

Abstract
Mitochondria play a pivotal role in the cascade of events associated with cell death pathways that are involved with several forms of neurodegeneration. Recent findings show that in the Bax/Bak-dependent pathway of apoptosis, the release of cytochrome c from mitochondria is a consequence of two carefully coordinated events: opening of crista junctions triggered by OPA1 oligomer disassembly and formation of outer-membrane pores. Both steps are necessary for the complete release of proapoptotic proteins. The remodeling of mitochondrial structure accompanies this pathway, including mitochondrial fission, and cristae and crista junction alterations. Yet, there is controversy surrounding the timing of certain remodeling events and whether they are necessary early events required for the release of pro-apoptotic factors or are simply a downstream after-effect. Here, we analyze the current knowledge of mitochondrial remodeling during cell death and discuss what structural alterations occur to this organelle during neurodegeneration, focusing on the higher resolution structural correlates obtained by electron microscopy and electron tomography.
Keywords: apoptosis, cardiolipin, crista junction, cristae remodeling, cytochrome c, electron tomography, mitochondria, neurodegeneration, OPA1
Mitochondrial Structure and Apoptosis
Recent research is building a consensus that mitochondrial structure and dynamics are actively and tightly controlled by cellular stimuli, signaling events, and perturbations inside this organelle (reviewed by Chan, 2006; Mannella, 2008; Zick et al., 2009), including programmed cell death and certain forms of neurodegeneration. Developmentally regulated programmed cell death in the brain uses the Bak/Bax-dependent pathway with tBid acting as a pro-apoptotic effector protein (Jemmerson et al., 2005). Apoptosis-like cell death is also involved in neurodegenerative diseases and stroke. A key event in many forms of neuronal degeneration and cell death is the release from mitochondria of proapoptotic effectors, such as cytochrome c, HtrA2/Omi, smac/DIABLO, and AIF, that initiate the caspases involved in downstream proteolytic processes for cellular digestion (Munoz-Pinedo et al., 2006). The release mechanism has not yet been elucidated completely but it is known to be regulated by the Bcl-2 protein family (Chipuk and Green, 2008).
A governing principle of mitochondrial architecture is that structure determines function. The three distinct mitochondrial compartments that separate functionality are involved in the major cell death pathways; these are the intermembrane space, intracristal space, and matrix, defined by three membrane systems: outer, inner boundary, and cristae. Recent interest in mitochondrial structure/function correlates has centered on cristae and crista junction shape, size, and abundance (Mannella, 2006b; Zick et al., 2009). Because electron transport chain molecules reside on the cristae membranes (Gilkerson et al., 2003; Vogel et al., 2006), the ratio of cristae/ mitochondrion surface area can be viewed as the ATP synthesizing capacity of the mitochondrion. When central nervous system (CNS) neurons are compared, this ratio does not vary much (mean between 1.6 and 1.7; table 1), and is significantly greater than the ratios for peripheral nervous system (PNS) mitochondria. The greatest ratio is found for the rod photoreceptor terminal (spherule) consistent with the high metabolic rate of retina (Johnson et al., 2007). Crista junctions are hypothesized to limit movement of signaling molecules, enzymatic substrates, products, and metabolites into or out of the intracristal space or between the inner boundary and cristae membranes (Frey et al., 2002; 2006; Mannella, 2006a; Mannella et al., 2001; Perkins et al., 1997; Perkins et al., 2001). Whereas the crista junction diameter is similar in neuronal mitochondria, the crista junction density varies substantially (table 1). The role that the crista junction diameter plays in cell death is discussed below. However, the significance of the variation in crista junction density has not been explored yet and may require further investigations, but one can speculate that it may affect the heterogeneity of mitochondrial components (Perkins and Ellisman, 2007).
Table 1. Cristae Surface Area and Crista Junction Measurements from Tomographic Volumes.
| Cell Type | Cristae/Mitochondrion Surface Area Ratio | Crista Junction Density (per μm2) | Crista Junction Diameter (nm) |
|---|---|---|---|
| Neurons - CNS | |||
| Rod Spherule | 5.2 ± .17& | 54 ± 17 (6) | 12 ± 4# (102) |
| Cone Pedicle | 2.0 ± .78 | 56 ± 45 (6) | 9 ± 2 (32) |
| Rod Inner Segment | 1.2 ± .47 | 72 ± 29 (5) | 17 ± 4 (148) |
| Cone Inner Segment | 2.6 ± .50 | 52 ± 17 (12) | 12 ± 3 (215) |
| Retinal Ganglion | 1.7 ± .40 | 25 ± 11 (7) | 14 ± 4 (184) |
| Cerebellum | 1.6 ± .28 | 136 ± 55 (3) | 16 ± 5 (85) |
| Hippocampus | 1.7 ± .79 | 25 ± 15 (3) | 14 ± 4 (38) |
| Striatum | 1.6 ± 1.0 | 65 ± 27 (6) | 14 ± 4 (43) |
| Cortex | 1.7 ± .32 | 37 ± 8 (6) | 14 ± 5 (26) |
| Spinal Cord | 1.6 ± .41 | 28 ± 10 (6) | 13 ± 3 (99) |
| Neurons and Astrocytes – PNS | |||
| Schwann Cell | 0.70 ± .18 | 80 ± 29 (6) | 13 ± 4 (40) |
| Axon (spinal root) | 0.41 ± .13 | 19 ± 10 (6) | 13 ± 4 (65) |
All values indicated as mean ± sd; number of samples indicated in parentheses. All measurements performed on tomographic reconstructions.
Inner diameter that is only the opening, i.e., the membrane width was excluded.
In recent years scientists have unraveled the structural alterations to mitochondria linked to the major apoptotic pathways. One such pathway is calcium-induced cytochrome c release, prevalent in neurons during stroke and ischemia (Jemmerson et al., 2005). Distinctive to this pathway is rupture of the mitochondrial outer membrane that can be blocked by inhibitors of the mitochondrial permeability transition. Another pathway uses pro-apoptotic proteins, such as Bax, Bak and tBid to initiate cytochrome c release independently of the permeability transition and notably without rupturing the mitochondrial outer membrane and may have no large-scale changes in mitochondrial ultrastructure (von Ahsen et al., 2000). Instead, it has been proposed that in order to release cytochrome c and other proteins during tBid-mediated apoptosis, pores must form on the mitochondrial outer membrane (Reed and Green, 2002). Pore formation is likely triggered by tBid-induced oligomerization of Bak or Bax (Lovell et al., 2008) but VDAC channels are not involved (Galluzzi and Kroemer, 2007). Supporting this model are the observations that in the absence of Bak or Bax oligomerization, there is no cytochrome c release (Kuwana et al., 2002; Scorrano et al., 2003; Wei et al., 2001; Yamaguchi et al., 2008). Once cytochrome c is released from mitochondria during apoptosis and activates apoptosomes in the cytosol, death is the certain outcome for the cell (Green and Reed, 1998; Li et al., 1997; Li et al., 2000; Liu et al., 1996). What happens to the cell's mitochondria, on the other hand, is turning out to be a complicated story. Adding to the complexity is the timing of structural perturbations to this organelle that may differ between apoptotic stimuli and the death pathway taken. Publications in the last few years have provided new insight into the timing and structural correlates of the tBid plus Bax/Bak pathway.
Cristae Remodeling During tBid-induced Apoptosis
Cristae remodeling is believed to be a necessary part of tBid-mediated cytochrome c release. This remodeling includes a fivefold widening of crista junctions and a reversal of the membrane curvature of cristae in certain regions, forming tubes that enclose the matrix instead of the intracristal space, a feature seen in ultracondensed mitochondria (Frezza et al., 2006; Scorrano et al., 2002). A remodeling to the condensed state with dilated crista junctions occurs during apoptosis induced by growth factor withdrawal and a subsequent decrease in the inner membrane potential (Gottlieb et al., 2003). Genetic manipulation has generated mitochondria with altered structure that are unable to release all of their cytochrome c (Arnoult et al., 2005; Cipolat et al., 2006; Frezza et al., 2006; Griparic et al., 2004; John et al., 2005; Lee et al., 2004), showing that this molecule can be trapped inside cristae. Once in the intermembrane space, cytochrome c can escape to the cytosol when pores are formed on the outer membrane. However, now new work questions whether this remodeling indeed is required for rapid and complete cytochrome c release or is simply a consequence of the released cytochrome c.
New data show that during tBid-induced apoptosis more subtle forms of structural changes take place inside mitochondria. One change was rather the narrowing instead of the widening of crista junction openings (Yamaguchi et al., 2008). The addition of the proteasome inhibitor MG132 blocked tBid-induced cytochrome c release by blocking Bak oligomerization and pore formation on the outer membrane. When these mitochondria and those treated with tBid alone were analyzed by electron tomography, the average diameter of crista junctions was about 9 nm, half the diameter of crista junctions in untreated mitochondria (average 16 nm). No cristae remodeling was observed, though. Remarkably, in the presence of MG132 plus tBid or tBid alone, crista junctions narrowed while the accessibility of cytochrome c to the outer membrane was increased. Thus, only a subtle modification to crista junctions occurred but was sufficient for complete cytochrome c mobilization and release. Other work did not note a change in crista junction diameters associated with cytochrome c release in cells treated with etoposide and zVAD (Sun et al 2007). In sum, given these differing data, further investigations are needed to investigate possible cristae remodeling during apoptosis induced by other mechanisms.
A New Role for OPA1--Maintaining Cristae Morphology
Optic atrophy 1 (OPA1) protein decrease by RNAi knockdown converts the tubular mitochondrial network in cell lines into many small and fragmented organelles, underscoring the role OPA1 plays in mitochondrial fusion (Olichon et al., 2003). Similarly, the yeast homologue of OPA1, Mgm1, mediates yeast mitochondrial inner membrane fusion (Meeusen et al., 2006). Of more recent interest is exactly how OPA1 maintains cristae structures and whether this function is related to its mitochondrial fusion properties. OPA1 is localized to the outer surface of the inner membrane throughout both inner boundary and cristae membranes (Griparic et al., 2004; Olichon et al., 2002). Mitochondria lacking OPA1 have altered cristae structures (Griparic et al., 2004). However, this finding is not unique as there are a number of proteins that affect cristae structures (Zick et al., 2009), one gaining increasing interest being mitofilin (John et al., 2005). OPA1 is a nuclear encoded gene and in humans, there are 8 splice variants of OPA1 (Griparic et al., 2004; Song et al., 2007). In most human and mouse cells, six isoforms of OPA1 are detectable by western blotting: two long membrane-bound isoforms and four short membrane-free isoforms. Presence of at least one short isoform and one long isoform is necessary for the generation of fusion competent mitochondria (Misaka et al., 2006; Song et al., 2007). Loss of membrane-bound isoforms causes aberrant cristae morphogenesis and impaired cellular proliferation (Merkwirth et al., 2008; Song et al., 2007).
There are two conditions under which the complete conversion of all long isoforms of OPA1 to short isoforms takes place: one, during tBid-induced apoptosis and two, during the treatment of cells with the mitochondrial uncoupler, CCCP. When isolated mitochondria are incubated with tBid, all the long isoforms of OPA1 are quickly converted to short isoforms and it is only the short isoforms that are released from mitochondria when OPA1 oligomers are disassembled (Yamaguchi et al., 2008). However, it is worth mentioning that the release of the short isoforms is not completed during the 30 minutes in which 100% of cytochrome c and Htra2/Omi molecules are released. This might be because the membrane-free short isoforms are still attached to a processing protease. In tissue culture cells, the transmembrane potential is lost within a few minutes of cytochrome c release in the absence of zVAD (Goldstein et al., 2005). CCCP-treatment of cells causes the long isoforms of OPA1 to disappear from mitochondria in 10-30 minutes (Griparic et al., 2007; Song et al., 2007) and the mitochondrial network in these cultured cells becomes fragmented. This treatment does not cause the immediate disassembly of OPA1 oligomers, nor does it cause the loss of cristae structures, arguing that the fusion function of OPA1 is separate from its maintenance of cristae structure. However, the long-term absence of OPA1 long isoforms seems to destabilize OPA1 complexes that eventually leads to the loss of cristae structures (Merkwirth et al., 2008). Thus, the changes to OPA1 brought on by two different treatments differ qualitatively – tBid-treatment causes immediate disassembly of OPA1 oligomers, whereas CCCP-treatment only destabilizes OPA1 complexes. Because the loss of transmembrane potential precedes (CCCP treatment) or is perhaps concurrent (tBid treatment) with the loss of the long isoforms of OPA1, further work is needed to determine what role the transmembrane potential plays in the stability of OPA1 oligomers.
What is the protease that cleaves OPA1? The current candidates are PARL (presenilin associated rhomboid like protein), the m-AAA protease, and Yme1 (Frezza et al., 2006; Griparic et al., 2007; Song et al., 2007). Cells with PARL knocked out display normal mitochondrial morphology, show no defects in mitochondrial fusion/fission, have no primary respiratory defects, and show only minor alterations in OPA1 processing (Cipolat et al., 2006). Likewise, cells with paraplegin (an m-AAA protease subunit) knocked out show minor changes in OPA1 processing (Ishihara, 2006; Griparic et al., 2007). Yme1 is the only protease that when knocked down affects OPA1 processing significantly (Griparic et al., 2007). It seems that PARL and paraplegin are not involved, or only involved peripherally, in OPA1 processing. Nevertheless, PARL-/- cells are more sensitive to certain apoptotic stimuli: etoposide, staurosporine, or hydrogen peroxide. This sensitivity is reversed by over-expression of OPA1 (Frezza et al., 2006). OPA1 and PARL can co-precipitate and it may be that cristae remodeling involves PARL (Pellegrini and Scorrano, 2007). More work is needed to determine how PARL is involved in cristae morphology. Loss of PARL may indeed alter cristae membranes, but the mechanism seems unlikely to be through OPA1.
Another piece to the puzzle is prohibitin. Prohibitins 1 and 2 are inner membrane proteins that form a large ring structure embedded in the inner membrane facing the intermembrane space, yet able to influence the matrix side Merkwirth and Langer (2008). In prohibitin-depleted cells, mitochondria are largely devoid of cristae and both membrane-bound long isoforms of OPA1 are missing, similar to OPA1-depleted cells. The expression of a long isoform of OPA1 suppresses these defects suggesting that prohibitins regulate the one or more serine proteases that cleave membrane-bound OPA1 isoforms.
How Might OPA1 Sequester Cytochrome c Inside Cristae?
A new structural alteration observed by independent groups during apoptosis is the disassembly of OPA1 protein complexes causing changes in the crista junction opening (Arnoult et al., 2005; Frezza et al., 2006; Yamaguchi et al., 2008). In isolated mitochondria, about 50% of OPA1 proteins are found in an oligomeric complex (Frezza et al., 2006; Yamaguchi et al., 2008). During tBid-treatment, oligomerized OPA1 complexes disappear rapidly, leaving only the monomeric form. A follow-on study of the Scorrano group to their 2002 report showed a correlation between disassembly of OPA1 oligomers and remodeling of cristae (Frezza et al., 2006). Yamaguchi and coworkers also observed that tBid and BH3 peptides caused the disassembly of OPA1 complexes. However, unlike the report from Frezza and colleagues, it was found that this disassembly required the presence of either Bak or Bax. As with cytochrome c, when isolated mitochondria were treated with MG132 and tBid, OPA1 was retained inside the organelle, but in the monomeric form. Further, mutant Bid with an inactive BH3 domain did not initiate OPA1 disassembly. Remarkably, the Bax/Bak dependent events at the inner membrane (OPA1 complex disassembly and crista remodeling) could be uncoupled from Bax/Bak-dependent events at the outer membrane (Bak oligomerization and outer membrane permeabilization).
Expression of a disassembly-resistant mutant OPA1 blocked both the full release of cytochrome c and apoptosis. Thus, it appears that OPA1 complex assembly and disassembly regulate cytochrome c sequestration inside cristae, perhaps like a dam. Under normal conditions, this dam would block the crista junction opening. It is envisioned that the dam would block soluble proteins, but not membrane proteins because of evidence that no physical barrier blocks the lateral movement of small inner membrane proteins across crista junctions (Vogel et al., 2006). Crista junctions in normal central and peripheral nervous systems mitochondria vary in length but have mean diameters typically between 10-20 nm (table 1). Oligomerized OPA1 complexes formed at this junction may exceed several hundred kDalton and so may be large enough to act as a dam, net, or scaffold. During apoptosis, though, the OPA1 dam is demolished by a process independent of caspases, leaving holes about 8 nm across, large enough to let pass 60-100 kDa proteins (Kinnally and Antonsson, 2007); hence it could allow passage of cytochrome c molecules.
When is Cytochrome c Released?
Green and colleagues observed that when HeLa cells stably expressing green fluorescent protein conjugated to cytochrome c (GFP-Cyt C) were stimulated with STS, Act D or UV to induce apoptosis, the entire content of GFP-Cyt C was released from all the mitochondria within 10 minutes (Goldstein et al., 2000; 2005). Several years later, Frey and colleagues used correlated light microscopy and electron tomography at key time points to examine the 3D structure of mitochondria in cells that released GFP-Cyt C and found that in the presence of the pan-caspase inhibitor z-VAD, HeLa cells released the content of GFP-Cyt C from mitochondria with the same kinetics as in the absence of z-VAD (Sun et al., 2007). Consistent with this report, more recent work showed that isolated mouse liver mitochondria released the entire content of cytochrome c in 10-15 minutes in the presence of 10-20 nM tBid or BimS (Yamaguchi et al., 2008). Remarkably, the narrowing of crista junctions still allowed for essentially all cytochrome c to exit the cristae and cross the outer membrane in as short as 10 minutes without large-scale structural alterations. There was an accumulation of vesicular inner membranes in the apoptotic mitochondria reported by Frey and coworkers, but no large-scale alterations of mitochondria took place in the presence of z-VAD. The vesiculated inner membrane appeared to have been caused by a widening of crista junctions into progressively longer slits that eventually circumscribed the mitochondrial interior, which was hypothesized to be a precursor to mitochondrial fragmentation. These new results suggest that large-scale mitochondrial structural alterations are secondary caspase-dependent events unnecessary for the efficient release of cytochrome c. However, caspase activation was not required for cristae remodeling of liver mitochondria (Scorrano et al., 2002), raising the possibility that the elongation of crista junctions might be triggered by loss of OPA1 (Arnoult et al., 2005; Frezza et al., 2006; Kanazawa et al., 2008; Olichon et al., 2007) or by alterations to cardiolipin initiated by tBid (discussed below).
Breaking the Cardiolipin-Cytochrome c Association: The Last Step in the Release of Cytochrome c
BH3-only proteins such as tBid induce outer membrane pore formation and open crista junctions by disassembling OPA1 oligomers, but does this suffice for the rapid release of cytochrome c? They are enough to release intermembrane space proteins, such as short membrane-free OPA1 and HtrA2/Omi (Yamaguchi et al., 2008), but there is an electrostatic force between cytochrome c and cardiolipin and unless this weak interaction is broken, cytochrome c remains inside mitochondria (Rytomaa and Kinnunen, 1995; Rytomaa et al., 1992; Tuominen et al., 2002). Cardiolipin is a charged lipid found almost exclusively in the mitochondrial inner membrane where it constitutes about 20% of the total lipid content (de Kroon et al., 1997). Several reports show that tBid associates with cardiolipin (Gonzalvez et al., 2005; Kim et al., 2004b; Kuwana et al., 2002; Lutter et al., 2000). It has been suggested that cardiolipin even draws tBid to the mitochondrial inner membrane (Epand et al., 2002; Kim et al., 2004b). Interestingly, tBid produced ultracondensed mitochondria showing a reversal of the curvature of cristae membranes (Frezza et al., 2006; Scorrano et al., 2002). Consistent with this report, a cardiolipin deficiency disease also produces reversed curvature of the cristae membranes (Acehan et al., 2007). It has even been proposed that cardiolipin anchors the apoptosis-initiating caspase, caspase-8, at contact sites between inner and outer mitochondrial membranes, where this caspase oligomerizes facilitating its self-activation (Gonzalvez et al., 2008). Renewed interest in contact sites and cardiolipin should better define transduction of signals across this structural site that bridges milieu outside the mitochondrion with the matrix.
Might calcium influx change the ionic strength of the intracristal space and thus disrupt the cardiolipin-cytochrome c association during tBid-induced apoptosis? Apoptotic agents such as STS and etoposide that generate tBid also trigger influx of calcium from the endoplasmic reticulum (ER) into mitochondria (Csordas et al., 1999). Bax deficient DU145 cells have very little store of calcium in either the ER or in mitochondria. STS-treatment induced almost no change in the amount of stored calcium in either organelle, but these cells released cytochrome c and died by apoptosis (Nutt et al., 2002; Rehm et al., 2003). Thus, the influx of calcium observed in STS and etoposide-treated normal cells does not appear to be required for tBid-induced disruption of the cardiolipin-cytochrome c association. Yet, calcium has been shown to play a critical role in one apoptotic pathway.
Unlike developmentally regulated programmed cell death that uses Bak/Bax-dependent apoptosis, external injuries to the brain that activate glutamate receptors, such as NMDAR, increase intracellular calcium levels by opening plasma membrane or ER ion channels. To distinguish the Bax/Bak-dependent intrinsic pathway of apoptosis from the ER-calcium-dependent cell death pathway, Scorrano and coworkers (2003) used Bak/Bax double knockout cells. Their experiments showed that there exist two independent pathways of programmed cell death yet STS and etoposide activate both pathways. Ceramide, arachidonic acid, and H2O2 stimulate an ER-calcium-dependent programmed cell death whereas the expression of tBid triggers an ER-calcium-independent, Bax/Bak-dependent apoptosis. There is one well-studied consequence of massive calcium influx into mitochondria: activation of the mitochondrial permeability transition (MPT), which opens a pore on the outer membrane. It is noteworthy that the MPT is dispensable for tBid-induced cytochrome c release (Rostovtseva et al., 2005). Recent work with ER-stress-induced apoptosis demonstrated that an MPT caused co-release of OPA1 and cytochrome c from both isolated mitochondria and mitochondria in situ, offering another mechanism for the remodeling of the crista junction (Zhang et al., 2008). There was also a loss of cristae with the MPT, which might be related to the release of OPA1, and an increase in the number of contact sites (He et al., 2003). These provide separate phenotypes to the cristae remodeling observed previously (Frezza et al., 2006).
Might loss or modification of cardiolipin disrupt cardiolipin-cytochrome c association during tBid-induced apoptosis? Cardiolipin can induce hexagonal structures on liposomes and for this reason, one can speculate that hexagonal structures are formed at mitochondrial contact sites, exposing cardiolipins to the cytosol and thus to tBid (Gonzalvez and Gottlieb, 2007). In support, Lutter and coworkers found that tBid was localized to contact sites (Lutter et al., 2000; 2001). Interrupting the binding of tBid to cardiolipin inhibits cytochrome c release (Kim et al., 2004b). It was reported using mass spectrometry that these mitochondria lost more than 50% of their cardiolipin content. Composition of other lipids remained largely unchanged. A 50% reduction in cardiolipin content was also observed in mitochondria treated with tBid-G94E as well as mitochondria treated with tBid plus Bcl-XL (Yamaguchi et al., 2008), which eliminate mitochondrial outer membrane pore formation and crista junction remodeling. Kim and colleagues interpreted the reduction in the amount of cardiolipin in their lipid scan as an indication of tight binding of tBid to this lipid (Kim et al., 2004b), perhaps facilitating the translocation of tBid to mitochondria, and providing further impetus for continued studies of cardiolipin and mitochondrial contact sites.
It may be that cardiolipin modification by reactive oxygen species (ROS) affects its binding to cytochrome c (Iverson and Orrenius, 2004). In STS-induced apoptosis, cardiolipin underwent oxidation as measured by mass spectroscopy (Tyurin et al., 2008). Furthermore, in actinomycin-D-induced apoptosis, oxidation of cardiolipin (6h) preceded cytochrome c release (8h), caspase3 and 7 activation (8h), annexin V positivity (9h) and decrease in the transmembrane potential (12-14h) (Kagan et al., 2006), suggesting that peroxidation of this lipid is not the consequence of cytochrome c release nor a drop in the transmembrane potential. Interestingly, OPA1 mutation showed enhanced sensitivity to elevated ROS (Kanazawa et al., 2007), consistent with the possibility that vesiculated cristae may have limited ability to clear ROS.
Structural Correlates in Mitochondrial Fission and Cell Death
New reviews of structural correlates to disease, osmotic perturbations, and metabolic states suggest that mitochondrial inner-membrane topology is influenced by a balance between fusion and fission (Knott and Bossy-Wetzel, 2008; Knott et al., 2008; Mannella, 2006a; 2008). The term “remodeling” when applied to mitochondria is used in the literature to describe both fission/fusion and cristae/crista junction alterations with references to fission/fusion greatly predominating over the latter. Upon apoptotic stimulation in many situations, mitochondria undergo fission, and this is often an early event (Lee et al., 2004). Since the first studies to describe the relationship between mitochondrial fission and cell death (Frank et al., 2001; Karbowski et al., 2002), the precise role of fission in cell death has not been fully elucidated. What is known is that Bax and Bak colocalize with mitochondrial fission and fusion GTPases (Karbowski et al., 2002). Bak appears to be the facilitator (Brooks and Dong, 2007). Through interaction with mitofusins, Bak blocks mitochondrial fusion and so induces fragmentation. In this process, Bak may collaborate with Bax. For example, neurons challenged with toxic levels of nitric oxide display Bax foci on mitochondria undergoing fission. Inhibiting Drp1 delays fission, Bax foci formation, and the loss of neurons (Barsoum et al., 2006; Goyal et al., 2007; Yuan et al., 2007). Interestingly, loss of mitofusin 2 has profound effects on mitochondrial structure and function, including increase in mitochondrial fragmentation and swelling, loss of mitochondrial DNA nucleoids, and cristae vesiculation or disorganization (Chen et al., 2007). Despite the correlation between mitochondrial fission and cell death, some studies have questioned the importance of fragmentation as a causative agent for apoptosis (Alirol et al., 2006; Estaquier and Arnoult, 2007; James et al., 2003; Lee et al., 2004; Parone et al., 2006; Wasiak et al., 2007), because the pro-apoptotic and fission-promoting functions of Drp1 or hFIS1 might be distinct. Also, Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release (Barsoum et al., 2006; Sheridan et al., 2008; Yuan et al., 2007). Thus, mitochondrial fragmentation per se might not cause apoptosis.
In addition to fission, structural alterations to the internal compartments and membranes of mitochondria can accompany cell death. Mitochondrial fission in a non-apoptotic, ‘necrosis-like’ cell-death pathway that is caspase-independent produced slight mitochondrial swelling and a transition to the condensed state (Bras et al., 2007). A more severe phenotype was revealed in neurons, where the cristae, which have normally tubular or lamellar components, became rounded, fragmented, and degenerated in fissioned mitochondria). Blebbing of the inner boundary membrane and extensive swelling of the matrix were occasionally observed leading to cristae depletion and outer membrane rupture. Release of OPA1 during apoptosis precipitates mitochondrial fragmentation (Arnoult et al., 2005). As noted above, different forms of crista vesiculation, indicative of unbalanced membrane fission, are observed in an OPA1 mutant (Frezza et al., 2006) and after etoposide treatment (Sun et al., 2007). Further, OPA1 appears to be a chaperone for subunit e of ATP synthase and Mgm1/OPA1 mutation inhibits the formation of ATP synthase dimers and abolishes normal tubular crista junctions (Amutha et al., 2004). Thus, evidence is accumulating that OPA1 plays a role not only in mitochondrial fusion, but also in the maintenance of normal inner membrane architecture (Ishihara et al., 2006; Olichon et al., 2007).
Mitochondrial Structural Alterations during Neurodegeneration
A new and growing research effort in neurodegeneration is to determine at what stage can mitochondrial structural alterations be detected because mitochondrial dysfunction is an early event in many neurodegenerative diseases (Lin and Beal, 2006). Recent findings also suggest that mitochondrial dysfunction plays a role in psychiatric disorders (Shao et al., 2008). Mitochondrial structural alteration is a common characteristic of most neurodegenerative diseases. However, there exists no systematic review or catalogue of the types of mitochondrial structural alterations commonly observed in neurodegenerative diseases. In table 2, we present this catalogue. The purpose of the catalogue is to provide a reference tool that correlates the type of neurodegeneration with mitochondrial structural features provided by electron microscopy. The catalogue does not include the growing body of evidence for mitochondrial fission, motility changes, or aggregation in neurodegeneration—alterations usually adequately studied with modern light microscopy techniques—but focuses on those alterations best studied with the higher resolution afforded by electron microscopy. With this catalogue, researchers working on a neurodegenerative disease will be able to compare the structural alterations observed in their system with those alterations previously catalogued for possible clues as to the mechanism causing the damage. For example, one of the best characterized mitochondrial structural alterations, and known for a number of years, is the MPT occurring during ischemia/reperfusion injury and stroke. The typical structural alterations accompanying the MPT are swollen mitochondria, ruptured outer membrane, and disruption of the cristae. Unsurprisingly, these alterations predominate table 2 for the ischemia entries. However, neuronal mitochondria undergoing transient ischemia followed by reperfusion exhibit condensed mitochondria with inclusion bodies, which is a pattern of injury significantly different from permanent ischemia, exhibiting the typical MPT alterations (Solenski et al., 2002). Thus, there may be multiple entries for the same neurodegeneration in table 2 to reflect the different mitochondrial structural manifestations. It is noteworthy that swollen mitochondria have been observed in many neurodegenerative diseases (table 2): Parkinson's, Alzheimer's, Huntington's, ischemia/stroke, amyotrophic lateral sclerosis, epilepsy, and peripheral neuropathy diseases, suggesting that the MPT might be more involved with neurodegeneration than previously anticipated. Another bit of data mining from table 2 reveals that vacuolated cristae have been observed in Parkinson's, Alzheimer's, spinal cord and peripheral neurodegeneration, and epilepsy. Some of the instances of vacuolated cristae are likely a manifestation of condensed mitochondria, indicating a more active functional state, as shown by biochemical findings demonstrating a high oxidative capacity coupled with marked phosphorylation associated with the condensed organelle (De Martino et al., 1979). It is anticipated that other researchers will use table 2 for additional data mining.
Table 2. Catalogue of Mitochondrial Structural Alterations from Neurodegeneration.
| Type of Neurodegeneration | Mitochondrial Structural Alteration | Reference1 | |
|---|---|---|---|
| Charcot-Marie-Tooth | vesiculated mitochondria and swollen mitochondria with disorganized cristae | Chen et al. (2007) | |
| Parkinson's and Alzheimer's diseases | loss of mitochondrial cardiolipin affecting the inner membrane | Pope et al. (2008) | |
| Parkinson's and Alzheimer's diseases | enlarged mitochondria with vacuolated cristae | Hsu et al. (2000) | |
| Parkinson's disease | swollen mitochondria and disruption of outer and inner membranes | Arriagada et al. (2004); Song et al. (2004) | |
| Parkinson's disease | enlarged mitochondria | Martin et al. (2006) | |
| Parkinson's disease | swollen mitochondria with loss of outer membrane | Park et al. (2006) | |
| Parkinson's disease | swollen mitochondria and loss of cristae | Sharma et al. (2003) | |
| Parkinson's disease | enlarged and vacuolated mitochondria with dense inclusion bodies | Yazdani et al. (2006) | |
| Alzheimer's disease | mitochondrial dense bodies | Baloyannis et al. (2000) | |
| Alzheimer's disease | swollen matrix | Moreira et al. (2002) | |
| Huntington's disease | swollen mitochondria and degenerated cristae | Yu et al. (2003) | |
| ischemia | loss of cristae | Tombol et al. (2002) | |
| transient spinal cord ischemia | ruptured mitochondria | Lee et al. (2005) | |
| cerebral ischemia | swollen mitochondria | Bossy-Wetzel et al. (2004); Rival et al. (2004); Schwartz-Bloom et al. (2000); Solenski et al. (2002); Yang et al. (2008) | |
| cerebral ischemia | condensed mitochondria with inclusion bodies | Solenski et al. (2002) | |
| brief cardiac arrest | swollen mitochondria | Hossmann et al. (2001) | |
| hypoxia-ischemia | swollen and ruptured mitochondria | Northington et al. (2007) | |
| hypoxia-ischemia | swollen ‘giant’ mitochondria and outer membrane rupture | Puka-Sundvall et al. (2000) | |
| traumatic brain injury | swollen mitochondria, disrupted cristae, rupture of outer membrane | Singh et al. (2006) | |
| amyotrophic lateral sclerosis | loss of cristae | Pullen et al. (2004) | |
| amyotrophic lateral sclerosis | ruptured mitochondria with damaged cristae | Gonzalez Deniselle et al. (2002b) | |
| amyotrophic lateral sclerosis | swollen mitochondria | Guo et al. (2007), Damiano et al. (2006) | |
| amyotrophic lateral sclerosis | inclusion bodies inside mitochondria, regularly spaced transverse processes in the intermembrane space, increased cristae and stubby mitochondria | Sasaki et al. (2007) | |
| amyotrophic lateral sclerosis | vacuolated mitochondria | Jaarsma et al. (2000; 2001) | |
| spinal cord neurodegeneration | vacuolated mitochondria | Gonzalez Deniselle et al. (2002a) | |
| epilepsy | rounded mitochondria with degenerated cristae | Fujikawa et al. (2000) | |
| epilepsy | vacuolated mitochondria | Jiang et al. (2007) | |
| epilepsy | swollen mitochondria and ruptured outer membrane | Chuang et al. (2004); Gao et al. (2007) | |
| epilepsy (Leigh syndrome) | mitochondria altered in shape and size, widened and disordered cristae | Leshinsky-Silver et al. (2003) | |
| epilepsy (HEADD syndrome) | mitochondria with concentric cristae | Fillano et al. (2002) | |
| epilepsy (MEHMO disorder) | enlarged mitochondria with concentric cristae and inclusion bodies | Leshinsky-Silver et al. (2002) | |
| hypoglycemic convulsion | vacuolated and “whorled” mitochondria | Gallyas et al. (2005) | |
| peripheral neuropathy from AIDS | enlarged mitochondria | Dalakas et al. (2001) | |
| peripheral neuropathy from paclitaxel chemotherapy | swollen and vacuolated mitochondria | Flatters & Bennett (2006); Jin et al. (2008) | |
| progressive motor neuropathy | swollen and vacuolated mitochondria | Sagot et al. (2000) | |
| glutamate and homocysteine neurotoxicity | swollen mitochondria | Zieminska et al. (2006) | |
| gentamicin exposure | enlarged mitochondria | Hirose et al. (2004) | |
| chronic toluene exposure | swollen mitochondria with degenerated cristae | Kanter (2008) | |
| acoustic overstimulation | swollen mitochondria or electron dense mitochondria with degenerated cristae | Kim et al. (2004a) | |
| rhizotomy | enlarged mitochondria | Wroblewski et al. (2000) | |
| late-onset motor neuron disease | vacuolated mitochondria | Marubuchi et al. (2005) | |
| Creutzfeldt-Jakob disease | swollen mitochondria with degenerated cristae | Liberski et al. (2005) | |
| aging human retina | mitochondria with cristae remnants and dense matrix | Nag et al. (2006) | |
| kainate-induced oxidative stress and senility | swollen mitochondria and electron-dense inclusions | Shin et al. (2008) | |
| neurodegeneration of postmenopausal females | swollen mitochondria with loss of cristae | Xu et al. (2008) | |
With apologies to the many excellent older structural studies of mitochondria and neurodegeneration, we limited our references to those in this millennium, consistent with the focus on “New Insights into Mitochondrial Structure”.
Conclusion
Here we focused on the new insights gained into the molecular mechanisms of tBid-induced remodeling of mitochondria during cell death and on the structural alterations to mitochondria manifested in neurodegenerative diseases. Accumulating evidence points to the intricate timing and complex interplay of three events necessary for the complete release of cytochrome c: (1) the opening of crista junctions triggered by disassembly of OPA1 oligomers, (2) the dissolution of the cardiolipin-cytochrome c association, and (3) the formation of mitochondrial outer membrane pores. Progress has been made in identifying the proteins and lipids involved, yet considerably more work is required to identify and characterize the signaling pathways and/or feedback mechanisms by which remodeling is regulated. Unresolved issues are (1) how OPA1 oligomers are assembled and disassembled and (2) how the association between cardiolipin and cytochrome c is broken. A larger issue still to be addressed is how mitochondrial cristae are generated and maintained. These are important questions because defects in the protein and lipid components found in cristae are known to be involved with premature aging and several neurodegenerative diseases (Chan, 2006).
Figure 1.
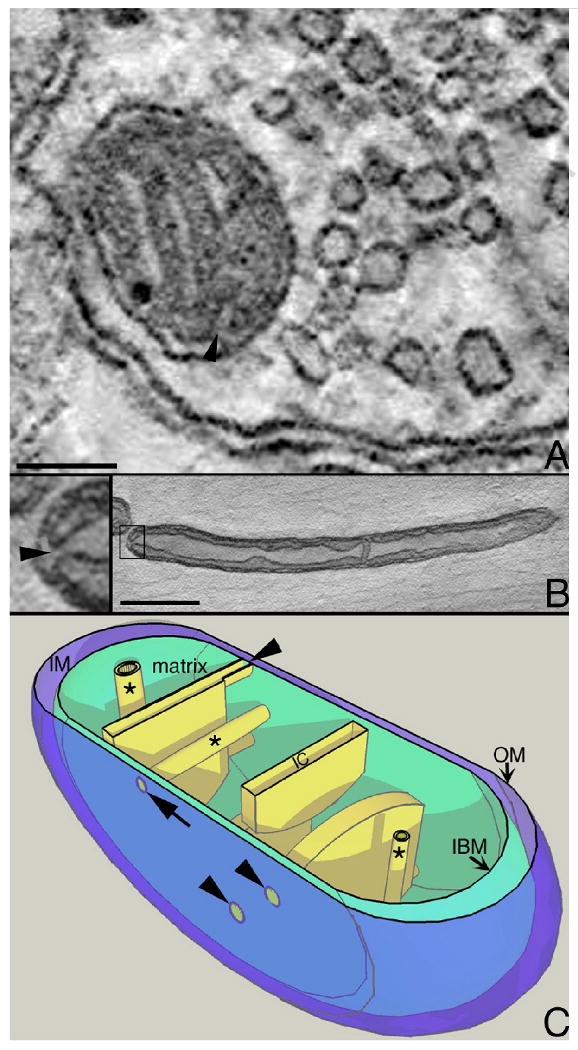
The crista junction architecture in neurons. A. A tomographic slice from the volume of a CNS mitochondrion found near a synapse in the striatum. The arrowhead points to a crista junction opening. Synaptic mitochondria in the CNS are typically small with mostly lamellar cristae. Even with lamellar cristae, the narrow, tubular crista junction architecture prevails in brain mitochondria and the opening is at the end and not on the side of the cristae. Scale = 100 nm. B. A tomographic slice from the volume of a PNS mitochondrion found in the axon of a spinal root. The crista junction is boxed and expanded 4× in the insert at left (arrowhead). PNS axonal mitochondria are typically elongated with longitudinally oriented cristae and often show a condensed matrix as seen here. Even with structural features different from CNS mitochondria, PNS axonal mitochondria nevertheless have the same crista junction architecture and again, the opening is at the end and not on the side of the cristae. Scale = 400 nm. C. Cut-away model of neuronal mitochondria. Tubular cristae (*) can be oriented in different directions and have different diameters in CNS mitochondria, but are typically longitudinal in PNS mitochondria and the crista junction is at the end of the tube (arrow). The crista junction is relatively uniform in size, even for lamellar cristae (arrowheads). Mitochondria have 3 membrane systems, the outer membrane (OM), inner boundary (IBM), and cristae, and 3 compartments, the matrix, intermembrane space (IM), and intracristal space (IC). The outer membrane was made translucent to better visualize the crista junctions.
Figure 2.
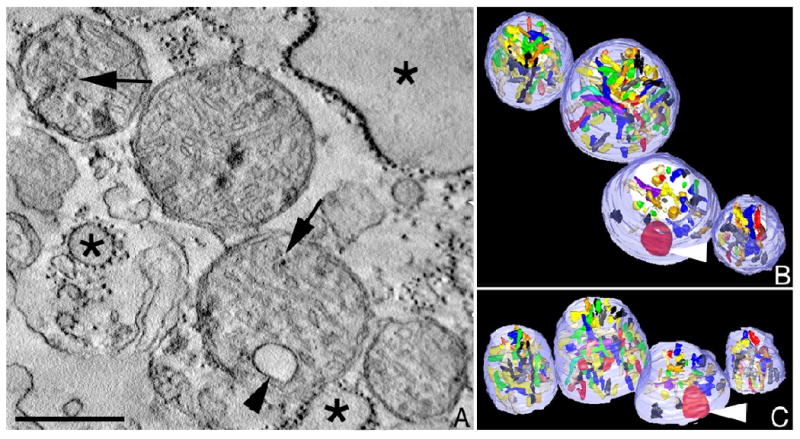
Mitochondrial fragmentation, cristae degeneration and vesiculation can occur in neurodegenerative diseases. When produced in excess, nitric oxide (NO) reacts with superoxide anion to form highly neurotoxic peroxynitrite, thought to contribute to the pathogenesis of several neurodegenerative disorders, including stroke, Parkinson's and Alzheimer's diseases (Barsoum et al. 2006). A. A slice through an electron tomographic volume showing four fragmented mitochondria in a neuronal process after exposure to a nitric oxide donor. Cristae vesiculation is seen (arrowhead) with some cristae degradation (arrows). Swollen endoplasmic reticulum is also observed (*). Scale = 400 nm. B. Top view of the surface-rendered volume after segmentation. The outer membrane is shown in translucent pale blue and the cristae are shown in various colors. Cristae fragmentation is evident from the smaller and regionally confined cristae. In contrast, the vesiculated crista is enlarged (arrowhead). C. Side view of the surface-rendered volume.
Acknowledgments
This publication was made possible by NIH NCRR grant P21 RR04050, grant ES010337 from the NIEHS, NIH grant DK54441 to MHE, and NIH grants R01 NS047456, R01EY016164, and R01NS055193 to EBW. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–4605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amutha B, Gordon DM, Gu Y, Pain D. A novel role of Mgm1p, a dynamin-related GTPase, in ATP synthase assembly and cristae formation/maintenance. Biochem J. 2004;381:19–23. doi: 10.1042/BJ20040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742–35750. doi: 10.1074/jbc.M505970200. [DOI] [PubMed] [Google Scholar]
- Arriagada C, Paris I, Sanchez de las Matas MJ, Martinez-Alvarado P, Cardenas S, Castaneda P, Graumann R, Perez-Pastene C, Olea-Azar C, Couve E, Herrero MT, Caviedes P, Segura-Aguilar J. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation: mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol Dis. 2004;16:468–477. doi: 10.1016/j.nbd.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Baloyannis SJ, Manolidis SL, Manolidis LS. Synaptic alterations in the vestibulocerebellar system in Alzheimer's disease--a Golgi and electron microscope study. Acta Otolaryngol. 2000;120:247–250. doi: 10.1080/000164800750001026. [DOI] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. Embo J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Bras M, Yuste VJ, Roue G, Barbier S, Sancho P, Virely C, Rubio M, Baudet S, Esquerda JE, Merle-Beral H, Sarfati M, Susin SA. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol Cell Biol. 2007;27:7073–7088. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Dong Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle. 2007;6:3043–3047. doi: 10.4161/cc.6.24.5115. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YC, Chang AY, Lin JW, Hsu SP, Chan SH. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia. 2004;45:1202–1209. doi: 10.1111/j.0013-9580.2004.18204.x. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio L, Derks C, Dejaegere T, Pellegrini L, D'Hooge R, Scorrano L, De Strooper B. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. Embo J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, Semino-Mora C, Leon-Monzon M. Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3′-dideoxycytidine (ddC) Lab Invest. 2001;81:1537–1544. doi: 10.1038/labinvest.3780367. [DOI] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Beal F, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- de Kroon AI, Dolis D, Mayer A, Lill R, de Kruijff B. Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim Biophys Acta. 1997;1325:108–116. doi: 10.1016/s0005-2736(96)00240-4. [DOI] [PubMed] [Google Scholar]
- De Martino C, Floridi A, Marcante ML, Malorni W, Scorza Barcellona P, Bellocci M, Silvestrini B. Morphological, histochemical and biochemical studies on germ cell mitochondria of normal rats. Cell Tissue Res. 1979;196:1–22. doi: 10.1007/BF00236345. [DOI] [PubMed] [Google Scholar]
- Epand RF, Martinou JC, Fornallaz-Mulhauser M, Hughes DW, Epand RM. The apoptotic protein tBid promotes leakage by altering membrane curvature. J Biol Chem. 2002;277:32632–32639. doi: 10.1074/jbc.M202396200. [DOI] [PubMed] [Google Scholar]
- Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Fillano JJ, Goldenthal MJ, Rhodes CH, Marin-Garcia J. Mitochondrial dysfunction in patients with hypotonia, epilepsy, autism, and developmental delay: HEADD syndrome. J Child Neurol. 2002;17:435–439. doi: 10.1177/088307380201700607. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Frey TG, Perkins GA, Ellisman MH. Electron Tomography of Membrane-Bound Cellular Organelles. In: Rees DC, Sheetz MP, Williamson JR, editors. Annual Review of Biophysics and Biomolecular Structure (Vol 35) Annual Reviews; Palo Alto, CA: 2006. pp. 199–224. [DOI] [PubMed] [Google Scholar]
- Frey TG, Renken CW, Perkins GA. Insight into mitochondrial structure and function from electron tomography. Biochim Biophys Acta. 2002;1555:196–203. doi: 10.1016/s0005-2728(02)00278-5. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98:41–53. doi: 10.1016/s0306-4522(00)00085-3. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol. 2007;9:487–489. doi: 10.1038/ncb0507-487. [DOI] [PubMed] [Google Scholar]
- Gallyas F, Csordas A, Schwarcz A, Mazlo M. “Dark” (compacted) neurons may not die through the necrotic pathway. Exp Brain Res. 2005;160:473–486. doi: 10.1007/s00221-004-2037-4. [DOI] [PubMed] [Google Scholar]
- Gao J, Chi ZF, Liu XW, Shan PY, Wang R. Mitochondrial dysfunction and ultrastructural damage in the hippocampus of pilocarpine-induced epileptic rat. Neurosci Lett. 2007;411:152–157. doi: 10.1016/j.neulet.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Gilkerson RW, Selker JM, Capaldi RA. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003;546:355–358. doi: 10.1016/s0014-5793(03)00633-1. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Gonzalez Deniselle MC, Lopez Costa JJ, Gonzalez SL, Labombarda F, Garay L, Guennoun R, Schumacher M, De Nicola AF. Basis of progesterone protection in spinal cord neurodegeneration. J Steroid Biochem Mol Biol. 2002a;83:199–209. doi: 10.1016/s0960-0760(02)00262-5. [DOI] [PubMed] [Google Scholar]
- Gonzalez Deniselle MC, Lopez-Costa JJ, Saavedra JP, Pietranera L, Gonzalez SL, Garay L, Guennoun R, Schumacher M, De Nicola AF. Progesterone neuroprotection in the Wobbler mouse, a genetic model of spinal cord motor neuron disease. Neurobiol Dis. 2002b;11:457–468. doi: 10.1006/nbdi.2002.0564. [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B, Diolez P, Manon S, Martinou JC, Goubern M, Wang X, Bernard S, Petit PX. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 2005;12:614–626. doi: 10.1038/sj.cdd.4401571. [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- Goyal G, Fell B, Sarin A, Youle RJ, Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–816. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein OPA1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- Guo Y, Liu Y, Xu L, Wu S, Yang C, Wu D, Wu H, Li C. Astrocytic pathology in the immune-mediated motor neuron injury. Amyotroph Lateral Scler. 2007;8:230–234. doi: 10.1080/17482960701278612. [DOI] [PubMed] [Google Scholar]
- He L, Perkins GA, Poblenz AT, Harris JB, Hung M, Ellisman MH, Fox DA. Bcl-xL overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc Natl Acad Sci U S A. 2003;100:1022–1027. doi: 10.1073/pnas.0333594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Westrum LE, Cunningham DE, Rubel EW. Electron microscopy of degenerative changes in the chick basilar papilla after gentamicin exposure. J Comp Neurol. 2004;470:164–180. doi: 10.1002/cne.11046. [DOI] [PubMed] [Google Scholar]
- Hossmann KA, Oschlies U, Schwindt W, Krep H. Electron microscopic investigation of rat brain after brief cardiac arrest. Acta Neuropathol. 2001;101:101–113. doi: 10.1007/s004010000260. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. Embo J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berlin) 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome C release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid Redox Signal. 2005;7:1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- Jiang W, Du B, Chi Z, Ma L, Wang S, Zhang X, Wu W, Wang X, Xu G, Guo C. Preliminary explorations of the role of mitochondrial proteins in refractory epilepsy: some findings from comparative proteomics. J Neurosci Res. 2007;85:3160–3170. doi: 10.1002/jnr.21384. [DOI] [PubMed] [Google Scholar]
- Jin HW, Flatters SJ, Xiao WH, Mulhern HL, Bennett GJ. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exp Neurol. 2008;210:229–237. doi: 10.1016/j.expneurol.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Jr, Perkins GA, Giddabasappa A, Chaney S, Xiao W, White AD, Brown JM, Waggoner J, Ellisman MH, Fox DA. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol Vis. 2007;13:887–919. [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurina YY, Bayir H, Chu CT, Kapralov AA, Vlasova II, Belikova NA, Tyurin VA, Amoscato A, Epperly M, Greenberger J, Dekosky S, Shvedova AA, Jiang J. The “pro-apoptotic genies” get out of mitochondria: oxidative lipidomics and redox activity of cytochrome c/cardiolipin complexes. Chem Biol Interact. 2006;163:15–28. doi: 10.1016/j.cbi.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, McDonald K, Mannella CA, van der Bliek AM. The C. elegans OPA1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter M. Protective effects of Nigella sativa on the neuronal injury in frontal cortex and brain stem after chronic toluene exposure. Neurochem Res. 2008;33:2241–2249. doi: 10.1007/s11064-008-9702-0. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Gross J, Potashner SJ, Morest DK. Fine structure of degeneration in the cochlear nucleus of the chinchilla after acoustic overstimulation. J Neurosci Res. 2004a;77:798–816. doi: 10.1002/jnr.20213. [DOI] [PubMed] [Google Scholar]
- Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004b;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283–292. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lee JC, Hwang IK, Park SK, Yoo KY, Seo K, Kang TC, Oh YS, Won MH. Histochemical and electron microscopic study on motor neurone degeneration following transient spinal cord ischaemia at normothermic conditions in rabbits. Anat Histol Embryol. 2005;34:252–257. doi: 10.1111/j.1439-0264.2005.00603.x. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and OPA1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshinsky-Silver E, Levine A, Nissenkorn A, Barash V, Perach M, Buzhaker E, Shahmurov M, Polak-Charcon S, Lev D, Lerman-Sagie T. Neonatal liver failure and Leigh syndrome possibly due to CoQ-responsive OXPHOS deficiency. Mol Genet Metab. 2003;79:288–293. doi: 10.1016/s1096-7192(03)00097-0. [DOI] [PubMed] [Google Scholar]
- Leshinsky-Silver E, Zinger A, Bibi CN, Barash V, Sadeh M, Lev D, Sagie TL. MEHMO (Mental retardation, Epileptic seizures, Hypogenitalism, Microcephaly, Obesity): a new X-linked mitochondrial disorder. Eur J Hum Genet. 2002;10:226–230. doi: 10.1038/sj.ejhg.5200791. [DOI] [PubMed] [Google Scholar]
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liberski PP, Streichenberger N, Giraud P, Soutrenon M, Meyronnet D, Sikorska B, Kopp N. Ultrastructural pathology of prion diseases revisited: brain biopsy studies. Neuropathol Appl Neurobiol. 2005;31:88–96. doi: 10.1111/j.1365-2990.2004.00595.x. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- Lutter M, Perkins GA, Wang X. The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol. 2001;2:22. doi: 10.1186/1471-2121-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella CA. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta. 2006a;1762:140–147. doi: 10.1016/j.bbadis.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006b;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Mannella CA. Structural diversity of mitochondria: functional implications. Ann N Y Acad Sci. 2008;1147:171–179. doi: 10.1196/annals.1427.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella CA, Pfeiffer DR, Bradshaw PC, Moraru II, Slepchenko B, Loew LM, Hsieh CE, Buttle K, Marko M. Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life. 2001;52:93–100. doi: 10.1080/15216540152845885. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marubuchi S, Wada Y, Okuda T, Hara Y, Qi ML, Hoshino M, Nakagawa M, Kanazawa I, Okazawa H. Polyglutamine tract-binding protein-1 dysfunction induces cell death of neurons through mitochondrial stress. J Neurochem. 2005;95:858–870. doi: 10.1111/j.1471-4159.2005.03405.x. [DOI] [PubMed] [Google Scholar]
- Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2008;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Misaka T, Murate M, Fujimoto K, Kubo Y. The dynamin-related mouse mitochondrial GTPase OPA1 alters the structure of the mitochondrial inner membrane when exogenously introduced into COS-7 cells. Neurosci Res. 2006;55:123–133. doi: 10.1016/j.neures.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Santos MS, Moreno A, Rego AC, Oliveira C. Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J Neurosci Res. 2002;69:257–267. doi: 10.1002/jnr.10282. [DOI] [PubMed] [Google Scholar]
- Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci U S A. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag TC, Wadhwa S, Chaudhury S. The occurrence of cone inclusions in the ageing human retina and their possible effect upon vision: an electron microscope study. Brain Res Bull. 2006;71:224–232. doi: 10.1016/j.brainresbull.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Zelaya ME, O'Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, Martin LJ. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007;149:822–833. doi: 10.1016/j.neuroscience.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O'Neil RG, McConkey DJ. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem. 2002;277:20301–20308. doi: 10.1074/jbc.M201604200. [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, Guillou E, Delettre C, Valette A, Hamel CP, Ducommun B, Lenaers G, Belenguer P. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–176. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- Olichon A, Landes T, Arnaune-Pelloquin L, Emorine LJ, Mils V, Guichet A, Delettre C, Hamel C, Amati-Bonneau P, Bonneau D, Reynier P, Lenaers G, Belenguer P. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. 2007;211:423–430. doi: 10.1002/jcp.20950. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Scorrano L. A cut short to death: Parl and OPA1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ. 2007;14:1275–1284. doi: 10.1038/sj.cdd.4402145. [DOI] [PubMed] [Google Scholar]
- Perkins G, Ellisman M. Mitochondrial architecture and heterogeneity. In: G GE, Dienel GA, editors. Handbook of neurochemistry and molecular neurobiology. Brain energetics : integration of molecular and cellular processes. 3rd. Springer; New York: 2007. p. 924. [Google Scholar]
- Perkins GA, Renken CW, Frey TG, Ellisman MH. Membrane architecture of mitochondria in neurons of the central nervous system. J Neurosci Res. 2001;66:857–865. doi: 10.1002/jnr.10050. [DOI] [PubMed] [Google Scholar]
- Perkins G, Renken C, Martone ME, Young SJ, Ellisman M, Frey T. Electron tomography of neuronal mitochondria: three-dimensional structure and organization of cristae and membrane contacts. J Struct Biol. 1997;119:260–272. doi: 10.1006/jsbi.1997.3885. [DOI] [PubMed] [Google Scholar]
- Pope S, Land JM, Heales SJ. Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim Biophys Acta. 2008;1777:794–799. doi: 10.1016/j.bbabio.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Puka-Sundvall M, Gajkowska B, Cholewinski M, Blomgren K, Lazarewicz JW, Hagberg H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res Dev Brain Res. 2000;125:31–41. doi: 10.1016/s0165-3806(00)00110-3. [DOI] [PubMed] [Google Scholar]
- Pullen AH, Demestre M, Howard RS, Orrell RW. Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2+ enhancement. Acta Neuropathol. 2004;107:35–46. doi: 10.1007/s00401-003-0777-z. [DOI] [PubMed] [Google Scholar]
- Reed JC, Green DR. Remodeling for demolition: changes in mitochondrial ultrastructure during apoptosis. Mol Cell. 2002;9:1–3. doi: 10.1016/s1097-2765(02)00437-9. [DOI] [PubMed] [Google Scholar]
- Rehm M, Dussmann H, Prehn JH. Real-time single cell analysis of Smac/DIABLO release during apoptosis. J Cell Biol. 2003;162:1031–1043. doi: 10.1083/jcb.200303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rival T, Soustelle L, Strambi C, Besson MT, Iche M, Birman S. Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Curr Biol. 2004;14:599–605. doi: 10.1016/j.cub.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK. Reversibility of the binding of cytochrome c to liposomes. Implications for lipid-protein interactions. J Biol Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Mustonen P, Kinnunen PK. Reversible, nonionic, and pH-dependent association of cytochrome c with cardiolipin-phosphatidylcholine liposomes. J Biol Chem. 1992;267:22243–22248. [PubMed] [Google Scholar]
- Sagot Y, Toni N, Perrelet D, Lurot S, King B, Rixner H, Mattenberger L, Waldmeier PC, Kato AC. An orally active anti-apoptotic molecule (CGP 3466B) preserves mitochondria and enhances survival in an animal model of motoneuron disease. Br J Pharmacol. 2000;131:721–728. doi: 10.1038/sj.bjp.0703633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Horie Y, Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007;114:633–639. doi: 10.1007/s00401-007-0299-1. [DOI] [PubMed] [Google Scholar]
- Schwartz-Bloom RD, Miller KA, Evenson DA, Crain BJ, Nadler JV. Benzodiazepines protect hippocampal neurons from degeneration after transient cerebral ischemia: an ultrastructural study. Neuroscience. 2000;98:471–484. doi: 10.1016/s0306-4522(00)00144-5. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, Bunney WE, Vawter MP. Mitochondrial involvement in psychiatric disorders. Ann Med. 2008;40:281–295. doi: 10.1080/07853890801923753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Carlson EC, Ebadi M. Neuroprotective actions of Selegiline in inhibiting 1-methyl, 4-phenyl, pyridinium ion (MPP+)-induced apoptosis in SK-N-SH neurons. J Neurocytol. 2003;32:329–343. doi: 10.1023/B:NEUR.0000011327.23739.1b. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Shin EJ, Jeong JH, Bing G, Park ES, Chae JS, Yen TP, Kim WK, Wie MB, Jung BD, Kim HJ, Lee SY, Kim HC. Kainate-induced mitochondrial oxidative stress contributes to hippocampal degeneration in senescence-accelerated mice. Cell Signal. 2008;20:645–658. doi: 10.1016/j.cellsig.2007.11.014. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helm GA. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke. 2002;33:816–824. doi: 10.1161/hs0302.104541. [DOI] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1065. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- Tombol T, Pataki G, Nemeth A, Hamar J. Ultrastructural changes of the neuromuscular junction in reperfusion injury. Cells Tissues Organs. 2002;170:139–150. doi: 10.1159/000046187. [DOI] [PubMed] [Google Scholar]
- Tuominen EK, Wallace CJ, Kinnunen PK. Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J Biol Chem. 2002;277:8822–8826. doi: 10.1074/jbc.M200056200. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Feng W, Mnuskin A, Jiang J, Tang M, Zhang X, Zhao Q, Kochanek PM, Clark RS, Bayir H, Kagan VE. Mass-spectrometric characterization of phospholipids and their primary peroxidation products in rat cortical neurons during staurosporine-induced apoptosis. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel F, Bornhovd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. 2006;175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski R, Roomans GM, Kozlova EN. Effects of dorsal root transection on morphology and chemical composition of degenerating nerve fibers and reactive astrocytes in the dorsal funiculus. Exp Neurol. 2000;164:236–245. doi: 10.1006/exnr.2000.7429. [DOI] [PubMed] [Google Scholar]
- Xu XW, Shi C, He ZQ, Ma CM, Chen WH, Shen YP, Guo Q, Shen CJ, Xu J. Effects of phytoestrogen on mitochondrial structure and function of hippocampal CA1 region of ovariectomized rats. Cell Mol Neurobiol. 2008;28:875–886. doi: 10.1007/s10571-008-9265-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. OPA1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang XJ, Yin J, Li LT. Brain damage related to hemorrhagic transformation following cerebral ischemia and the role of K ATP channels. Brain Res. 2008;1241:168–175. doi: 10.1016/j.brainres.2008.08.083. [DOI] [PubMed] [Google Scholar]
- Yazdani U, German DC, Liang CL, Manzino L, Sonsalla PK, Zeevalk GD. Rat model of Parkinson's disease: chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+) Exp Neurol. 2006;200:172–183. doi: 10.1016/j.expneurol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J Biol Chem. 2008;283:3476–3486. doi: 10.1074/jbc.M707528200. [DOI] [PubMed] [Google Scholar]
- Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta. 2009;1793:5–19. doi: 10.1016/j.bbamcr.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Zieminska E, Matyja E, Kozlowska H, Stafiej A, Lazarewicz JW. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: role of calcium and mitochondrial alterations. Neurochem Int. 2006;48:491–497. doi: 10.1016/j.neuint.2005.12.023. [DOI] [PubMed] [Google Scholar]