

Abstract
Many methods exist for reconstructing phylogenies from molecular sequence data, but few phylogenies are known and can be used to check their efficacy. Simulation remains the most important approach to testing the accuracy and robustness of phylogenetic inference methods. However, current simulation programs are limited, especially concerning realistic models for simulating insertions and deletions. We implement a portable and flexible application, named INDELible, for generating nucleotide, amino acid and codon sequence data by simulating insertions and deletions (indels) as well as substitutions. Indels are simulated under several models of indel-length distribution. The program implements a rich repertoire of substitution models, including the general unrestricted model and nonstationary nonhomogeneous models of nucleotide substitution, mixture, and partition models that account for heterogeneity among sites, and codon models that allow the nonsynonymous/synonymous substitution rate ratio to vary among sites and branches. With its many unique features, INDELible should be useful for evaluating the performance of many inference methods, including those for multiple sequence alignment, phylogenetic tree inference, and ancestral sequence, or genome reconstruction.
Keywords: indels, insertion, deletion, simulation, codon models, nonstationary process
Introduction
A variety of methods and computer programs are available for aligning multiple sequences, reconstructing phylogenetic trees, and estimating evolutionary parameters. Because true phylogenetic relationships are rarely known with certainty (cf. Hillis et al. 1992; Sousa et al. 2008), simulated data are used to investigate the accuracy and efficiency of phylogenetic reconstruction methods (e.g., Gaut and Lewis 1995; Huelsenbeck 1995), ancestral sequence reconstruction methods (e.g., Blanchette et al. 2004), or methods of sequence alignment (e.g., Nuin et al. 2006). They can also be used in parametric bootstrap analysis to calculate confidence intervals for parameter estimates or to estimate the null distribution for hypothesis testing (e.g., Goldman 1993). Simulation can also be used to examine the robustness of the analytical method to model misspecification, by simulating data under a complex model and analyzing them under a simplistic incorrect model (e.g., Lemmon and Moriarty 2004). When the simulation does not incorporate indels, there will be no need for sequence alignment and thus an important step that may contribute significantly to errors in inference is ignored.
However, existing programs for simulating molecular sequence evolution are often found lacking, especially concerning simulation of insertions and deletions. Two widely used programs, Seq-Gen (Rambaut and Grassly 1997) and Evolver (Yang 1997), do not include indels at all. Rose (Stoye et al. 1998) has an unrealistic model of indel formation and EvolveAGene (Hall 2008) is inflexible and allows the use of the spontaneous mutational spectrum of Escherichia coli only. Similarly, GSimulator (Varadarajan et al. 2008) does not use continuous branch lengths or implement commonly used substitution models; it must be “trained” before it can be used and only comes pretrained with estimates based on the Drosophila genome. DAWG (Cartwright 2005) cannot simulate amino acid or codon sequences, whereas SIMPROT (Pang et al. 2005) and indel-Seq-Gen (Strope et al. 2007) cannot simulate nucleotide or codon sequences. Evolver (Yang 1997) is the only program that can simulate under codon models, whereas only MySSP (Rosenberg 2005) can simulate under nonstationary and nonhomogenous models. Thus, we have developed INDELible to fill those gaps and to provide a flexible and powerful tool for simulating molecular sequence evolution.
Material and Methods
Outline of the Simulation Algorithm
The main difficulty in dealing with insertions and deletions, especially in developing a likelihood model for inference (e.g., Bishop and Thompson 1986; Thorne et al. 1991), lies in the lack of independence of data among sites in the sequence. However, if we view the entire sequence (instead of one nucleotide, amino acid, or codon in the sequence) as the unit of evolution, the change from one sequence to another is described by a Markov chain, with the whole sequence being the state of the chain. Thus, sequence evolution through insertions and deletions as well as substitutions can be simulated by using the standard algorithm for simulating Markov chains, that is, by generating exponentially-distributed waiting times and sampling from the jump chain (Yang 2006, pp. 303–304). This is also known as Gillespie's algorithm (Gillespie 1977).
Consider the simulation of evolution of a sequence along a branch on the phylogeny, with the sequence at the start of the branch as well as the branch length (t) given. Let λ = I + D + S be the total event rate for the current sequence, with I, D, and S to be the total insertion, deletion, and substitution rates, respectively. We generate the waiting time s1 until the next event by sampling from the exponential distribution with mean 1/λ. If s1 > t, no event occurs before the end of the branch. Otherwise an event occurs at time s1, and it is randomly drawn to be an insertion, deletion, or substitution with probabilities I/λ, D/λ, or S/λ, respectively. The location of the event is similarly determined by random sampling with probabilities proportional to the rates. If the event is an insertion or deletion (indel), the location is drawn uniformly from the pool of all possibilities, whereas the length of the indel is drawn from the indel-length distribution (see below). If the event is a substitution, a site is chosen at random with the probability proportional to the substitution rate at the site, and the new state at the site is chosen using the transition matrix of the jump chain J (see below). Thus, the new sequence at time s1 is generated, and the sequence length L and the rates for the new sequence are updated. The time remaining for the branch (= t – s1) is calculated. We then generate the next waiting time s2 based on the rate for the current sequence. The procedure is repeated until the end of the branch is reached, that is, until s1 + s2 + ⋯ > t.
Ideally the sequence length L at the root should be sampled from the distribution of sequence lengths implied by the model of insertions and deletions (Thorne et al. 1991). However, sampling from this distribution is complicated because of the arbitrary nature of the indel-size distribution accepted by INDELible. Instead, we require L to be specified by the user. The sequence at the root is then generated by sampling L characters (nucleotides, amino acids, or codons) at random from the equilibrium distribution under the substitution model at the root. For models of rate heterogeneity among sites, the rates at sites are generated from the rate distribution. The Gillespie algorithm is then used to simulate the evolution of the sequence from the root along the branches toward the tips of the tree. Sequences at the tips of the tree constitute a replicate data set.
The models we have implemented assume that the insertion and deletion rates are constant among sites in the sequence. As a result, the substitution process is independent of insertions and deletions, and substitutions can be simulated separately from insertions and deletions. Thus, an alternative procedure is to use the Gillespie algorithm to simulate indels only, with substitutions simulated afterward by sampling from the transition probability matrix for the branch (Yang 2006, p. 303). This is the method used by Cartwright (2005), and will be referred to in this paper as method 1. The method described above, of simulating waiting times for substitutions as well as insertions and deletions, is referred to as method 2. For most models, method 1 is more efficient than method 2 but the opposite is true for models of continuous rate variation among sites. Method 2, however, provides a way of simulating sequences under more complex models in which the insertion and deletion rates may depend on the local sequence context and vary along the sequence (see Discussion).
Simulation of Substitutions
Substitutions are assumed to be independent among sites, and are described by a continuous-time Markov chain, characterized by the matrix of instantaneous rates
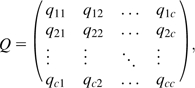 |
(1) |
where the number of characters c is equal to 4, 20, and 64 for nucleotides, amino acids, and codons, respectively. The off-diagonal elements of the matrix are specified by the model, whereas the diagonal elements are defined as . Rate matrices are rescaled by INDELible such that the branch lengths represent the expected number of substitutions per site (or the average expected number of substitutions per site under a heterogeneous-sites model).
Method 1 requires the transition probability matrix for a branch of length t. For reversible models, this is calculated by numerical computation of the eigenvalues and eigenvectors of Q (Yang 1995), whereas for nonreversible models, it is calculated by repeated matrix squaring (Yang 2006, pp 68–70).
Method 2 requires the calculation of substitution rates at individual sites. Given Q, the rate “away” from state i is qi = −qii. The total substitution rate for the entire sequence is thus where i(k) is the state at site k and rk is the relative rate at site k. Given that a substitution occurs at site k, the resulting state is sampled using the transition matrix of the jump chain, , where mij = qij/qi if i ≠ j and mij = 0 otherwise (Yang 2006, eq. 9.7). In other words, if the site is currently in state i, the probability that the new state is j is simply mij.
Nucleotide Substitution Models
The most general model of nucleotide substitution places no constraint on the rate matrix Q. This is the UNREST model of Yang (1994a), and in INDELible is specified by using 11 relative rate parameters (the off-diagonal elements of the rate matrix Q). The equilibrium frequencies (πi) are then calculated by solving the system of simultaneous equations for all j, subject to the constraint (e.g., Yang 2006, p. 32). Note that this model is often described and implemented incorrectly in the literature (e.g., Swofford et al. 1996).
INDELible also includes the general time-reversible model (GTR or REV, Tavaré 1984; Yang 1994a) and many commonly used models that are its special cases, such as JC69 (Jukes and Cantor 1969), K80 (Kimura 1980), K81 (Kimura 1981), F81 (Felsenstein 1981), F84 (Felsenstein, DNAML program since 1984, PHYLIP Version 2.6), HKY85 (Hasegawa et al. 1984, 1985), T92 (Tamura 1992), and TN93 (Tamura and Nei 1993). The rates under GTR can be written as qij = sijπj, with sij = sji, where sij is also known as the exchangeability between i and j (Whelan and Goldman 2004). Thus, GTR is specified using the exchangeability parameters sij and the nucleotide frequencies πj.
Amino Acid Substitution Models
INDELible currently incorporates 15 empirical amino acid substitution models, derived from analysis of protein alignments from a variety of sources (table 2). All of those models are time reversible and are specified using the amino acid exchangeabilities sij and the stationary amino acid frequencies πj (see the description above). It is also possible for the user to supply a time-reversible substitution rate matrix. INDELible also implements the Poisson model of protein evolution, which assumes that the substitution rates between any two amino acids are the same.
Table 2.
Empirical Amino Acid Substitution Models Implemented in INDELible
| Model | Source of Alignment | Reference |
| DAYHOFF | Nuclear proteins | Dayhoff et al. (1978) |
| JTT | Nuclear proteins | Jones et al. (1992) |
| WAG | Nuclear proteins | Whelan and Goldman (2001) |
| VT | Nuclear proteins | Müller and Vingron (2000) |
| DAYHOFF (DCMUT) | Nuclear proteins | Kosiol and Goldman (2005) |
| JTT (DCMUT) | Nuclear proteins | Kosiol and Goldman (2005) |
| LG | Nuclear proteins | Le and Gascuel (2008) |
| BLOSUM62 | Nuclear proteins | Henikoff and Henikoff (1992) |
| MTMAM | Mammalian mitochondrial proteins | Yang et al. (1998) |
| mtREV | Vertebrate mitochondrial proteins | Adachi and Hasegawa (1996) |
| MtArt | Arthropod mitochondrial proteins | Abascal et al. (2007) |
| CpREV | Chloroplast proteins | Adachi et al. (2000) |
| RtREV | Viral reverse transcriptase proteins | Dimmic et al. (2002) |
| HIVb and HIVw | HIV-1 viral genes | Nickle et al. (2007) |
Among-Site Heterogeneity
INDELible incorporates a number of random-sites models for simulating rate heterogeneity among sites in a sequence. Under these models, the relative rates are independent and identically distributed among sites, and unless a nonhomogeneous process is being simulated, the relative rate at each site is held constant throughout the simulation with daughter sites inheriting the rate of their parent. (Under nonhomogeneous models, different branches may have different models, and thus the rate for a site may change as a result of the changed model.) For nucleotide and amino acid simulations, variable substitution rates among sites can be simulated using any of the following models: 1) a constant rate for all sites, 2) a proportion of invariable sites plus a constant rate for all other sites (+I, Hasegawa et al. 1985), 3) a continuous or discrete-gamma distribution of rates among sites (the “+Γ” and “+Γ5” models) (Yang 1993; 1994b), and 4) a proportion of invariable sites plus gamma-distributed rates for other sites (“+I + Γ” and “+I + Γ5” models) (Gu et al. 1995).
Codon Substitution Models
For codon models, the state space consists of the sense codons of the genetic code, for example, 61 sense codons for the universal code and 60 for the vertebrate mitochondrial code. Because stop codons are not allowed inside a functional protein, they are not considered in the chain. INDELible currently supports 17 genetic codes: codes 1–6, 9–16, and 21–23 listed in GenBank. The basic codon model specifies the instantaneous rate of substitution from codon i to j as
 |
(2) |
where κ is the transition–transversion rate ratio, ω is the nonsynonymous–synonymous rate ratio, and πj is the equilibrium frequency of codon j (Goldman and Yang 1994; Yang and Nielsen 1998). INDELible also allows the use of two empirical codon models (ECMs, Kosiol et al. 2007). The first (ECMrest) was constructed under the assumption that only one codon position can change instantaneously, as in equation (2). The second (ECMunrest) was constructed allowing instantaneous doublet and triplet changes as well.
Several advanced models of codon substitution are implemented, which allow the selective pressure on the protein-coding gene, measured by the nonsynonymous–synonymous rate ratio ω, to vary among sites (codons) in the gene, among branches in the tree, or among both sites and branches (see Anisimova and Kosiol 2009 for a recent review). The site models allow ω to vary among sites (Nielsen and Yang 1998; Yang et al. 2000). All the site models are special cases of model M3 (discrete), which assumes a general discrete distribution for ω (Yang et al. 2000). This is implemented in INDELIble by specifying the number of site classes, and the proportions and ω ratios for the site classes. A small script is included with INDELible, which calculates the discrete ω values from the parameters under models M4–M13 of Yang et al. (2000).
The branch models (Yang 1998) and branch-site models (Yang and Nielsen 2002; Yang et al. 2005; Zhang et al. 2005) are implemented in INDELible as well. The latter allows the ω ratio to vary both among branches and among sites. Although the branch-site model described by Yang et al. (2005) allows only two types of branches (the foreground and background branches) and four site classes, INDELible allows an arbitrary number of site classes and branch types.
Those codon models are widely used in likelihood ratio tests of natural selection affecting the evolution of protein-coding genes. Implementation of those models in INDELible makes it possible for the first time to evaluate the impact of alignment errors and of insertions and deletions on the robustness of those methods.
Nonstationary and Nonhomogeneous Processes
Most models currently used in phylogenetic analysis assume homogeneity and stationarity of the substitution process across the whole tree, that is, substitutions occur according to the same rate matrix Q, and nucleotide, amino acid or codon frequencies have remained more or less constant during the course of evolution. Sequences from distantly related species are often noted to have different nucleotide or amino acid frequencies, which is a clear indication of violation of those assumptions. Few attempts have been made to implement nonhomogeneous models (Yang and Roberts 1995; Galtier and Gouy 1998; Blanquart and Lartillot 2006) for phylogenetic inference. Therefore, data simulated under nonstationary and nonhomogeneous conditions should be useful for testing the robustness of phylogenetic reconstruction methods.
The branch and branch-site models of codon substitution mentioned above may be considered examples of nonhomogeneous models, in which the ω ratio and thus the rate matrix Q vary among branches. INDELible allows any parameter or any aspect of the evolutionary model to change along branches in the tree. Each branch may have its own insertion–deletion rates and size distributions, equilibrium frequencies, or level of rate heterogeneity among sites. Parameters are also allowed to change at arbitrary points within a branch; this is achieved by specifying a tree with an internal node having only one daughter branch.
Simulation of Insertions and Deletions
Indel Formation
INDELible treats insertions and deletions as separate processes, each with its own instantaneous rate and its own size distribution. The model assumes that insertions and deletions occur at the fixed rates λI and λD, respectively, at every site in the sequence. We define one time unit as one expected substitution per site, so that λI and λD are the expected numbers of indels per substitution. In simulation under codon models, a site refers to a codon, and indels of whole codons only are allowed.
Insertions are relatively simple to simulate. A sequence with L sites has L + 1 possible positions for insertion (including both ends of the sequence). The total rate of insertions is thus I = λI (L + 1). Insertions at the two ends of the sequence are allowed, and the sequence has an “immortal link” at the beginning (Thorne et al. 1991). When an insertion occurs, the insertion-size distribution is used to generate the size of the insertion (u). Then, u characters (nucleotides, amino acids, or codons) are generated by sampling at random from the equilibrium distribution of the substitution model to form the sequence to be inserted. For site-heterogeneous models, the rates for the u sites are generated by sampling from the rate distribution.
Deletions are more complex to simulate as one has to make somewhat arbitrary decisions concerning deletions at the ends of the sequence. We follow the procedure of Cartwright (2005) and consider that the simulated sequence, of length L, lies within a larger sequence, of length N, with N ≫ L. Let the maximum deletion length be M, with M ≪ N. A deletion of size u in the larger sequence will delete some of the smaller sequence if it occurs at any of the L sites of the smaller sequence or any of the u − 1 sites preceding the smaller sequence. As deletions are assumed to occur uniformly in the larger sequence, the probability that a deletion of size u in the larger sequence deletes some sites in the smaller sequence is (u − 1 + L)/N. Thus, the probability that a deletion in the larger sequence deletes some sites in the smaller sequence is , where is the mean deletion size (Cartwright 2005). The total rate of deletion in the larger sequence is NλD where λD is the rate of deletion per site, so that the total rate of deletion in the smaller sequence is . This is independent of N.
Indel-Size Distributions
INDELible uses two separate distributions to model the sizes of insertions and deletions. For simplicity, here, we use indel-size distribution to refer to both. Several indel-size distributions are implemented in INDELible.
The first is the negative binomial distribution, by which the probability that the indel has size u is
| (3) |
where the parameters are the integer r and probability q. This distribution has mean and variance rq/(1 − q)2. If r = 1, the distribution reduces to the geometric distribution.
The second model is the Zipfian distribution or a power law, by which indel length u has probability
| (4) |
where a > 1 is a parameter of the distribution and is the Riemann Zeta function. This distribution has a very heavy tail, and the mean is infinite if a < 2 and the variance is infinite if a < 3. If a > 2, the mean is , and if a > 3, the variance is . Empirical estimates of a range from 1.5 to 2, with infinite variance (Benner et al. 1993; Gu and Li 1995; Zhang and Gerstein 2003; Chang and Benner 2004; Yamane et al. 2006; Cartwright 2009). There is evidence that parameter a, which is inversely related to indel size, differs for insertions and deletions (Gu and Li 1995; Zhang and Gerstein 2003), so the ability of INDELible to allow different length distributions for insertions and deletions may be useful.
The third model is the Lavalette distribution, by which the probability for size u is
| (5) |
where a is a parameter and M is the maximum indel size (Lavalette 1996; Popescu et al. 1997; Popescu 2003). The proportionality constant is determined such that the probabilities sum to 1. This model was first proposed to explain the distribution of journal impact factors. It has two desirable features. First, the mean and variance are finite because of the maximum length M. Second, it can approximate the Zipf distribution arbitrarily well by the use of a large M. This is because, apart from the normalizing constants, the two distributions differ only by the factor ϕ = [M/(M − u + 1)]−a, which is ≈1 when M ≫ 1. Figure 1 shows the distribution for a few different values of M.
FIG. 1.—

The Lavalette distribution of indel length plotted for different values of the maximum indel length M, with a = 0.5 fixed (see eq. 5). Note that u can take integer values 1, 2, … , M only.
Besides the three models above, INDELible also allows the user to define an indel-size distribution.
A number of authors have attempted to estimate empirical indel-size distributions. Gu and Li (1995) suggested that the power-law model fitted the data much better than the geometric model, which was found to be inadequate. Many other studies also found that the power law fitted a variety of data sets reasonably well (Benner et al. 1993; Zhang and Gerstein 2003; Chang and Benner 2004; Yamane et al. 2006). Qian and Goldstein (2001) used a mixture of four exponential distributions to describe indel lengths, which was adapted into a distance-dependent indel-length distribution for use in the simulation program SIMPROT (Pang et al. 2005). This distribution appears to be more complicated than necessary.
Program Validation
We conducted extensive simulations to confirm the validity of the simulation program. To validate the implementation of the substitution model, we simulated larger and larger data sets (with 106 or 107 sites, say) and analyzed them under the same model using BASEML and CODEML in the PAML package (Yang 1997), to confirm that the parameter estimates are close to the true values, relying on the consistency of maximum likelihood estimates. It is more difficult to validate our simulation under the models of insertions and deletions, as correct analytical results are lacking. We compared the observed indel-size distribution in the simulated data sets with the true distribution and found that they matched each other closely. We simulated data sets on trees of 2, 8, or 40 taxa with insertions only, with deletions only, and with both insertions and deletions, using many different rates, parameters and length distributions. The proportions of columns in the true alignment that have 0, 1, 2, … gaps were calculated and compared with the correct proportions generated using a small simulation program that keeps track of the sequence lengths only. In all combinations investigated, there was good agreement between the two.
Our extensive comparison with DAWG revealed a few problems with DAWG version 1.1.2 and earlier. For example, two biological mechanisms can generate columns with all gaps in the true alignment: 1) deleted insertions, that is, deletion of part of an earlier insertion on the same branch, and 2) parallel deletions, that is, deletion of the same nucleotides along different lineages. DAWG keeps track of 2) but not of 1). Furthermore, the true alignment produced by DAWG may be incorrect with nucleotides from parallel insertions misaligned. Those bugs will be fixed in a new release of the program (Cartwright R, personal communication).
Results
The simulation program that is most similar to INDELible is DAWG (Cartwright 2005). Although DAWG does not have some of the advanced features of INDELible, it is possible to simulate data under the same nucleotide-substitution models to make a fair comparison. Thus, we conducted a computer simulation to examine the computational efficiency of the two simulation programs. Sequence data were simulated under the HKY model, with κ = 2 and base frequencies 0.4 (T), 0.3 (C), 0.2 (A), and 0.1 (G). In the basic model, we set the insertion and deletion rates to λI = λD = 0.1 per substitution, with the indel length following a negative binomial distribution with r = 1 and q = 0.25 (the geometric distribution). The phylogenetic tree was symmetric with 32 taxa, with all branch lengths set to 0.1 substitutions per site. Substitution rates over sites were either constant or follow the gamma distribution with shape parameter α = 1. The number of replicate data sets is 100. We then explored several variations of the basic simulation scheme to examine the impact of various factors on the simulation efficiency, such as the number of taxa, the insertion–deletion rate ratio λI/λD, the amount of evolution measured by the branch length, the average indel length, and the sequence length at the root. INDELible (methods 1 and 2) and DAWG were used to generate the data. The results are shown in figure 2.
FIG. 2.—

Speed comparison between DAWG and INDELible, with and without continuous gamma rate heterogeneity. The basic simulation model is specified by the settings in figure 3, whereas one factor is varied to see its impact in each plot. INDELible1 and INDELible2 refer to INDELible simulation under methods 1 and 2, respectively. The tests were carried out on a SunFire Opteron X4600M2 server running Linux.
DAWG is faster than INDELible in simple circumstances, such as simulating short sequences with low insertion rate and small insertions on small trees with few taxa and short branches. However, with the increase in the complexity of the simulation, the time taken by DAWG increases much faster than by INDELible. The exception to this pattern is simulation using INDELible method 2, which is sensitive to the average branch length as longer branches mean simulation of more rounds of exponential-waiting times in the algorithm. However, method 2 has a speed advantage over method 1 and DAWG for simulation under the continuous gamma model of variable rates among sites. Under this model, every site has a distinct rate, so that the transition probability matrix P(t) needs to be calculated for every site on every branch. In contrast, the transition matrix of the jump chain (M in method 2) is the same for all sites and does not need to be calculated for every site, leading to an increase in computational efficiency.
Speed differences between INDELible and DAWG are largely a matter of programing design. Both programs are written in C++, and both programs store sequence information in the vector container from the standard template library. INDELible implements insertions via a modified lookup table whose execution time is mostly independent of the complexity of the simulation but can be slow in very simple simulations. In contrast, DAWG implements insertions via the C++ function vector::insert, the speed of which is proportional to the number of elements inserted (copying) plus the number of elements between the insertion position and the end of the vector (moving).
Discussion
Features of INDELible
INDELible is driven by a control data file (fig. 3). The program is designed to be flexible, and a wide range of options can be specified to control different aspects of the simulation, including the substitution model, indel models and indel-size distributions, heterogeneous-rates model, and the underlying phylogeny. The tree with branch lengths (measured by the expected number of substitutions per site) may be specified by the user or created at random from the birth–death process with species sampling (Yang and Rannala 1997). No constraints are placed on the size and structure of the tree, the sequence length, or the values of model parameters.
FIG. 3.—

An example input file for INDELible. The substitution model has been set to HKY + Γ with a transition–transversion rate ratio of κ = 2, stationary base frequencies of 0.4 (T), 0.3 (C), 0.2 (A), and 0.1 (G), and continuous gamma rate variation with shape parameter α = 1. Insertions and deletions have both been set to have an instantaneous rate of 0.1 (relative to an average substitution rate of 1) and the same geometric length distribution with a mean length of 4. Then, the phylogeny with branch lengths is specified. In the simulations for the speed tests, a 32-taxa, symmetric, strictly bifurcating tree with all branch lengths equal to 0.1 is used instead. This simulation creates 100 replicate data sets each containing one partition with a randomly created root sequence of 1,000 bases.
INDELible also offers the ability to simulate data in multiple partitions where different partitions may have different substitution models, indel lengths, or heterogeneous rate distributions and may evolve on different trees (e.g., to simulate gene-tree/species-tree conflict). Deletions are not allowed to span different partitions; different partitions must have the same data type (nucleotide, amino acid, or codon); and the tree must have the same number of leaves. Apart from those restrictions, every other parameter or setting is allowed to vary between partitions. The history of insertions and deletions is maintained during the course of the simulation. Inserted bases/residues are stored in separate memory containers to those in the original sequence at the root, and deletions are not removed from the computer memory but are simply marked as deletions and ignored during the remainder of the simulation. Thus, at the end of the simulation, sites are recognizable as either core sites that evolved from the root, deleted core sites, insertions, or deleted insertions, and the true alignment can be assembled and output easily. INDELible also offers the option to print inserted residues in lowercase and print core residues that evolved from the root in uppercase, and codon sequences can also be translated into amino acid sequences for output.
A summary of features of INDELible in comparison with other simulation programs is provided in table 1. INDELible is unique in its implementation of codon models and nonstationary and nonhomogeneous models among programs of indel simulation.
Table 1.
Comparison of Simulation Programs
| Feature | Seq-Gen v1.3.2 | Evolver v4 | Rose v1.3 | DAWG v1.1.2 | MySSP v1.0 | Indel-Seq-Gen v1.0.3 | EvolveAGene v3 | GSimulatior v1.1 | SIMPROT v1.01 | INDELible v1.0 |
| GTR | x | x | x | x | x | |||||
| UNREST | x | |||||||||
| Empirical amino acid models | 6 | 10a | 3 | 3 | 15a | |||||
| ECMs | 2 | |||||||||
| Codon “site” model | x | x | ||||||||
| Codon “branch” model | x | x | ||||||||
| Codon “branch-site” model | x | x | ||||||||
| Non-stationary models | x | x | ||||||||
| Discrete gamma | x | x | x | |||||||
| Continuous gamma | x | x | x | x | x | x | ||||
| Proportion of invariant sites | x | x | x | x | ||||||
| Indels | x | x | x | x | x | x | x | x | ||
| Ancestral sequences | x | x | x | x | x | x | x | x | x | |
| Batch mode | x | x | x | x | ||||||
| Multi-gene mode | x | x | x | x | x | |||||
| Platform | ||||||||||
| Unix | x | x | x | x | x | x | x | x | x | |
| Mac OS X | x | x | x | x | x | x | x | |||
| Win32 | x | x | x | x | x | x | x | |||
Evolver and INDELible can also use user-defined amino acid substitution models.
Correct Simulation under a Model and Biological Realism
We consider it important for an indel-simulation program to simulate data correctly under a model of insertions, deletions, and substitutions, that is, to generate data sets with the correct probability distribution under such a model. Most existing indel-simulation programs do not appear to have achieved this goal, as they often involve somewhat arbitrary manipulations of the simulation process that cannot be justified under any model. Those manipulations were often claimed to improve the biological realism of the generated data. One common mistake is to fix the sequence at the root of the tree to be a real sequence rather than generating a sequence at random. In a model of insertions, deletions and substitutions, the sequence at the root is a random realization of the model and should be allowed to vary among data sets.
Although it is important for the simulation to represent real-data scenarios, this goal should be achieved by using representative values of parameters in the model, such as substitution rates, base or amino acid frequencies, sequence length, the size and shape of the tree, etc. Most parameters (such as substitution rates, stationary frequencies, or heterogeneous rate distributions) are easily estimated via maximum likelihood using standard phylogenetic software (e.g., PAML: Yang 1997), but parameters for indel formation and indel-length distributions are more of a problem. INDELible is a simulation program and does not include methods for estimating model parameters from the real data, which is the remit of an inference tool. A number of studies have produced estimates of the insertion and deletion rates (λI and λD) relative to the substitution rate (λS), with λS/(λI + λD) estimated to be around 13–15 (Silva and Kondrashov 2002; Britten et al. 2003; Ogurtsov et al. 2004). Estimates also suggest that deletions occur more often than insertions, with λD/λI ranging from 1.3 to 4 (Gu and Li 1995; Zhang and Gerstein 2003; Arndt and Hwa 2004), although Mills et al. (2006) estimated λD/λI ≈ 1 in a comparison of human and chimpanzee genomes. Thus, the ability of INDELible to specify separate insertion and deletion rates (λI, λD) and separate insertion and deletion size distributions, and to permit those parameters to change on the tree, may be important for realistic simulation of molecular sequence evolution.
Extending the Evolutionary Model
INDELible could be improved upon in a number of ways, by incorporating important features of sequence or genome evolution. Indeed, the current version of INDELible is mainly aimed at generating sequences suitable for phylogenetic comparisons and does not include models of genome rearrangements such as duplication, inversion, and translocation. To evaluate methods that attempt to reconstruct ancestral genomes (Blanchette et al. 2004), it may be important to simulate such large-scale events. Also, repetitive elements appear to have very high insertion and deletion rates. The ALU sequence in humans is about 300 bp long and recurs 300,000 times throughout the DNA. This causes a spike in the observed indel-size distribution around ≈300 bp when the human genome is compared with other genomes (Kent et al. 2003). Even shorter sequences may be repeated as many as 106 times. Such repetitive sequences create indel hotspots and clearly violate the assumption of uniform insertion–deletion rates.
Similarly, substitution or mutation rate is known to depend on the local sequence context. The most dramatic instance of such context effect is found in the so-called CpG dinucleotide “hotspots” (e.g., Ehrlich and Wang 1981). Codon models consider the context effect to some extent by accounting for dependence between positions of the codon triplet but cannot deal with context effects across codon boundaries (Pedersen et al. 1998; Siepel and Haussler 2004). There is also evidence that rates of substitutions, insertions, and deletions are positively correlated, so that genomic regions with high substitution rates also show high insertion and deletion rates (Waterston et al. 2002).
It should be straightforward to extend INDELible to simulate genome-rearrangement events, to accommodate insertions and deletions of repetitive elements, substitutional context effects, or correlated substitution and indel rates, as long as precise models for those processes can be formulated. Note that simulation of the evolutionary process by Gillespie's algorithm (INDELible method 2 but not method 1 or DAWG) is possible as long as one can generate the sequence at the root of the tree and calculate the instantaneous rates; there is no need for matrix-exponential solutions to the transition probabilities, contra Varadarajan et al. (2008). Even with dependence among sites in the sequence, the evolution from one sequence to another is described by a Markov chain, the instantaneous rates of various events are easy to calculate and thus it should be straightforward to simulate the process. Nevertheless, such processes are currently poorly understood, and lack of suitable inference tools to analyze real data makes it difficult to obtain reliable parameter estimates under such models.
Implementation Details and Program Availability
INDELible is written in standard ANSI C++ and tested on Windows, Mac OS X, and Linux systems. Precompiled executables are provided for Windows and Mac OS X, whereas the C++ source code is provided for compilation on UNIX systems. The program is distributed free of charge for academic use at the web site http://abacus.gene.ucl.ac.uk/software/indelible/.
Acknowledgments
We thank three anonymous referees for suggestions, which led to improvement of the manuscript. We thank Reed Cartwright for promptly answering our queries about DAWG. W.F. is financially supported by an EPSRC/MRC Doctoral Training Centre studentship and Z.Y. is funded by a grant from the BBSRC.
References
- Abascal F, Posada D, Zardoya R. MtArt: a new Model of amino acid replacement for Arthropoda. Mol Biol Evol. 2007;24:1–5. doi: 10.1093/molbev/msl136. [DOI] [PubMed] [Google Scholar]
- Adachi J, Hasegawa M. MOLPHY version 2.3: programs for molecular phylogenetics based on maximum likelihood. Comput Sci Monogr. 1996;28:1–150. [Google Scholar]
- Adachi J, Waddell PJ, Martin W, Hasegawa M. Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J Mol Evol. 2000;50:348–358. doi: 10.1007/s002399910038. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- Arndt PF, Hwa T. Regional and time-resolved mutation patterns of the human genome. Bioinformatics. 2004;20:1482–1485. doi: 10.1093/bioinformatics/bth105. [DOI] [PubMed] [Google Scholar]
- Benner SA, Cohen MA, Gonnet GH. Empirical and structural models for insertions and deletions in the divergent evolution of proteins. J Mol Biol. 1993;229:1065–1082. doi: 10.1006/jmbi.1993.1105. [DOI] [PubMed] [Google Scholar]
- Bishop MJ, Thompson EA. Maximum likelihood alignment of DNA sequences. J Mol Biol. 1986;190:159–165. doi: 10.1016/0022-2836(86)90289-5. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquart S, Lartillot N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol Biol Evol. 2006;23:2058–2071. doi: 10.1093/molbev/msl091. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Rowen L, Williams J, Cameron RA. Majority of divergence between closely related DNA samples is due to indels. Proc Natl Acad Sci USA. 2003;100:4661–4665. doi: 10.1073/pnas.0330964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright RA. DNA assembly with gaps (Dawg): simulating sequence evolution. Bioinformatics. 2005;21(iii):31–38. doi: 10.1093/bioinformatics/bti1200. [DOI] [PubMed] [Google Scholar]
- Cartwright RA. Problems and solutions for estimating indel rates and length distributions. Mol Biol Evol. 2009;26:473–480. doi: 10.1093/molbev/msn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MSS, Benner SA. Empirical analysis of protein insertions and deletions determining parameters for the correct placement of gaps in protein sequence alignments. J Mol Biol. 2004;341:617–631. doi: 10.1016/j.jmb.2004.05.045. [DOI] [PubMed] [Google Scholar]
- Dayhoff MO, Schwartz RM, Orcutt BC. 1978. A model of evolutionary change in proteins. Pp. 345–352. Atlas of protein sequence and structure. Vol 5, Suppl. 3. National Biomedical Research Foundation, Washington (DC). [Google Scholar]
- Dimmic MW, Rest JS, Mindell DP, Goldstein RA. RArtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J Mol Evol. 2002;55:65–73. doi: 10.1007/s00239-001-2304-y. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science. 1981;212:1350–1357. doi: 10.1126/science.6262918. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Galtier N, Gouy M. Inferring pattern and process: maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol Biol Evol. 1998;15:871–879. doi: 10.1093/oxfordjournals.molbev.a025991. [DOI] [PubMed] [Google Scholar]
- Gaut BS, Lewis PO. Success of maximum likelihood phylogeny inference in the four-taxon case. Mol Biol Evol. 1995;12:152–162. doi: 10.1093/oxfordjournals.molbev.a040183. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- Goldman N. Statistical tests of models of DNA substitution. J Mol Evol. 1993;36:182–198. doi: 10.1007/BF00166252. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Gu X, Fu YX, Li WH. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol Biol Evol. 1995;12:546–557. doi: 10.1093/oxfordjournals.molbev.a040235. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH. The size distribution of insertions and deletions in human and rodent pseudogenes suggests the logarithmic gap penalty for sequence alignment. J Mol Evol. 1995;40:464–473. doi: 10.1007/BF00164032. [DOI] [PubMed] [Google Scholar]
- Hall BG. EvolveAGene 3: a DNA coding sequence evolution simulation program. Mol Biol Evol. 2008;25:688–695. doi: 10.1093/molbev/msn008. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Yano T, Kishino H. A new molecular clock of mitochondrial DNA and the evolution of Hominoids. Proc Japan Acad B. 1984;60:95–98. [Google Scholar]
- Henikoff S, Henikoff J. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis DM, Bull JJ, White ME, Badgett MR, Molineux IJ. Experimental phylogenetics: generation of a known phylogeny. Science. 1992;255:589–592. doi: 10.1126/science.1736360. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP. The performance of phylogenetic methods in simulation. Syst Biol. 1995;44:17–48. [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. CABIOS. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. 1969. Evolution of protein molecules. [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci USA. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Kimura M. Estimation of evolutionary distances between homologous nucleotide sequences. Proc Natl Acad Sci USA. 1981;78:454–458. doi: 10.1073/pnas.78.1.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C, Goldman N. Different versions of the Dayhoff rate matrix. Mol Biol Evol. 2005;22:193–199. doi: 10.1093/molbev/msi005. [DOI] [PubMed] [Google Scholar]
- Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24:1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- Lavalette D. Facteur d'impact: impartialité ou impuissance? Orsay (France): Institut Curie—Recherche, Bât. 112, Centre Universitaire, 91405; 1996. [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Lemmon AR, Moriarty EC. The importance of proper model assumption in Bayesian phylogenetics. Syst Biol. 2004;53:265–277. doi: 10.1080/10635150490423520. [DOI] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Vingron M. Modeling amino acid replacement. J Comput Biol. 2000;7:761–776. doi: 10.1089/10665270050514918. [DOI] [PubMed] [Google Scholar]
- Nickle DC, Heath L, Jensen MA, Gilbert PB, Mullins JI, Kosakovsky Pond SL. HIV-specific probabilistic models of protein evolution. PLoS ONE. 2007;2:e503. doi: 10.1371/journal.pone.0000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuin PAS, Wang Z, Tillier ERM. The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinform. 2006;24:471. doi: 10.1186/1471-2105-7-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogurtsov AY, Sunyaev S, Kondrashov AS. Indel-based evolutionary distance and mouse-human divergence. Genome Res. 2004;14:1610–1616. doi: 10.1101/gr.2450504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang A, Smith AD, Nuin PAS, Tillier ERM. SIMPROT: using an empirically determined indel distribution in simulations of protein evolution. BMC Bioinform. 2005;27:236. doi: 10.1186/1471-2105-6-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen A-MK, Wiuf C, Christiansen FB. A codon-based model designed to describe lentiviral evolution. Mol Biol Evol. 1998;15:1069–1081. doi: 10.1093/oxfordjournals.molbev.a026006. [DOI] [PubMed] [Google Scholar]
- Popescu I-I. On a Zipf's law extension to impact factors. Glottometrics. 2003;6:83–93. [Google Scholar]
- Popescu I-I, Ganciu M, Penache MC, Penache D. On the Lavalette ranking law. Romanian Rep Phys. 1997;49:3–27. [Google Scholar]
- Qian B, Goldstein RA. Distribution of indel lengths. Proteins: structure, Function. Genetics. 2001;45:102–104. doi: 10.1002/prot.1129. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Grassly NC. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. CABIOS. 1997;13:235–238. doi: 10.1093/bioinformatics/13.3.235. [DOI] [PubMed] [Google Scholar]
- Rosenberg MS. MySSP: non-stationary evolutionary sequence simulation, including indels. Evol Bioinf. 2005;1:81–83. [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Silva JC, Kondrashov AS. Patterns in spontaneous mutation revealed by human–baboon sequence comparison. Trends Genet. 2002;18:544–547. doi: 10.1016/s0168-9525(02)02757-9. [DOI] [PubMed] [Google Scholar]
- Sousa A, Zé-Zé L, Silva P, Tenreiro R. Exploring tree-building methods and distinct molecular data to recover a known asymmetric phage phylogeny. Mol Phylogenet Evol. 2008;48:563–573. doi: 10.1016/j.ympev.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Stoye J, Evers D, Meyer F. ROSE: generating sequence families. Bioinformatics. 1998;14:157–163. doi: 10.1093/bioinformatics/14.2.157. [DOI] [PubMed] [Google Scholar]
- Strope CL, Scott SD, Moriyama EN. Indel-Seq-Gen: a new protein family simulator incorporating domains, motifs, and indels. Mol Biol Evol. 2007;24:640–649. doi: 10.1093/molbev/msl195. [DOI] [PubMed] [Google Scholar]
- Swofford DL, Olsen GJ, Waddell PJ, Hillis DM. 1996. Phylogeny inference. [Google Scholar]
- Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C content biases. Mol Biol Evol. 1992;9:678–687. doi: 10.1093/oxfordjournals.molbev.a040752. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tavaré S. Lines of descent and genealogical processes, and their applications in population genetics models. Theor Popul Biol. 1984;26:119–164. doi: 10.1016/0040-5809(84)90027-3. [DOI] [PubMed] [Google Scholar]
- Thorne JL, Kishino H, Felsenstein J. An evolutionary model for maximum likelihood alignment of DNA sequences [Erratum in J. Mol. Evol. 1992, 34:91] J Mol Evol. 1991;33:114–124. doi: 10.1007/BF02193625. [DOI] [PubMed] [Google Scholar]
- Varadarajan A, Bradley RK, Holmes IH. Tools for simulating evolution of aligned genomic regions with integrated parameter estimation. Genome Biol. 2008;9:R147. doi: 10.1186/gb-2008-9-10-r147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, et al. (222 co-authors) Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Whelan S, Goldman N. Estimating the frequency of events that cause multiple-nucleotide changes. Genetics. 2004;167:2027–2043. doi: 10.1534/genetics.103.023226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Yano K, Kawahara T. Pattern and rate of indel evolution inferred from whole chloroplast intergenic regions in Sugarcane, maize and rice. DNA Res. 2006;13:197–204. doi: 10.1093/dnares/dsl012. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol Biol Evol. 1993;10:1396–1401. doi: 10.1093/oxfordjournals.molbev.a040082. [DOI] [PubMed] [Google Scholar]
- Yang Z. Estimating the pattern of nucleotide substitution. J Mol Evol. 1994a;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994b;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Yang Z. On the general reversible Markov-process model of nucleotide substitution: a reply to Saccone et al. J Mol Evol. 1995;41:254–255. [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z. Computational molecular evolution. Oxford: Oxford University Press; 2006. [Google Scholar]
- Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–418. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A-MK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Hasegawa M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol Biol Evol. 1998;15:1600–1611. doi: 10.1093/oxfordjournals.molbev.a025888. [DOI] [PubMed] [Google Scholar]
- Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov chain Monte Carlo Method. Mol Biol Evol. 1997;14:717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]
- Yang Z, Roberts D. On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol Biol Evol. 1995;12:451–458. doi: 10.1093/oxfordjournals.molbev.a040220. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003;31:5338–5348. doi: 10.1093/nar/gkg745. [DOI] [PMC free article] [PubMed] [Google Scholar]