

Abstract
Maurocalcine is a scorpion venom toxin of 33 amino acid residues that bears a striking resemblance to the domain A. This domain belongs to the II-III loop of Cav1.1 which is implicated in excitation-contraction coupling. Besides the structural homology, maurocalcine also modulates RyR1 channel activity in a manner akin to a synthetic peptide of domain A. Owing to these similarities, we hypothesized that maurocalcine and domain A may bind onto an identical region(s) of RyR1. Using a set of RyR1 fragments, we demonstrate that peptide A and maurocalcine bind onto two discrete RyR1 regions: fragments 3 and 7 encompassing amino acid residues 1021-1631 and 3201-3661, respectively. The binding onto fragment 7 is of greater importance and was thus further investigated. We found that the amino acid region 3350-3501 of RyR1 (fragment 7.2) is sufficient for these interactions. Proof that peptide A and maurocalcine bind onto the same site is provided by competition experiments in which binding of fragment 7.2 to peptide A is inhibited by preincubation with maurocalcine. At the functional level, deletion of fragment 7 abolishes the maurocalcine induced stimulation of [3H]-ryanodine binding onto microsomes of COS-7 cells transfected with RyR1 without affecting the caffeine response.
Keywords: Adenosine Triphosphate; chemistry; Animals; Binding Sites; Binding, Competitive; COS Cells; Calcium Channels, L-Type; chemistry; Caveolin 1; Caveolins; chemistry; Chromatography; Cloning, Molecular; Cryoelectron Microscopy; Microscopy, Fluorescence; Muscle, Skeletal; metabolism; Peptides; chemistry; Plasmids; metabolism; Protein Binding; Protein Structure, Tertiary; Recombinant Fusion Proteins; chemistry; Ryanodine; chemistry; Ryanodine Receptor Calcium Release Channel; chemistry; Scorpion Venoms; chemistry; pharmacology; Transfection
INTRODUCTION
In skeletal muscle cells, the activation of the plasma membrane dihydropyridine receptor induces, through the ryanodine receptor (RyR1), a massive release of the calcium stored in the terminal cisternae of the sarcoplasmic reticulum (SR). The set of events, that starts with the depolarization of the plasma membrane, sensed by the DHPR, and ends with the opening of the RyR1, is called excitation-contraction (EC) coupling. Several results have led to the conclusion that the skeletal muscle EC coupling is based on a mechanical process that is made possible by the physical interactions of the two Ca2+ channels (1). The transmission of information from the DHPR to the RyR1 has been termed orthograde signal. In addition, the same physical interactions, which occur between RyR1 and DHPR, have also been proposed to be responsible for a retrograde signal in which the DHPR calcium channel activity is controlled by RyR1 (2,3). The identification of protein domains involved in these interactions between both DHPR and RyR1 and of proteins regulating these interactions remains today an important field of research.
The DHPR is composed of four different subunits α1, β, α2δ and γ (4). The α1 subunit has been shown not only to form the pore region and to carry the voltage sensitivity of the channel but also to be directly involved in the functional coupling of the DHPR with RyR1 (5). Indeed, the cytoplasmic loop that links the domains II and III of the α1 subunit has been proposed to be responsible for the mechanical coupling between the DHPR and the RyR1 (6). Within this cytoplasmic loop, two regions (domains A and C, respectively) have been shown to regulate ryanodine binding and channel gating of RyR1 in vitro (7). Other regions of the α1 subunit have been found to interact with RyR1 but the functional role of these interactions is less essential or remains to be established (8).
Ryanodine receptors are formed by the homotetramer assembly of a 560 kDa polypeptide. RyR1 activity is regulated in vitro by a number of chemical compounds such as ATP, Ca2+, Mg2+, and proteins such as calmodulin and FK506 binding protein (FKBP12) (9). Nevertheless, due to i) the extremely large size of RyR1, ii) the small number of high affinity effectors and iii) the absence of atomic resolution structure, the mapping of the functional sites of RyR1 is still far from completion. Expression in dyspedic cells (lack RyR1) of chimeric RyR1 channels, that carry sequences of different RyR isoforms, allowed the identification of domains engaged in the EC coupling process (10–12). At the same time, binding sites for different effectors and partner proteins have been identified. As part of the search for molecules able to strongly modify RyR1 properties, toxins isolated from scorpion venoms have been described as being able to induce important modifications in RyR1 channel behavior. Nowadays, these toxins represent the most effective effectors of RyR1. Among them, imperatoxin A (IpTx A) and maurocalcine (MCa) have been extensively characterized both in terms of their effects on RyR1 and their three-dimensional structure (13–18). Interestingly, these two peptide toxins present some amino acid sequence homology with the domain A of DHPR α1 subunit and structural studies strongly suggest that the beta-sheet structure of these toxins could mimic that of the domain A of the DHPR α1 subunit (16,17). In a previous work, we demonstrated that nanomolar concentrations of MCa induces a dramatic conformational change of RyR1, witnessed by the increase in [3H]-ryanodine binding (7-fold) and the induction of long-lasting channel openings (13,14). The observed homologies in amino acid sequences and in 3-D solution structure have prompted different authors to make the hypothesis that IpTx A and MCa could share a common binding site with the domain A of the DHPR α1 subunit on the RyR1. We have therefore undertaken the identification of the amino acid region of RyR1 that carries the binding site for MCa and/or the domain A of the skeletal DHPR α1 subunit.
In this work, a set of polypetides, covering the entire sequence of RyR1, was expressed in vitro and tested individually for their ability to interact with either MCa or peptide Ask (a synthetic peptide corresponding to the domain A of the skeletal DHPR α1 subunit). We identified a single domain of RyR1 interacting with both MCa and peptide Ask. We also showed that MCa and peptide Ask compete for the interaction on this domain. In order to asses the functional relevance of this domain for the effect of MCa on RyR1, we expressed a RyR1 channel that lacks the MCa binding domain in COS-7 cells and measured the effect of this deletion on the activation of ryanodine binding by MCa. Taken together, our results clearly establish that MCa and domain A of the DHPR α1 subunit share a common binding site on RyR1.
MATERIAL AND METHODS
Purification of RyR1
The skeletal heavy SR vesicles were prepared from rabbit leg and back muscles by differential centrifugation, as previously described (19). Vesicles were then solubilized in the presence of CHAPS, and RyR1 was purified by centrifugation of the solubilized proteins on a sucrose density gradient (20) and stored in liquid nitrogen.
Peptide syntheses
Peptides used in this work were synthesized as previously described (14,21), with the addition of an exogenous biotin on the C-terminal amino acid of i) MCa synthetic peptide, ii) peptide Ask, corresponding to residues Thr671-Lys690 of the DHPR α1s subunit, and iii) peptide Csk, corresponding to the residues Glu724-Pro760 of the DHPR α1s subunit.
Pull-down experiments
Polystyrene magnetic beads (500 μg) coated with streptavidine (Dynal) were incubated for 30 min at room temperature in the presence of biotinylated peptide (100 μg/ml in buffer A; 150 mM NaCl, 2 mM EGTA, 2 mM CaCl2, pCa5 and 20 mM HEPES, pH 7.4). Beads were then washed three times with buffer A and incubated for 2 h at room temperature with purified RyR1 (100 nM in buffer A). After washing twice with buffer A, proteins bound to the immobilized peptide were eluted by boiling the beads for 5 min in 2.5% SDS and analyzed by Western blot using antibodies directed against RyR1 after separation on 8% SDS-PAGE.
Cloning and expression of RyR1 fragments
RyR1 fragments F1-F8 were cloned in pSG5 vector (Stratagene) by a PCR-based method using specific primers with a linker for further enzymatic digestion and insertion into the plasmid backbone (22). Fragment F1 (from aminoacid 1 to 526) was cloned in EcoRI and NotI restriction sites; F2 (aminoacids 527-1021) in BamHI-NotI sites; F3 (aminoacids 1022-1631), F4 (aminoacids 1632-2169), F5 (aminoacids 2170-2697), F6 (aminoacids 2698-3200) and F7 (aminoacids 3201-3661) were cloned between EcoRI and BamHI restriction sites; F8 (aminoacids 3662-4244) was cloned between BamHI and NotI. Fragment F9 (aminoacids 4007-5038) was produced by cutting the original pCIneo-RyR1 plasmid (kindly provided by Dr. Allen) with XhoI, followed by self-ligation.
35S-labeled RyR1 fragments were expressed using a coupled in vitro transcription and translation system (TNT™ kit, Promega), according to the manufacturer’s instructions. For pull-down assays, 5 μl of the translation product were incubated with polystyrene magnetic-beads coated with the peptide of interest as described above. After overnight incubation at 4°C, beads were washed with 1 volume of buffer A. The associated proteins were eluted in denaturing buffer and analyzed by autoradiography after separation on SDS-PAGE.
Expression of His-tagged RyR1 fragments in bacteria
RyR1-F7 fragment was sub-cloned into three shorter segments (F7.1, F7.2 and F7.3) by a PCR-based approach. Briefly, the cDNA was amplified using primers containing a linker encompassing a NcoI site (in the 5′ end of the forward primer) and a XbaI (in the 3′ end of the reverse primer). The PCR product was cloned using the NcoI and XbaI sites in the pMR78 plasmid that possesses a His-tag sequence upstream of the polylinker (23). E. Coli. BL21 pLys(DE3) bacteria were transformed with RyR1-F7(1–3) containing plasmid. Protein synthesis was induced by the addition of 1 mM IPTG (Sigma) to the culture media. 3 h after induction, the bacteria were collected by centrifugation (3,000 g for 10 min at 4°C) and suspended in MCAC buffer (20 mM Tris/HCl, 500 mM NaCl, 10% v/v glycerol, pH 8.0) supplemented with protease inhibitors (Complete™, Boehringer Mannheim). Cell lysis was induced by adding Triton X-100 (0.1% v/v), DNAse I (50 μg) and MgCl2 (10 mM). After 10 min incubation at room temperature, insoluble material was removed by centrifugation (15 min at 20,000 g and 4°C). Solubilized proteins were then passed through a column of Ni-NTA agarose beads (Qiagen) equilibrated with MCAC buffer. After wash with four volumes of MCAC buffer containing 60 mM imidazole, his-tagged RyR fragments were eluted in 0.5 ml fractions with MCAC buffer containing 250 mM imidazole and detected by Western blot analysis with antibodies directed against the histidine tag (Sigma) used at a dilution factor of 1/10,000.
Transfection of COS-7 cells with RyR1 constructs and cytoimmunofluorescence
Wild-type RyR1 (RyR1wt) construct was cloned into a modified pCI-neo (Promega). Briefly, NdeI (nt 387) and NheI (nt 1085) sites on the pCI-neo vector were destroyed. Within the RyR1 cDNA, EcoRV (nt 9726) and NheI (nt 10014) sites were introduced and used for cloning into the modified pCI-neo vector, for further expression studies in mammalian cells. RyR1?F7 construct was generated from the pCI-neo/RyR1 clone (containing unique EcoRV, NheI and NdeI restriction sites), by cutting out the sequence between nucleotides 9730 and 10983.
COS-7 cells were transfected with the RyR1wt or RyR1ΔF7 containing plasmid using the linear polyethylenimine jetPEI™ reagent according to the manufacturer’s instructions (Qbiogene). Expression of RyR1wt and RyR1ΔF7 was controlled by immunofluorescence using antibodies directed against the C-terminal domain of RyR1 (19). 48 h after transfection, cells were fixed in 4% paraformaldehyde for 10 min, washed 3 times with PBS and permeabilized with 0.1% Triton X-100. Cells were then incubated for 1 h at room temperature in the presence of 1% BSA and 1% goat serum before adding anti-RyR1Cter antibodies (dilution 1:200 in PBS-0.1% Tween 20, PBS-T). After 2 h, the cells were washed four times with PBS-T and incubated for 30 min at room temperature with Alexa488-conjugated anti-rabbit IgG (dilution 1:800 in PBS-T). Cells were then washed four times with PBS-T and mounted. Samples were analyzed by confocal laser scanning microscopy, using a Leica TCS-SP2 operating system. Alexa488, and DsRed fluorescence were excited and collected sequentially (400 Hz line by line) by using the 488 nm line of an argon laser for Alexa488, and the 543 nm line of an helium-neon laser for DsRed excitation. Fluorescence emission was collected from 598 to 541 nm for Alexa488, and from 554 to 625 nm for DsRed.
Microsomes preparation
COS-7 cells were harvested in ice cold buffer containing 320 mM sucrose, 5 mM Hepes pH 7.4 and protease inhibitors, using a cell-scraper. Cells were then homogenized using a Teflon potter and centrifuged for 5 min at 7,000 g at 4°C. Microsomes were then collected by centrifugation at 100,000 g for 1 h at 4°C and resuspended in the same buffer. After quantification of protein concentration by Bradford assay, samples were stored in liquid nitrogen.
[3H]-ryanodine binding assay
Microsomes preparation (100 μg of proteins) obtained from control non-transfected COS-7 cells, and RyR1 or RyR1?F7-expressing cells were incubated at room temperature for 1 h in presence of 1 μM recombinant FKBP12 in a buffer containing 150 mM NaCl, 2 mM EGTA and 2 mM CaCl2 (pCa 5), 20 mM Hepes, pH 7.4. Microsomes were then incubated for 150 min at 37°C in the presence of [3H]-ryanodine (10 nM) and caffeine (0 or 6 mM) or MCa (0 or 30 nM). Free and bound [3H]-ryanodine were separated by filtration of the microsomes on Whatmann GF/B filters and [3H]-ryanodine measured by liquid scintillation. Non-specific [3H]-ryanodine binding was measured in the same conditions in the presence of 10 μM unlabeled ryanodine.
RESULTS
Figure 1A presents the primary structure of MCa, the domain A and the domain C of the II-III loop of the skeletal muscle α1 DHPR. Biotinylated peptides, named MCab, peptide-ASk (pAsk) and peptide-CSk (pAsk) corresponding to these different sequences were synthesized and immobilized on polystyrene beads coated with streptavidine. Figure 1B shows the SDS PAGE analysis of the bound RyR1 fraction following incubation of beads covered with each of the peptide with purified RyR1 as described in Materials. A polypeptide band corresponding to RyR1 is only observed in the bound fraction obtained with beads covered with MCab, peptide-ASk. No signal was detected when beads were covered with peptide-CSk or biotin. These results confirm the specificity of the interaction between the skeletal sequence of domain A and RyR1 and represent the first biochemical evidence of a direct interaction of MCa with RyR1.
Figure 1. Identification of RyR1 fragments interacting with MCa and domain A.
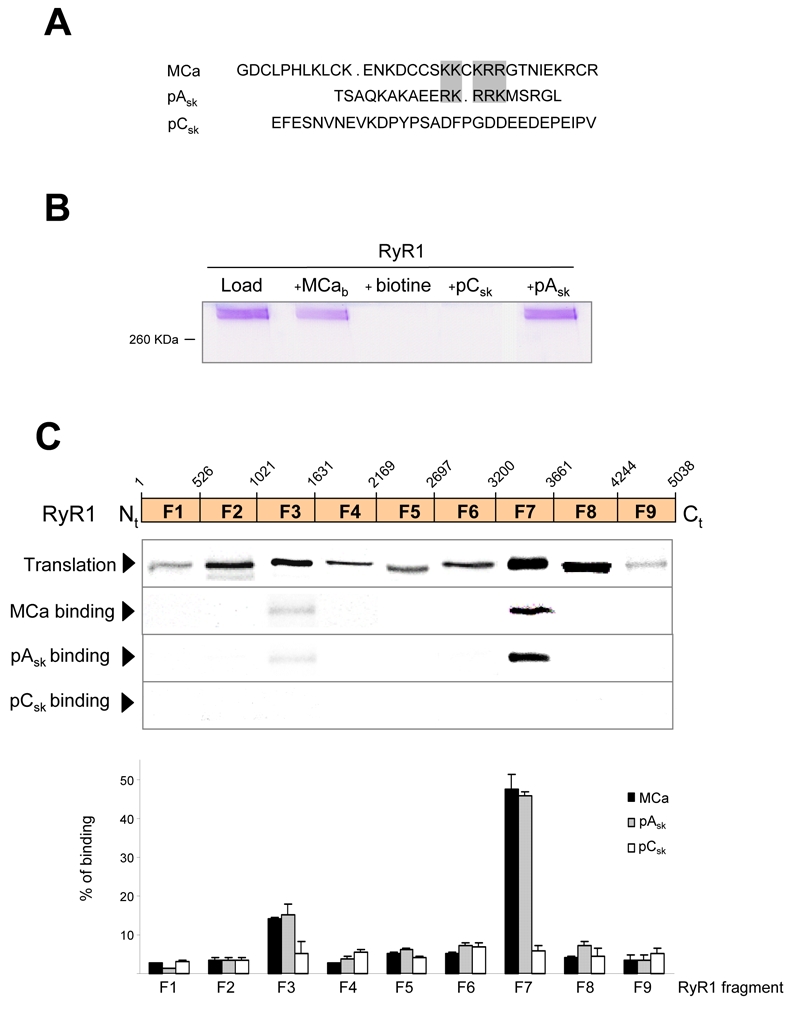
A, Sequence alignment of maurocalcine (MCa), domain A (pAsk) and domain C (pCsk) of the II-III loop of the α1 subunit of the DHPR. Grey boxes indicate the location of conserved clusters of basic amino acid residues in MCa and pAsk primary structures. In contrast, pCsk does not exhibit this repeat of positively charged residues.
B, Physical interaction between purified RyR1 and MCa, domain A or domain C. Biotinylated-MCa (MCab, at 40 μM), biotinylated-domain A (pAsk, at 40 μM) and biotinylated-domain C (pCsk, at 40 μM), immobilized on streptavidine-coated magnetic beads, were used for pull-down experiments. In these conditions, MCab (lane 2) and domain A (lane 5) showed an interaction with full-length purified RyR1, whereas no interaction was observed with domain C-coated beads (lane 4) or biotin-coated beads (lane 3).
C, Upper panel, Autoradiogram of MCab interaction with RyR1 fragments. After the generation of a set of clones spanning the full-length cDNA of RyR1 (dashed box indicates overlapping residues between fragments F8 and F9), in vitro translated fragments of RyR1 were assessed for their ability to interact either with 40 μM of MCab, pAsk or pCsk immobilized on streptavidine-magnetic beads.
Lower panel, Histogram representing the percentage of binding of in vitro translated RyR1 fragments to MCab, estimated by densitometry and expressed as the means ± S.E.M. from at least three independent experiments.
In order to identify the amino acid sequences of RyR1 involved in the interaction with MCab and peptide-Ask, we generated a contig of nine clones encompassing the full length RyR1 cDNA in plasmids (Figure 1C). Each of these clones was expressed separately in an in vitro cell-free translation system in the presence of radiolabeled [35S]-methionine. RyR1 fragments (F1–9) were incubated in the presence of beads covered either with MCab, peptide-Ask, peptide-Csk or biotin. The fragments interacting with the different peptides were then detected by autoradiography after separation on SDS-PAGE. Figure 1C shows that among the nine translated fragments, F7 fragment encompassing RyR1 amino acid residues 3201-3661 interacts strongly with MCab and with peptide-Ask. Fragment F3 encompassing the RyR1 amino acid residues 1021-1631 consistently shows a weak interaction with both MCab and peptide-Ask. In agreement with what is observed with purified RyR1, no significant interaction with peptide-Csk was observed with any of the nine RyR1 fragments. Similarly, no interaction was observed of any fragment with beads covered with biotin (not shown). Statistical analysis of the amount of complex formed with each of the fragment shows that about 50% of the F7 fragment interacts with MCab or peptide-Ask, while only about 15% of the F3 fragment was retained by MCab or peptide-Ask. For all other fragments, the amount of complex formed with MCab or peptide-Ask was not significantly different from what observed with peptide-Csk and represented less than 5% of the input.
To further define the RyR1 sequence that interacts with MCab and peptide-ASk, the cDNA corresponding to F7 was subcloned into three smaller fragments, named F7.1, F7.2 and F7.3, encompassing RyR1 amino acid residues 3201-3350, 3351-3501 and 3502-3661, respectively (Figure 2A). Corresponding polypeptides were then expressed in E. coli, affinity-purified trough their histidine tags, and tested for their interaction with MCab and peptide-Ask. Interacting fragments were detected by immunolabeling with antibodies directed against the his-tag, after separation on SDS-PAGE. Figure 2A shows that only the fragment F7.2, encompassing amino acid residues 3351-3501, retains the ability to interact with both MCab and peptide-Ask. No interaction was observed with fragment F7.1 or F7.3. The specificity of the interaction of the F7.2 fragment with MCab and peptide-Ask was confirmed by the lack of signal with beads covered with peptide-Csk or biotin (data not shown). We therefore investigated the ability of MCab to inhibit the interaction of fragment F7.2 with peptide-Ask. Incubation of fragment F7.2 for 2 h in the presence of non-biotinylated MCa induces a complete loss of interaction of this fragment with immobilized peptide-Ask (Figure 2B) strongly suggesting that MCa and peptide-Ask interact on the same site on the F7.2 fragment.
Figure 2. Maurocalcine and domain A of the II-III loop of the α1 subunit bind onto the same site of RyR1.
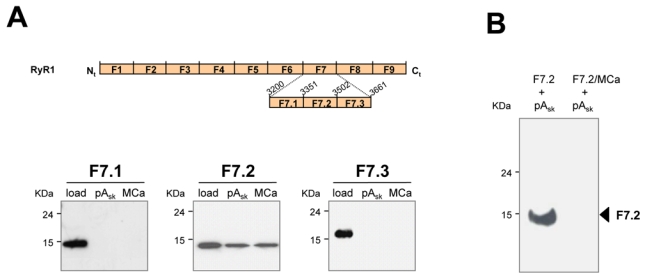
A, Fine dissection of RyR1 fragments that interact with MCa and pAsk. Upper panel, schematic representation of the RyR1-F7 sub-fragments (F7.1, F7.2, F7.3) cloned into pMR78-his tag vector for bacteria expression and affinity chromatography purification. Lower panel, Western blot with anti-his antibodies, showing the specific pull-down of the F7.2 fragment of RyR1 with both MCab and pAsk.
B, Western blot resulting from a competition experiment, where the RyR1-F7.2 fragment was preincubated with an excess of MCa (1 μM) and then incubated with 16 nM of pAsk previously immobilized on streptavidine-magnetic beads. While the fragment F7.2 was able to bind to pAskb (left lane), the presence of MCa abolished this interaction (right lane), showing a competition of these two peptides for the same binding site on RyR1.
We next investigated the importance of the F7 region in the sensitivity of RyR1 to MCa. For this purpose, COS-7 cells were co-transfected with two plasmids encoding the DsRed protein and the entire RyR1 (RyR1wt) or the RyR1 deleted of residues 3241-3661 (RyR1ΔF7) (Figure 3). Immunolabeling of DsRed-positive cells with antibodies directed against the C-terminal domain of RyR1 demonstrates that RyR1wt and RyR1ΔF7 exhibit a similar cell distribution. The distribution essentially consists in a cytoplasmic labeling most likely representing the endoplasmic reticulum (Figure 3A). Fluorescence analysis showed that about 10% of cells co-express DsRed and RyRwt or DsRed and RyRΔF7. A weak labeling with anti-RyR1 antibodies was also observed in cells that do not express DsRed protein, suggesting that some cells could express exogenous RyR1 without DsRed or that they possess a low level of endogenous RyR1 expression (however barely detected by Western blot analysis, Figure 3B).
Figure 3. Deletion of the F7 region abolishes the effect of maurocalcine on [3H]-ryanodine binding onto RyR1 expressed in COS-7 cells.
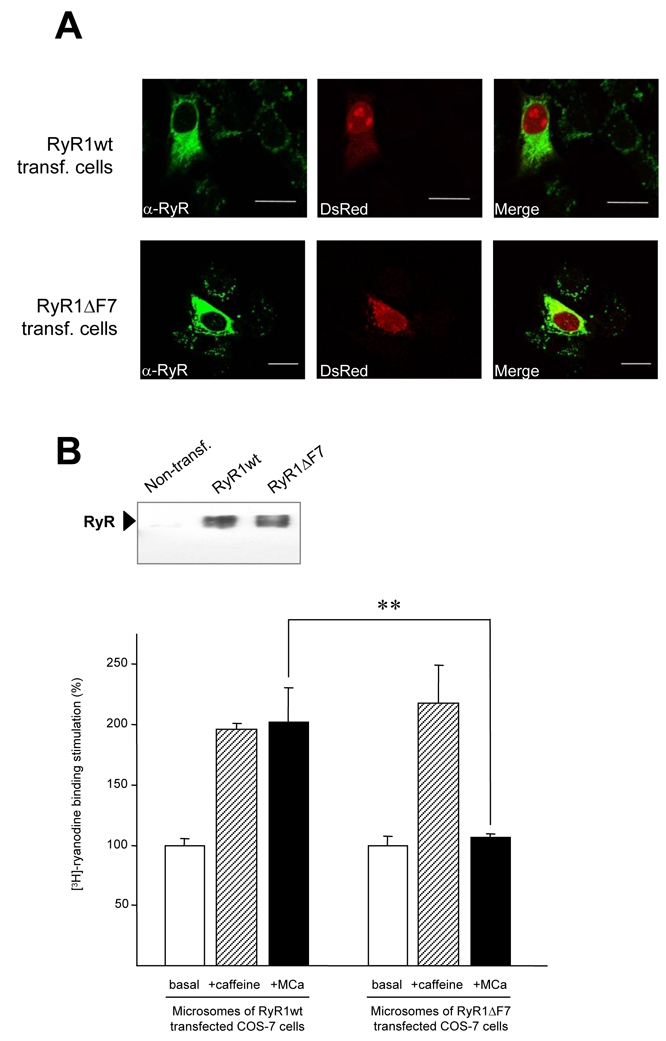
A, RyR expression pattern in COS-7 cells cotransfected either with DsRed and RyR1wt (upper panels) or with DsRed and RyR1-ΔF7 (lower panels) in COS-7 cells. Immunofluorescent signal in left panels (green signal) show the distribution pattern of RyR1 and red signal in central panels correspond to DsRed protein. Right panels correspond to merged images.
B, Expression of RyR1 constructs in COS-7 cells. Western blot with anti-RyR1 (C-terminal) antibodies, showing a lack of endogenous RyR1 expression (lane 1) in control cells and equal protein amounts of RyR1 wt (lane 2) and RyR1-ΔF7 (lane 3) in 50 μg of microsomes, 48h after transfection.
C, Functional effect of the F7 deletion on RyR1 binding properties. [3H]-ryanodine binding experiments on purified cell microsomes showed that, while both constructs responded to caffeine administration (6 mM), the deletion of the F7 fragment abolished MCa effect (30 nM).
In order to establish the functional relevance of the F7 region in RyR1 regulation by MCa or peptide-Ask, we characterized the binding of [3H]-ryanodine on RyR1wt or RyR1ΔF7 expressed in COS-7 cells and tested the effects of both caffeine and MCa on this binding. For this purpose, we prepared microsomes from cells transfected with either RyR1wt or RyR1ΔF7. Immunoblot analysis of the different microsome preparations using antibodies directed against RyR1 (Figure 3B upper panel) supports the localization of expressed RyR1wt and RyR1ΔF7 in reticulum membrane of transfected cells. These results also show that RyR1wt and RyR1ΔF7 are expressed at a similar level and remain stable in transfected cells. Finally, endogenous RyR1, if present, is barely measurable. Using these microsome preparations, we then measured [3H]-ryanodine binding on expressed RyR1wt and RyR1ΔF7 in the presence or absence of caffeine or MCa. No significant specific [3H]-ryanodine binding was observed on microsomes obtained from non-transfected cells (Figure 3B). In addition, no effect of caffeine or MCa was observed on these control microsomes (data not shown). This data confirmed immunological and Western blot data showing a low level if any of endogenous RyR1 in COS-7 cells. In contrast, [3H]-ryanodine binding was observed on microsomes populations obtained from COS-7 cells transfected with RyR1 constructs. Specific binding was 116 ± 57 fmol/mg and 113 ± 31 fmol/mg for microsomes obtained from RyR1wt and RyR1ΔF7 transfected cells, respectively. Taken together with the Western blot analysis, these results indicate a similar [3H]-ryanodine binding ability of expressed RyR1wt and RyR1ΔF7. Activation of [3H]-ryanodine binding by caffeine and MCa is presented in Figure 3B. In the presence of 6 mM caffeine, we observed a 197 ± 5% and 218 ± 32% increase of [3H]-ryanodine binding on RyR1wt and RyR1ΔF7 microsomes, respectively. In contrast, 30 nM MCa induced a 202 ± 28% increase of [3H]-ryanodine binding on RyR1wt microsomes but no significant change of [3H]-ryanodine binding on RyR1ΔF7 microsomes (107 ± 3%). These results clearly demonstrate that deletion of amino acids 3241 to 3661 of RyR1, included in fragment F7, induces a complete loss of RyR1 sensitivity to MCa while it does not modify its sensitivity to caffeine.
DISCUSSION
Recently, we characterized the effect of MCa on RyR1 (13,14). MCa is a 33 amino acid toxin initially isolated from the venom of the scorpion Scorpio maurus palmatus, and subsequently produced by chemical peptide synthesis (24). Based on sequence similarities, MCa and IpTxA (another scorpion toxin interacting with RyR1) were proposed to share with the domain A of the skeletal DHPR α1 subunit a common binding site(s) on RyR1. In this work, we identified two sequences of RyR1, fragment F3 encompassing amino acids 1021-1631 and fragment F7 comprising the residues 3201-3661, that are able to interact in vitro with both MCa and domain A of the II-III loop of the DHPR α1 subunit. Among these two sequences, fragment F7 displays the strongest interaction with MCa and peptide-Ask. We then generated a RyR1 construct that lacks the amino acid region corresponding to fragment F7. This RyR1 construct with its internal deletion displays an expression profile and a caffeine sensitivity that is perfectly similar to the wild-type RyR1. In contrast, deletion of the F7 region completely abolished the stimulatory effect of MCa on ryanodine binding, highlighting the critical role of this domain for MCa regulation. A molecular dissection of fragment F7 allowed us to identify a discrete domain corresponding to amino acids 3351-3501 (fragment F7.2) responsible for the interaction of MCa and peptide-Ask with RyR1. These results clearly demonstrate that, in agreement with earlier expectations, MCa and domain A interact with the same RyR1 region within the 3351-3501 stretch of amino acid residues. Binding of MCa and peptide-Ask to the same RyR1 site was confirmed by the complete inhibition of the interaction of peptide-Ask with the fragment F7.2 in the presence of an excess of MCa.
Using an affinity chromatography approach, Leong and MacLennan identified a domain of RyR1 that interacts with the entire II-III loop of the DHPR α1 subunit (25). This sequence (amino acid residues 1076 to 1112) is included within our fragment F3 that we found to interact more weakly with both peptide-Ask and MCa. In contrast, these authors did not observe any significant interaction of the entire II–III loop with a polypeptide comprising amino acid residues 3158-3724 (which includes fragment F7). Our previous results, together with those presented here, showing that domain C of α1 subunit does not interact with any sequence of RyR1, suggest that domain Ask is the only sequence of II–III loop interacting with RyR1 (21). The difference between the two sets of results may reflect an effect of the distal part of the II–III loop on its conformation thereby possibly modifying the accessibility of domain Ask to RyR1.
Several studies using either RyR1/RyR2 or RyR1/RyR3 chimeras led to the identification of different domains of RyR1 involved in functional and structural interaction of DHPR with RyR1 (11,12). Interestingly, fragment F7 carrying the MCa and peptide-Ask binding domain is included in one of these domains formally defined as domain R9 or Ch-9. Similarly, the deletion of residues 1272-1455 of RyR1 (included in the F3 fragment) has been shown to induce a loss of depolarization-induced Ca release (12). Therefore, regardless of the approach, the two regions of RyR1 spanning fragments F3 and F7 have been shown to play important roles in the functional communication between RyR1 and DHPR. In agreement with previous studies suggesting that noncontiguous regions of RyR1 may act together to control the functional coupling between RyR1 and DHPR, the F3 and F7 fragments may form a unique functional domain. This possibility is supported by three-dimensional reconstruction of RyR1 indicating that regions including the F3 and F7 fragments could be close from each other in the RyR1 tetramer (26).
In conclusion, we identified the binding domains of MCa on RyR1. We also introduce the first biochemical evidence that MCa and domain A of the DHPR α1 subunit interact with identical sequences on RyR1. In view of the important effects of MCa on RyR1 gating properties (13,14), the RyR1 sequences that we identified in this report ought to play an important function in RyR1 calcium channel behavior. The fact that domain A of the II–III loop of DHPR binds onto the same RyR1 sequences re-opens the question of the role of the domain A in the cont rol of RyR1 calcium channel activity in the context of the DHPR/RyR1 complex. Our results suggest that the two sequences of RyR1 (F3 and F7), that are involved in the binding of MCa, could fold together to create a unique MCa binding site. Cryoelectron microscopy studies of the RyR1-MCa complex should permit to test this hypothesis and to refine the correspondence between primary structure and 3-D reconstruction images of RyR1.
Acknowledgments
This work was supported by the European Commission Training and Mobility of Researchers Network Grant ERBFMRXCT960032 (to M.R.). X.A. is recipient of a fellowship from the European Commission. This work was also financially supported by INSERM, CEA and Université Joseph Fourier.
The abbreviations used are
- Cav1.1
Calcium channel, voltage-dependent 1.1
- CACNA1S
Calcium channel, voltage-dependent, L type, alpha-1s subunit
- SR
sarcoplasmic reticulum
- RyR1
ryanodine receptor
- DHPR
dihydropyridine receptor
- EC
excitation-contraction
- MCa
maurocalcine, MCab, biotinylated maurocalcine
- IpTxA
imperatoxin A
- pAsk
biotinylated peptide corresponding to the domain A of the II-III loop of the α1s subunit of the DHPR
- pCsk
biotinylated peptide corresponding to the domain C of the II-III loop of the α1s subunit of the DHPR
- EGTA
Ethylene Glycol-bis(beta-aminoethyl-ether)-N,N,N′,N′-TetraAcetate
- HEPES
N-[2-Hydroxyethyl]piperazine-N-[2-ethanesulfonic acid]
- IPTG
Isopropylthio-β-D-galactopyranoside
- PBS
Phosphate-Buffer Saline
- SDS-PAGE
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis
References
- 1.Chandler WK, Rakowski RF, Schneider MF. J Physiol. 1976;254:213–316. doi: 10.1113/jphysiol.1976.sp011233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 3.Chavis P, Fagni L, Lansman JB, Bockaert J. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- 4.Catterall WA. Science. 1991;253:1499–1500. doi: 10.1126/science.1654596. [DOI] [PubMed] [Google Scholar]
- 5.Tanabe T, Beam KG, Adams BA, Nicodome T, Numa S. Nature. 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 6.Nakai J, Tanabe T, Konno T, Adams B, Beam KG. J Biol Chem. 1998a;273:24983–24986. doi: 10.1074/jbc.273.39.24983. [DOI] [PubMed] [Google Scholar]
- 7.El-Hayek R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. J Biol Chem. 1995;270:22116–22118. doi: 10.1074/jbc.270.38.22116. [DOI] [PubMed] [Google Scholar]
- 8.Slavik KJ, Wang JP, Aghdasi B, Zhang JZ, Mandel F, Malouf N, Hamilton SL. Am J Physiol. 1997;272:C1475–C1481. doi: 10.1152/ajpcell.1997.272.5.C1475. [DOI] [PubMed] [Google Scholar]
- 9.MacKrill JJ. Biochem J. 1999;337:345–361. [PMC free article] [PubMed] [Google Scholar]
- 10.Nakai J, Sekiguchi N, Rando TA, Allen PD, Beam KG. J Biol Chem. 1998b;273:13403–13406. doi: 10.1074/jbc.273.22.13403. [DOI] [PubMed] [Google Scholar]
- 11.Protasi F, Paolini C, Nakai J, Beam KG, Franzini-Armstrong C, Allen PD. Biophys J. 2002;83:3230–3244. doi: 10.1016/S0006-3495(02)75325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez CF, Mukherjee S, Allen P. J Biol Chem. 2003;278:39644–39652. doi: 10.1074/jbc.M305160200. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Estève E, Sabatier JM, Ronjat M, De Waard M, Allen PD, Pessah IN. J Biol Chem. 2003;278:16095–16106. doi: 10.1074/jbc.M209501200. [DOI] [PubMed] [Google Scholar]
- 14.Esteve E, Smida-Rezgui S, Sarkozi S, Szegedi C, Regaya I, Chen L, Altafaj X, Rochat H, Allen PD, Pessah IN, Marty I, Sabatier JM, Jona I, De Waard M, Ronjat M. J Biol Chem. 2003;278:37822–37831. doi: 10.1074/jbc.M305798200. [DOI] [PubMed] [Google Scholar]
- 15.Gurrola GB, Arévalo C, Sreekumar R, Lokuta AJ, Walker JW, Valdivia HH. J Biol Chem. 1999;274:7879–7886. doi: 10.1074/jbc.274.12.7879. [DOI] [PubMed] [Google Scholar]
- 16.Lee CW, Lee EH, Takeuchi K, Takahashi H, Shimada I, Sato K, Shin SY, Kim DH, Kim JI. Biochem J. 2004;377:385–394. doi: 10.1042/BJ20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green D, Pace S, Curtis SM, Sakowska M, Lamb GD, Dulhunty AF, Casaroto MG. Biochem J. 2003;370:517–527. doi: 10.1042/BJ20021488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moshba A, Kharrat R, Fajloun Z, Renisio JG, Blanc E, Sabatier JM, Al Ayeb M, Darbon H. Proteins Struct Funct Genet. 2000;40:436–442. doi: 10.1002/1097-0134(20000815)40:3<436::aid-prot90>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 19.Marty I, Robert M, Villaz M, De Jongh KS, Lai Y, Catterall WA, Ronjat M. Proc Natl Acad, Sci, USA. 1994;91:2270–2274. doi: 10.1073/pnas.91.6.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 21.O’Reilly F, Robert M, Jona I, Szegedi C, Albrieux M, Geib S, De Waard M, Villaz M, Ronjat M. Biophys J. 2002;82:145–155. doi: 10.1016/S0006-3495(02)75381-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng W, Carbonneau L, Keys L, Altafaj X, Ronjat M, Coronado R. Biophys J. 2004;86:220a. [Google Scholar]
- 23.Arnaud N, Cheynet V, Oriol G, Mandrand B, Mallet F. Gene. 1997;199:149–156. doi: 10.1016/s0378-1119(97)00362-4. [DOI] [PubMed] [Google Scholar]
- 24.Fajloun Z, Kharrat R, Chen L, Lecomte C, di Luccio E, Bichet D, El Ayeb M, Rochat H, Allen PD, Pessah IN, De Waard M, Sabatier JM. FEBS Lett. 2000;469:179–185. doi: 10.1016/s0014-5793(00)01239-4. [DOI] [PubMed] [Google Scholar]
- 25.Leong P, MacLennan DH. J Biol Chem. 1998;273:29958–29964. doi: 10.1074/jbc.273.45.29958. [DOI] [PubMed] [Google Scholar]
- 26.Wagenknecht T, Samso M. Front Biosci. 2002;7:d1464–d1474. doi: 10.2741/A853. [DOI] [PubMed] [Google Scholar]