)(data not shown). Interestingly, stromal expression of CCN2, a close homolog of CCN1, has been shown to promote prostate cancer angiogenesis and tumorigenesis (21). Both CCN1 and CCN2 are potent angiogenic inducers (14;44), and may promote tumorigenesis through angiogenesis. Although CCN1 expression may confer a growth advantage in prostatic carcinoma cell lines, it may also enhance tumor suppression through TRAIL-induced apoptosis. It has been observed that CCN1 is down regulated in some prostate carcinomas (45), suggesting the possibility that low CCN1 expression may help evade immune surveillance in some instances.
Since TRAIL preferentially targets a variety of cancer cells over normal cells for elimination, its deployment has emerged as a promising approach of cancer therapy (7-9). TRAIL shows no systemic toxicity in non-human primates, and phase I clinical trials using agonistic mAbs against DR4 and DR5 have also found no significant toxicity, supporting the notion that inducing TRAIL signaling may be a useful therapeutic strategy (46). Thus, understanding the molecular basis of TRAIL sensitivity may be of potential therapeutic significance. Our findings indicate that CCN proteins may function as novel matrix regulators of TRAIL-induced apoptosis, providing contextual cues for the induction of cell death. Thus, the expression status of CCN1 may be an important consideration in assessing the efficacy of TRAIL-dependent cancer therapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by a grant from the NIH (CA46565 and GM78492) to L.F.L., and pre-doctoral fellowships from the American Heart Association to V.T. and V.J.
References
- 1.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006 Mar 15;12(6):1665–71. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 2.Damber JE, Aus G. Prostate cancer. Lancet. 2008 May 17;371(9625):1710–21. doi: 10.1016/S0140-6736(08)60729-1. [DOI] [PubMed] [Google Scholar]
- 3.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39(78):1462–75. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002 Feb 1;168(3):1356–61. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 5.Smyth MJ, Takeda K, Hayakawa Y, Peschon JJ, van den Brink MR, Yagita H. Nature's TRAIL--on a path to cancer immunotherapy. Immunity. 2003 Jan;18(1):1–6. doi: 10.1016/s1074-7613(02)00502-2. [DOI] [PubMed] [Google Scholar]
- 6.Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL-/- mice. Nat Immunol. 2003 Mar;4(3):255–60. doi: 10.1038/ni894. [DOI] [PubMed] [Google Scholar]
- 7.Ashkenazi A, Herbst RS. To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest. 2008 Jun;118(6):1979–90. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008 Oct;8(10):782–98. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 9.Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene. 2008 Oct 20;27(48):6207–15. doi: 10.1038/onc.2008.298. [DOI] [PubMed] [Google Scholar]
- 10.Lorenzo PI, Arnoldussen YJ, Saatcioglu F. Molecular mechanisms of apoptosis in prostate cancer. Crit Rev Oncog. 2007 Aug;13(1):1–38. doi: 10.1615/critrevoncog.v13.i1.10. [DOI] [PubMed] [Google Scholar]
- 11.Uzzo RG, Haas NB, Crispen PL, Kolenko VM. Mechanisms of apoptosis resistance and treatment strategies to overcome them in hormone-refractory prostate cancer. Cancer. 2008 Apr 15;112(8):1660–71. doi: 10.1002/cncr.23318. [DOI] [PubMed] [Google Scholar]
- 12.Chen CC, Lau LF. Functions and Mechanisms of Action of CCN Matricellular Proteins. Int J Biochem Cell Biol. 2008 doi: 10.1016/j.biocel.2008.07.025. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002 Oct;14(5):608–16. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 14.Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, product of a growth factor-inducible immediate-early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci U S A. 1998;95:6355–60. doi: 10.1073/pnas.95.11.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) Is Essential for Placental Development and Vascular Integrity. Mol Cell Biol. 2002 Dec;22(24):8709–20. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mo FE, Lau LF. The matricellular protein CCN1 is essential for cardiac development. Circulation Research. 2006;99:961–9. doi: 10.1161/01.RES.0000248426.35019.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menendez JA, Mehmi I, Griggs DW, Lupu R. International Congress on Hormonal Steroids and Hormones and Cancer: The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr Relat Cancer. 2003 Jun;10(2):141–52. doi: 10.1677/erc.0.0100141. [DOI] [PubMed] [Google Scholar]
- 18.O'Kelly J, Koeffler HP. Thr role of CCN1 in tumorigenesis and cancer progression. In: Perbal B, Takigawa M, editors. CCN proteins: a new family of cell growth and differentiation regulators. London: Imperial College Press; 2005. pp. 273–91. [Google Scholar]
- 19.Sakamoto S, Yokoyama M, Aoki M, Suzuki K, Kakehi Y, Saito Y. Induction and function of CYR61 (CCN1) in prostatic stromal and epithelial cells: CYR61 is required for prostatic cell proliferation. Prostate. 2004 Dec 1;61(4):305–17. doi: 10.1002/pros.20098. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu T, Okayama A, Inoue T, Takeda K. Analysis of gene expression during staurosporine-induced neuronal differentiation of human prostate cancer cells. Oncol Rep. 2005 Aug;14(2):441–8. [PubMed] [Google Scholar]
- 21.Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005 Oct 1;65(19):8887–95. doi: 10.1158/0008-5472.CAN-05-1702. [DOI] [PubMed] [Google Scholar]
- 22.Leu SJ, Chen N, Chen CC, Todorovic V, Bai T, Juric V, et al. Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J Biol Chem. 2004;279:44177–87. doi: 10.1074/jbc.M407850200. [DOI] [PubMed] [Google Scholar]
- 23.Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001 Mar 30;276:10443–52. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 24.Leers MP, Kolgen W, Bjorklund V, Bergman T, Tribbick G, Persson B, et al. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J Pathol. 1999 Apr;187(5):567–72. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 25.Schutte B, Henfling M, Kolgen W, Bouman M, Meex S, Leers MP, et al. Keratin 8/18 breakdown and reorganization during apoptosis. Exp Cell Res. 2004 Jul 1;297(1):11–26. doi: 10.1016/j.yexcr.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 26.Tong X, Li H. eNOS protects prostate cancer cells from TRAIL-induced apoptosis. Cancer Lett. 2004 Jul 8;210(1):63–71. doi: 10.1016/j.canlet.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 27.Lin SB, Wu LC, Huang SL, Hsu HL, Hsieh SH, Chi CW, et al. In vitro and in vivo suppression of growth of rat liver epithelial tumor cells by antisense oligonucleotide against protein kinase C-alpha. J Hepatol. 2000 Oct;33(4):601–8. doi: 10.1034/j.1600-0641.2000.033004601.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen N, Leu SJ, Todorovic V, Lam SCT, Lau LF. Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem. 2004;279:44166–76. doi: 10.1074/jbc.M406813200. [DOI] [PubMed] [Google Scholar]
- 29.Leu SJ, Lam SCT, Lau LF. Proangiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J Biol Chem. 2002;277:46248–55. doi: 10.1074/jbc.M209288200. [DOI] [PubMed] [Google Scholar]
- 30.Todorovic V, Chen CC, Hay N, Lau LF. The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol. 2005;171:559–68. doi: 10.1083/jcb.200504015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CC, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF. Cytotoxicity of TNFα is regulated by Integrin-Mediated Matrix Signaling. EMBO J. 2007;26:1257–67. doi: 10.1038/sj.emboj.7601596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu R, Mandlekar S, Ruben S, Ni J, Kong AN. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in androgen-independent prostate cancer cells. Cancer Res. 2000 May 1;60(9):2384–9. [PubMed] [Google Scholar]
- 33.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008 Oct 20;27(48):6194–206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 34.Leussink B, Brouwer A, el Khattabi M, Poelmann RE, Gittenberger-de Groot AC, Meijlink F. Expression patterns of the paired-related homeobox genes MHox/Prx1 and S8/Prx2 suggest roles in development of the heart and the forebrain. Mech Dev. 1995 Jul;52(1):51–64. doi: 10.1016/0925-4773(95)00389-i. [DOI] [PubMed] [Google Scholar]
- 35.Izeradjene K, Douglas L, Tillman DM, Delaney AB, Houghton JA. Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res. 2005 Aug 15;65(16):7436–45. doi: 10.1158/0008-5472.CAN-04-2628. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003 Sep 5;278(36):33753–62. doi: 10.1074/jbc.M303313200. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez-Guerrico AM, Kazanietz MG. Phorbol ester-induced apoptosis in prostate cancer cells via autocrine activation of the extrinsic apoptotic cascade: a key role for protein kinase C delta. J Biol Chem. 2005 Nov 25;280(47):38982–91. doi: 10.1074/jbc.M506767200. [DOI] [PubMed] [Google Scholar]
- 38.Couchman JR, Vogt S, Lim ST, Lim Y, Oh ES, Prestwich GD, et al. Regulation of inositol phospholipid binding and signaling through syndecan-4. J Biol Chem. 2002 Dec 20;277(51):49296–303. doi: 10.1074/jbc.M209679200. [DOI] [PubMed] [Google Scholar]
- 39.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005 Mar 18;96(5):488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 40.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002 Mar;3(3):221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 41.Woods A, Couchman JR. Syndecan-4 and focal adhesion function. Curr Opin Cell Biol. 2001 Oct;13(5):578–83. doi: 10.1016/s0955-0674(00)00254-4. [DOI] [PubMed] [Google Scholar]
- 42.Lahn M, Sundell K, Moore S. Targeting protein kinase C-alpha (PKC-alpha) in cancer with the phosphorothioate antisense oligonucleotide aprinocarsen. Ann N Y Acad Sci. 2003 Dec;1002:263–70. doi: 10.1196/annals.1281.029. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Yamashita M, Uetsuki H, Kakei Y. Localization of Cyr61 mRNA and Protein in Prostate Cancer. Japanese Journal of Urology. 2002;93(2):331. [Google Scholar]
- 44.Babic AM, Chen CC, Lau LF. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 1999;19:2958–66. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pilarsky CP, Schmidt U, Eissrich C, Stade J, Froschermaier SE, Haase M, et al. Expression of the extracellular matrix signaling molecule Cyr61 is downregulated in prostate cancer. Prostate. 1998 Jul 1;36(2):85–91. doi: 10.1002/(sici)1097-0045(19980701)36:2<85::aid-pros3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 46.Cretney E, Shanker A, Yagita H, Smyth MJ, Sayers TJ. TNF-related apoptosis-inducing ligand as a therapeutic agent in autoimmunity and cancer. Immunol Cell Biol. 2006 Feb;84(1):87–98. doi: 10.1111/j.1440-1711.2005.01413.x. [DOI] [PubMed] [Google Scholar]
- 47.Chen CC, Mo FE, Lau LF. The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J Biol Chem. 2001;276:47329–37. doi: 10.1074/jbc.M107666200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.