

Abstract
The substrate specificities of yeast alcohol dehydrogenases I and II from Saccharomyces cerevisiae (SceADH1 and SceADH2) and Saccharomyces carlsbergensis (ScbADH1) were studied. For this work, the gene for the S. carlsbergensis ADH1 was cloned, sequenced and expressed. The amino acid sequence of ScbADH1 differs at four positions as compared to SceADH1, including substitutions of two glutamine residues with glutamic acid residues, and has the same sequence as the commercial yeast enzyme, which apparently is prepared from S. carlsbergensis. The electrophoretic mobilities of ScbADH1, SceADH2 and commercial ADH are similar. The kinetics and specificities of ScbADH1 and SceADH1 acting on branched, long-chain and benzyl alcohols are very similar, but the catalytic efficiency of SceADH2 is about 10 to 100-fold higher on these substrates. A three dimensional structure of SceADH1 shows that the substrate binding pocket has Met-270, whereas SceADH2 has Leu-270, which allows larger substrates to bind. The reduction of a series of p-substituted benzaldehydes catalyzed by SceADH2 is significantly enhanced by electron-withdrawing groups, whereas the oxidation of p-substituted aromatic alcohols may be only slightly affected by the substituents. The substituent effects on catalysis generally reflect the effects on the equilibrium constant for the reaction, where electron-withdrawing substituents favor alcohol. The results are consistent with a transition state that is electronically similar to the alcohol, supporting previous results obtained with commercial yeast ADH.
Keywords: Alcohol dehydrogenase, Substrate specificity, Enzyme kinetics, Quantitative structure-activity relationships, Transition state, X-ray crystallography
1. Introduction
The zinc-containing, medium-chain alcohol dehydrogenases catalyze the interconversion of ethanol and NAD+ to acetaldehyde and NADH, and they are commonly found in bacteria, yeasts, plants and animals [1–3]. Mammalian ADHs react efficiently with a great variety of alcohols, aldehydes, and ketones, but yeast ADHs have more restricted specificities and are most active on ethanol and acetaldehyde [4,5]. The yeast S. cerevisiae produces three ADH isoenzymes - ADH1, ADH2, and ADH3. ADH1 is the constitutive, cytoplasmic form that reduces acetaldehyde during fermentation of glucose [6–8]. A three-dimensional structure of this enzyme has been determined (2HCY.pdb). ADH2 is also a cytoplasmic form, but it is repressed by glucose and functions to oxidize ethanol in aerobic metabolism [9]. ADH3 is a mitochondrial form, perhaps involved as a shuttle for reducing equivalents in NADH [10]. The kinetic constants of the S. cerevisiae enzymes are similar, except that ADH2 has about a 20-fold lower Km and 10-fold higher catalytic efficiency (V1/Km) for ethanol [11].
The mechanism of hydrogen transfer catalyzed by yeast ADH was studied extensively in a series of significant papers, using substituted benzyl alcohols and benzaldehydes [12–16]. However, the studies led to conflicting conclusions about the structure of the transition state. Quantitative structure-activity relationship studies with p-substituted benzyl alcohols and aldehydes suggested that the transition state resembled alcohol with little or no charge development at C-7 [14]. In contrast, the α-secondary kH/kT isotope effects for oxidation of benzyl alcohols were within error the same as the equilibrium isotope effects, suggesting that the transition state resembles aldehyde [15]. Further studies of the isotope effects suggested that hydride is transferred with quantum mechanical tunneling, with coupled motion and a transition state similar to alcohol [16]. The commercial enzyme used for these studies has much less activity on the aromatic substrates than on ethanol or acetaldehyde, but the cloned isoenzyme I from S. cerevisiae (SceADH1) had “no detectable activity” on benzyl alcohol, i.e., less than 1/105 of the activity on ethanol [4]. Thus, the origin of the activity on benzyl alcohol is uncertain, and we sought to determine which ADHs are active on benzyl alcohol and benzaldehyde.
The protein sequence of commercial yeast ADH is microheterogeneous [6], and it was suggested that the preparations contain ADH1 and ADH2 [17]. We noted that commercial preparations of yeast ADH are electrophoretically heterogeneous, with major components migrating with mobilities similar to that of the major enzyme isolated from S. carlsbergensis, brewer’s yeast, and to SceADH2, but different than SceADH1. S. cerevisiae is a top fermenting yeast for brewing ales, and S. calsbergensis is a bottom fermenting yeast for brewing lager beers [18]. These are related species [19], but S. carlsbergensis is also named S. pastorianus and may be a hybrid between S. cerevisiae and S. monacensis [20] or between S. cerevisiae and S. bayanus [21]. We cloned and expressed the gene for ADH1 from S. carlsbergensis and compared its activities on various substrates with the activities of SceADH1 and SceADH2. SceADH2 was identified as the isoenzyme most active on the aromatic substrates, and it was used to repeat the studies on the p-substituted benzyl alcohols and benzaldehydes.
2. Experimental Procedures
2.1. Materials
All alcohols were obtained from Aldrich or Fisher Scientific and redistilled before use. DEAE-Sepharose CL-4B and Octyl-Sepharose CL-4B were purchased from Pharmacia P-L Biochemicals. LiNAD+ and Na2NADH were the best grades available from Boehringer Mannheim, and pyrazole was purchased from Eastman Kodak. Commercial yeast ADH was obtained from Boehringer Mannheim Corporation, Cooper Labs, or Sigma Chemical Co.
2.2. Cloning and Expression
Genomic DNA was isolated from S. carlsbergensis strain Y379-50 by standard procedures [22]. DNA fragments of about 1600 base-pairs produced by restriction enzyme SphI were identified by hybridization with the ADH1 gene from S. cerevisiae and subcloned into the yeast expression plasmid YEp13, which was transformed into E. coli strain DH5α [23]. Colonies containing the ADH gene were identified by hybridization with the SceADH1 gene, and the plasmid was purified and characterized by restriction mapping and DNA sequencing [24]. One strand of the plasmid was sequenced with four different primers, providing the complete sequence for the coding region and 283 nucleotides of the 5′ and 186 nucleotides of the 3′ flanking regions. (The DNA sequence for the Saccharomyces carlbergensis ADH1 has been deposited in the GenBank/Embl Data Bank with accession number FJ195977.) Yeast strain 302-21#2, which does not produce ADHs [11], was transformed with the plasmid, and cells with the YEp13 plasmid were selected on synthetic medium lacking leucine.
2.3 Enzyme purification and characterization
Yeast transformed with plasmids expressing the ADH1 and ADH2 genes from S. cerevisiae and the ADH1 gene from S. carlsbergensis were grown in rich media, and the recombinant enzymes were purified as described previously [11,25]. The final enzyme preparations appeared to be at least 90 % pure by polyacrylamide gel electrophoresis in the presence or absence of sodium dodecyl sulfate [26]. Enzyme concentrations were determined by titration of the active sites with NAD+ in the presence of 10 mM pyrazole [27].
2.4. Enzyme kinetics
Activities on various substrates was determined in 83 mM potassium phosphate, 40 mM KCl, and 0.25 mM EDTA buffer, pH 7.3, at 30 °C, conditions that are similar to those in living yeast. The initial velocities for the change of absorbance at 340 nm on CARY 118C spectrophotometer were estimated by a linear or parabolic fit to the progress curve. Because the enzyme activities on many of the alcohols (other than linear aliphatic alcohols) studied here were relatively low, large amounts of enzyme (up to 0.1 mg/ml) were used. Contamination of solutions by readily oxidized alcohols was minimized by using redistilled alcohols and purified water. Activities were always at least 5 times higher than the activity determined in the absence of added alcohol. For assays with poor substrates, progress curves were obtained for about 5 min, and any small “burst” of activity (due to contaminants) was ignored in the analysis of the initial velocity. As a test of the procedures, tert-butyl alcohol was tested as a substrate and “activity” was less than 1/10th of the activity on the poorest substrate. Concentrations of alcohols were used that did not produce substrate inhibition, and the highest concentrations are reported in the results. The kinetic data were fitted to the appropriate equations using the programs HYPER for fitting to the Michaelis-Menten equation when one substrate is varied, SEQUEN or PING PONG for fitting initial velocity data for studies when coenzyme and substrate concentrations were each varied over a 9-fold range for a total of 25 combinations of concentrations, and COMP or NONCOMP for product inhibition studies [28]. The standard errors of the kinetic constants were usually less than 25 % of the values, indicating good precision and fits. In general, the values should be reproducible within a factor of 2.
The products of oxidation of benzyl alcohol in a reaction mixture containing 2 mM NAD+, 50 mM benzyl alcohol, and 10 to 30 μg/ml of enzyme at pH 7.3 and 30 °C were also determined. The production of NADH was followed at 340 nm. Production of benzaldehyde was determined by HPLC. Aliquots of 200 μl were taken at various times of reaction (up to 90 or 180 min) and immediately quenched in 800 μl of acetonitrile/acetic acid/water (30:1:69) containing 1 mM p-bromobenzoic acid as a internal standard. Samples were chromatographed on an Altex Ultrasphere Octyl column (4.6 mm × 25 cm) developed with acetonitrile/acetic acid/water (30:1:69) at 1 ml/min, with detection of the products at 254 nm [29].
3. Results
3.1. Characterization of S. carlsbergensis ADH1
As compared to the gene for SceADH1, the DNA sequence for ScbADH1 differs at 12 positions in the coding region and 2 in the 3′-flanking region. These changes would result in four substitutions in the amino acid sequence, as listed in Table 1. In three out of these four positions, ScbADH1 has the same amino acid sequence as does SceADH2 and commercial ADH, thought to be from S. cerevisiae [6], but SceADH1 and SceADH2 differ at 23 amino acid residues [9]. We conclude that ScbADH1 is the homolog of SceADH1. The ScbADH1 gene differs at 41 of 1044 nucleotides in the coding region of the S. pastorianus ADH1 gene. The sequence of the SceADH1 gene [8] showed five differences as compared to the protein sequence determined from the commercial enzyme, but preparations of commercial enzyme may be microheterogeneous [17]. Our sequencing of the SceADH1 gene [11] and X-ray crystallography (2HCY.pdb) gave the same sequence and showed that residue 20 is tyrosine, not histidine. Mass spectrometry of tryptic peptides of high quality commercial yeast ADH confirmed the amino acid compositions predicted from the DNA sequence for ScbADH1 (data not shown). The results show that commercial yeast enzyme apparently is prepared from S. carlsbergensis.
Table 1.
Comparison of Amino Acid Sequences of Yeast Alcohol Dehydrogenases
| Residue no. | SceADH1 | ScbADH1 | comADH | SceADH2 | SceADH3 |
|---|---|---|---|---|---|
| 58 | Val | Thr | Thr | Thr | Val |
| 127 | Gln | Glu | Glua | Glu | Gln |
| 147 | Gln | Glu | Glu | Glu | Glu |
| 151 | Ile | Val | Val | Ile | Ile |
The differences in amino acid sequences of the ADHs are also apparent on gel electrophoresis under non-denaturing conditions, at pH 8.4, where enzyme is located by activity or protein stain (Table 2). ScbADH1 migrates faster than SceADH1, slower than SceADH3, and at about the same rate as SceADH2 or commercial enzymes (which are heterogeneous and have a broad band). All enzymes migrate with the same relative molecular weight under denaturing conditions. The increased mobility of ScbADH1 relative to SceADH1 can be explained by the two additional negative charges due to the glutamates at positions 127 and 147. Electrophoretic mobilities are also altered with site-directed mutations that change one charge [25,30,31]. However, the change in mobility is not simply proportional to the change in charge, and although SceADH1 and SceADH2 have the same calculated net charge they migrate differently.
Table 2.
Relative Electrophoretic Mobilities of the Yeast Alcohol Dehydrogenases
| SceADH1 | ScbADH | SceADH2 | SceADH3 |
|---|---|---|---|
| 0.29 | 0.45 | 0.50 | 0.09 |
The Rf values are calculated relative to the dye front in electrophoresis in 6% polyacrylamide gel under non-denaturing conditions [26].
The kinetic constants of ScbADH1 obtained from initial velocity studies are most similar to those for SceADH1 for the oxidation of ethanol and reduction of acetaldehyde (Table 3). ScbADH1 appears to have about a 2-fold higher turnover number for reduction of acetaldehyde than SceADH1 does, which might explain the vigorous fermentative ability of S. carlsbergensis. SceADH2 has lower turnover numbers than the ADH1s, but catalytic efficiency is higher because the Kb (Michaelis constant for ethanol) and Kp (Michaelis constant for acetaldehyde) are 7 to 20-fold smaller. The kinetic mechanism for yeast ADHs acting on ethanol and acetaldehyde is predominantly ordered Bi Bi, as shown by previous experiments [11,32,33].
Table 3.
Kinetic Constants for Yeast Alcohol Dehydrogenases
| Kinetic constanta | SceADH1b | ScbADH1 | SceADH2c |
|---|---|---|---|
| Ka (μM) | 160 | 190 | 110 |
| Kb (μM) | 21,000 | 18,000 | 810 |
| Kp (μM) | 740 | 1080 | 90 |
| Kq (μM) | 94 | 210 | 50 |
| Kia (μM) | 950 | 1300 | 580 |
| Kiq (μM) | 31 | 50 | 11 |
| V1/Et (s−1) | 360 | 480 | 130 |
| V2/Et (s−1) | 1800 | 3500 | 1000 |
| Activity (s−1)d | 400 | 500 | 220 |
| Keq (pM)e | 12 | 16 | 12 |
Activity was measured at 30 °C in 83 mM potasium phosphate buffer, pH 7.3, containing 40 mM KCl, and 0.25 mM EDTA. Ka, Kb, Kp and Kq are the Michaelis constants for NAD+, ethanol, acetaldehyde and NADH, respectively; Ki values are the inhibition constants, and V1/Et and V2/Et are the turnover numbers for ethanol oxidation and acetaldehyde reduction. The kinetic constants for SceADH2 were determined from product inhibition studies. The errors were less than 15 % of the values.
Data from [25].
Data from [11].
The turnover number in the standard assay [49] is based on titration of active sites.
The equilibrium constants calculated using the Haldane equation, Keq = (V1KpKiq[H+])/(V2KbKia), are similar to the established value of 12 pM at 30 °C [50].
3.2. Substrate Specificities
The kinetic constants describing the activities of these ADHs on various alcohols are compared in Table 4. SceADH1 and ScbADH1 show about the same specificities, but SceADH2 has significantly higher activity (V1/Et and V1/Kb) for all of the alcohols. These results again confirm that the cloned S. carlsbergensis gene codes for ADH1. The secondary and branched chain alcohols are oxidized with much lower turnover numbers and catalytic efficiencies than the linear, primary alcohols. The variation in turnover numbers is consistent with hydride transfer being rate-limiting for catalysis, as has been confirmed by the significant deuterium isotope effect on V1/Et for oxidation of 2-propanol [34,35]. All of these enzymes prefer the S-isomer of butanol as compared to the R-isomer. Similar results were obtained earlier with commercial yeast ADH [5,36]. In general, increasing the size of the alcohol or introducing branching with additional methyl groups decreases catalytic efficiency for SceADH1 [4,11]. The yeast enzyme is inactive towards secondary alcohols where both alkyl groups are ethyl or larger [5], and we detected no activity with cyclohexanol as a substrate (although we found it to be a competitive inhibitor against ethanol with a Ki value of 7.8 mM).
Table 4.
Survey of Activities on Branched and Long Chain Aliphatic Alcoholsa
| Enzyme | 2-Propanol | R-2- Butanol | S-2- Butanol | 2-Methyl-1-propanol | S-2- Methyl-1-butanol | R,S-2- Methyl-1-butanol | 3- Methyl-1-butanol | Pentanol | Octanol |
|---|---|---|---|---|---|---|---|---|---|
| V1/Et (s−1) | |||||||||
| SceADH1 | 4.1 | 0.026 | 0.92 | 0.022 | 0.0018 | 0.0036 | 0.042 | 6.0 | 2.5 |
| ScrlADH | 5.1 | 0.030 | 1.2 | 0.026 | 0.003 | 0.02 | 0.046 | 9.1 | 9.3 |
| SceADH2 | 23 | 0.44 | 13 | 0.70 | 0.20 | 0.11 | 0.042 | 93 | 51 |
| Kb (mM) | |||||||||
| SceADH1 | 150 | 59 | 55 | 41 | 14 | 24 | 29 | 24 | 2.1 |
| ScrlADH | 110 | 53 | 43 | 31 | 13 | 60 | 46 | 40 | 5.2 |
| SceADH2 | 130 | 110 | 96 | 55 | 28 | 21 | 26 | 7.5 | 0.50 |
| V1/Kb (M−1s−1) | |||||||||
| SceADHI | 27 | 0.48 | 14 | 0.54 | 0.13 | 0.14 | 0.14 | 250 | 1200 |
| ScrlADH | 28 | 0.58 | 28 | 0.84 | 0.22 | 0.42 | 1.0 | 230 | 1800 |
| SceADH2 | 180 | 4.0 | 140 | 13 | 0.71 | 5.2 | 16 | 12000 | 100000 |
| Highest alcohol concentrations (mM) | |||||||||
| SceADHI | 500 | 200 | 100 | 100 | 100 | 100 | 100 | 70 | 1.6 |
| ScrlADH | 500 | 200 | 100 | 100 | 100 | 100 | 100 | 70 | 1.6 |
| SceADH2 | 300 | 200 | 50 | 100 | 100 | 200 | 100 | 70 | 1.6 |
The NAD+ concentration was fixed at 1 mM, and substrate concentrations were varied over a 5-fold range of Kb with the highest concentrations used indicated in the table. The kinetic constants are apparent because the concentration of NAD+ was not saturating.
In order to determine which yeast ADHs are active on benzyl alcohols and aldehydes, we surveyed the activities of the three isoenzymes. Table 5 shows that the activities (V/Km) on these aromatic substrates are 3 to 5 orders of magnitude lower than the activities on ethanol and acetaldehyde (Table 3). SceADH1 and ScbADH1 show similar substrate specificities, but SceADH2 is about one order of magnitude more active (V/Et and V/Km) than SceADH1 and ScbADH1. The activity of SceADH1 on benzyl alcohol is about 1/105 of that on ethanol, confirming the previous conclusion [4]. Significantly for these studies, SceADH2 is about 100 times more active than SceADH1 is on benzyl alcohol.
Table 5.
Survey of Activities on Benzyl Alcohols and Benzaldehydesa
| Enzyme | Benzyl alcohol | p-Methoxy- benzyl alcohol | Benzaldehyde | p-Methoxy- benzaldehyde |
|---|---|---|---|---|
| V/Et (s−1) | ||||
| SceADH1 | 0.012 | 0.058 | 0.91 | 0.075 |
| ScbADH1 | 0.011 | 0.11 | 0.52 | 0.071 |
| SceADH2 | 1.9 | 0.87 | 12 | 0.86 |
| Km (mM) | ||||
| SceADH1 | 34 | 11 | 33 | 2.9 |
| ScbADH1 | 24 | 22 | 23 | 3.4 |
| SceADH2 | 49 | 6.9 | 14 | 5.9 |
| V/Km (M−1s−1) | ||||
| SceADH1 | 0.32 | 5.3 | 27 | 26 |
| ScbADH1 | 0.48 | 4.5 | 23 | 21 |
| SceADH2 | 38 | 130 | 860 | 150 |
| Highest Concentrations of Alcohols and Aldehydes (mM) | ||||
| SceADH1 | 25 | 15 | 32 | 5 |
| ScbADH1 | 25 | 15 | 32 | 5 |
| SceADH2 | 25 | 35 | 55 | 5 |
Assays used 2 mM NAD+ and different concentrations of NADH (250 μM NADH for benzaldehyde and 100 μM NADH for p-methoxybenzaldehyde). V is V1 for alcohols and V2 for aldehydes. Km is the Kb for the alcohols and Kp for the aldehydes. The kinetic constants are apparent because the concentrations of coenzymes were not saturating.
Since the rates of oxidation of benzyl alcohol as determined by the change in NADH concentration are so slow, it is important to ascertain that benzyl alcohol is being oxidixed to benzaldehyde. The formation of benzaldehyde from benzyl alcohol upon oxidation with NAD+ as catalyzed by SceADH1, ScbADH1 and SceADH2 was confirmed by HPLC analyses. The concentrations of the products benzaldehyde and NADH were very similar throughout the course of the reaction, indicating a stoichiometric reaction. The rates of the reaction were comparable to those determined by steady-state kinetics. No benzoic acid was detected, suggesting that yeast ADH does not catalyze the dismutation of benzaldehyde, in contrast to the action of horse liver ADH [29].
The activity of a commercial preparation of yeast ADH on benzyl alcohol gave a V1/Et of 0.058 s−1 and a V1/Kb of 1.7 M−1s−1, which is about 4 times larger than the values obtained for the cloned SceADH1 or ScbADH1 (compare to Table 5). Since the commercial preparation is electrophoretically heterogeneous, it may contain a mixture of ADH1 and ADH2. Using the kinetic constants (V1/Kb) for the cloned enzymes, we can estimate that the commercial enzyme has about 3 % ADH2, which would contribute about 75 % of the activity on benzyl alcohol. The percentage may vary with different preparations, of course.
3.3. Quantitative structure-activity relationships
The relationships for the reactions of the p-substituted benzyl alcohols and benzaldehydes were studied previously with commercial yeast enzyme [12,14], but it seemed prudent to repeat these studies using a single enzyme form. The kinetic constants for SceADH2 acting on p-substituted aromatic alcohols and aldehydes were determined by varying the concentrations of both coenzyme and substrate and fitting the data to the equation for a sequential bi-substrate reaction, which provides true kinetic constants. As shown in Table 6, the p-substitutions have relatively small effects on alcohol oxidation, in particular on V1/Et and V1/Kb, which report on the hydrogen transfer step and the catalytic efficiency, respectively. The kinetic constants are similar to those observed previously [14], except that the turnover numbers are somewhat higher in the present study. It is difficult to compare the values directly because alcohol oxidation is faster at pH above a pK of 8.25 [13], and the previous study was done at pH 8.5 and 25 °C and the present study at pH 7.3 and 30 °C.
Table 6.
Kinetic Constants for the Oxidation of p-Substituted Benzyl Alcohols by SceADH2a
| Alcohols | Ka (mM) | Kb (mM) | Kia (mM) | V1/Et (s−1) | V1/Kb (M−1s−1) |
|---|---|---|---|---|---|
| CH3O- | 1.0 | 9.4 | 0.20 | 1.3 | 140 |
| CH3- | 0.66 | 4.1 | 1.20 | 2.3 | 570 |
| CH(CH3)2- | 1.2 | 3.6 | 0.56 | 2.0 | 540 |
| H- | 7.5 | 16 | 3.6 | 1.5 | 94 |
| Cl- | 1.2 | 7.2 | 0.42 | 3.8 | 530 |
| Br- | 1.3 | 8.4 | 0.37 | 5.9 | 700 |
| CF3- | 4.2 | 7.7 | 2.3 | 1.5 | 190 |
Data were fitted with SEQUEN program to the equation v = VAB/(KiaKb + KaB + KbA +AB), where A and B are substrate concentrations; Ka and Kb are the Michaelis constants for NAD+ and ethanol; Kia is the inhibition constant for NAD+; V1/Et is the turnover number and V1/Kb (= V1/EtKb) is the catalytic efficiency for alcohol oxidation.
For the reduction of the aldehydes, in contrast, the p-substituents have large effects, with electron-withdrawing substituents increasing V2/Et and V2/Kp by about 100-fold over the range studied (Table 7). The overall magnitude of the electronic effects on V2/Et is very similar to the previous results [12], but the turnover numbers are about 30-fold larger in the present study, in part due to the lower pH used in the present study. The Michaelis constants for the benzaldehydes are similar in magnitude to those in the previous study, but the dependence of V2/Kp on the electronic effects differs. The internal consistency of the kinetic constants was checked by calculating the Haldane relationship, Keq = V1KpKiq[H+]/V2KbKia, which gave reasonably good agreement with the values determined by Klinman [14] for the p-CH3O-, CH3-, Cl-, and Br- compounds.
Table 7.
Kinetic Constants for Reduction of p-Substituted Benzaldehydes by SceADH2a
| Aldehyde | Kq (μM) | Kp (mM) | Kiq (μM) | V2/Et (s−1) | V2/Kp (M−1s−1) |
|---|---|---|---|---|---|
| CH3O- | 17 | 5.5 | 5.6 | 0.83 | 150 |
| CH3- | 89 | 19 | 1.7 | 2.6 | 140 |
| F- | 24 | 17 | 11 | 16 | 930 |
| H- | 240 | 26 | b | 23 | 860 |
| Cl- | 84 | 11 | 20 | 59 | 5400 |
| Br- | 11 | 7.2 | 12 | 73 | 10000 |
| CN- | 400 | 16 | 6.7 | 250 | 15000 |
Data were fitted with the SEQUEN program. Kq and Kp are the Michaelis constants for NADH and acetaldehyde. Kiq is the inhibition constant for NADH. V2/Et is the turnover number and V2/Kp is the catalytic efficiency for aldehyde reduction.
Value not determined as the data fit best to the equation for a Ping Pong mechanism, where Kiq = 0.
The classical approach for analyzing the electronic effects of substituents uses the Hammett equation, supplemented by consideration of the hydrophobicity, molar refractivity, and van der Waals radii of the substituents, which may be factors in determining the enzyme specificity [37,38]. Multiple linear regression of the data for benzyl alcohol oxidation showed no significant correlation of the kinetic constants with any of these factors, because the rate constants were essentially not affected by the substituents. Fig. 1 shows the relationship of V1/Et and V1/Kb. with σ+, which describes reactions in which electron donation by resonance to the reaction center is important [37]. In contrast, the reduction of the benzaldehydes as measured by V2/Et or V2/Kp showed a good correlation with σ+ with a ρ value (slope of the dependence) of 1.9 or 1.7 (Fig. 2). The substituent effects on Kp alone are relatively small, as shown by inspection of the data in Table 7 and the similar slopes of the lines in Fig. 2. Combinations with additional factors resulted in modestly better fits, but more data points (additional substituted benzaldehydes) would be required to justify including the other factors.
Fig. 1.

Hammett plots of the quantitative structure-activity relationship for oxidation of p-substituted benzyl alcohols by SceADH2. The data are from Table 6, where V1/Et (●) and V1/Kb (■) are plotted against the σ+ values for CH3O-, CH3- CH(CH3)2- H-, Cl-, Br-CF3-, from left to right. The lines were calculated by linear regression, but the slopes were not significantly determined: for log (V1/Et) slope is (0.19 ± 0.25)σ+, and for log (V1/Kb) slope is (0.06 ± 0.36)σ +.
Fig. 2.

Hammett plots of the quantitative structure-activity relationship for reduction of p-substituted benzaldehydes by SceADH2. The data are from Table 7, where V2/Et (●) and V2/Kp (■) are plotted against the σ+ values for CH3O-, CH3-, H-, Cl-, Br- CN-, from left to right. and lines were calculated by linear regression with the following results.
4. Discussion
4.1. Enzyme structure and substrate specificities
The cloning and sequencing of the ADH1 gene from S. carlsbergensis shows that ScbADH1 differs from SceADH1 by four amino acid residues, which change the electrophoretic mobility of the proteins, but cause only small effects on the kinetic constants for enzymatic activity on various substrates. It appears that commercial yeast ADH is produced from S. carlsbergensis and contains predominantly ADH1. The commercial ADH does not appear to be produced from S. pastorianus, which is the name also used for S. carlsbergensis. However, commercial ADH preparations are usually heterogeneous and may also contain ADH2. The ADH2 gene from S. carlsbergensis should also be cloned so that ScbADH2 can be characterized. SceADH2 has an electrophoretic mobility similar to that ScbADH1, but clearly differs by having higher activity (V1/Et and V1/Km) on aliphatic, branched chain, and aromatic alcohols.
SceADH2 is more active than SceADH1 on the larger alcohols. These enzymes differ in 24 amino acid residues (if position 20 is considered), but the only difference in the substrate binding site is Leu-270 in SceADH2 as compared to Met-270 in SceADH1 and ScbADH1. SceADH3 also has Leu-270 [10], and its activity on benzyl alcohol should be determined. The M270L substitution in SceADH1 increases catalytic efficiency with butanol, pentanol, and hexanol, but not on ethanol or propanol [11]. Residues distant from the active site must be implicated in the 10-fold higher catalytic activity of SceADH2 as compared to SceADH1 on ethanol (Table 3), but these have not been identified.
A three-dimensional structure of SceADH1 complexed with NAD+ and 2,2,2-trifluoroethanol has been determined by X-ray crystallography (2HCY.pdb), which makes it possible to explain the higher activity of SceADH2 as compared to SceADH1 on benzyl alcohol. A model of the yeast enzyme was constructed by replacing the trifluoroethanol with the benzyl alcohol, positioning the hydroxymethyl group so that the pro-R hydrogen is directed toward C4 of the nicotinamide ring, and rotating the benzene ring into the position with optimized contacts with the amino acid residues (Fig. 3). SceADH1 has Thr-45, Trp-54, Trp-92, Met-270 and Tyr-294 in close contact with the substrate, but Met-270 makes a bad contact (1.8 Å) that is not relieved by changing rotamers. The M270L substitution would relieve some of the steric hindrance as illustrated in Fig. 3. Nevertheless, some close contacts remain, and we must assume that the enzymes are somewhat flexible if benzyl alcohol can bind and be oxidized.
Fig. 3.
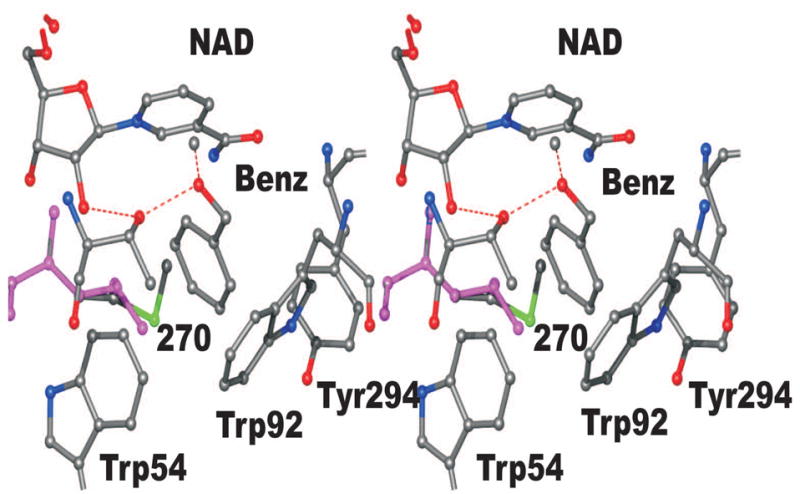
Model of the active site of yeast alcohol dehydrogenase with benzyl alcohol bound. The coordinates are based on the structure of the SceADH1 complexed with NAD+ and 2,2,2-trifluoroethanol (2HCY.pdb), with benzyl alcohol replacing trifluoroethanol. In this model, the oxygen of benzyl alcohol binds the catalytic zinc and the hydroxyl group of Thr-45. The benzene ring makes some unfavorable steric contacts with the three labeled aromatic amino acid residues and especially with Met-270. By substituting Met-270 with Leu-270 (which has the compact isopropyl group) these bad contacts are relieved, but the other close contacts remain.
Horse liver ADH has a large substrate binding site and readily accommodates and efficiently oxidizes benzyl alcohol, cyclohexanol, and branched chain alcohols [39–41]. The horse enzyme has much higher activity than SceADH1 does on branched chain alcohols [4]. The lower activity of SceADH1 with larger alcohols apparently reflects the restricted substrate binding pocket. Enlarging the substrate binding pocket in SceADH1 with the T45S and W92A substitutions produced enzymes that were more active on branched chain and benzyl alcohols as compared to wild-type SceADH1, and the double substitution inverted the relative specificity for ethanol as compared to hexanol by a factor of 5000 [4]. The horse enzyme has Ser-48, Phe-93 and Val-294 corresponding to Thr-45, Trp-92 and Met/Leu-270 in yeast ADH. It remains to be determined why SceADH1 is twice as active as horse ADH on ethanol, whereas horse ADH is 24,000 times more active than SceADH1 on benzyl alcohol.
4.2. Transition State Analysis
Klinman previously studied structure-activity relationships with commercial yeast ADH [12,14]. The major observations were a substantial electronic effect on the turnover for benzaldehyde reduction (ρ value of 2.1 against σ+ for V2/Et), no electronic effect on turnover for benzyl alcohol oxidation, a modest electronic effect on benzaldehyde binding (ρ value of −0.9 for 1/Kp), and a ρ value of 1.5 on the equilibrium constant for aldehyde reduction. The hydride transfer steps were predominantly rate-limiting as the average kinetic isotope effects (using NADD or NADH) were 3.6 on V2/Et (kcat) for aldehyde reduction and 4.0 on V1/Et for oxidation of dideutero alcohols. The isotope effects could also be analyzed to show that the Michaelis constants for aldehyde or alcohol approximate the dissociation constants. Because the electronic effects on V2/Et were offset by the effects on binding of aldehyde so that the net value of ρ of 1.2 calculated for V2/Kp was similar to the effect on the equilibrium constant, it was concluded that the electronic effects are on the ground state, rather than on the transition state. The lack of an electronic effect on benzyl alcohol oxidation then led to the conclusion “that there is little or no development of charge at C-7 at the transition state relative to the alcohol in the ground state” [14]. The implication that the transition state resembles the alcohol is puzzling because subsequent studies on the secondary isotope effects suggested that bond hybridization at the transition state resembles the aldehyde [15,42]. However, the interpretation of the isotope effects is complicated by the fact that hydride transfer occurs with quantum mechanical tunneling [16,43], and interpretation of secondary isotope effects can be affected by a lack of synchrony in rehybridization of the donor and acceptor carbons [44].
Structure-activity relationships were also studied with horse liver ADH for which benzyl alcohols and benzaldehydes are excellent substrates [45]. However, hydride transfer is not rate-limiting for steady-state reactions with wild-type enzyme, and a chemically activated form of liver ADH, for which hydride transfer is at least partially limiting, was used. Significant substituent effects were found with four p-substituted benzaldehydes, with a ρ value of 1.1 on V2/Et and an average deuterium kinetic isotope effect of 2.3, and a ρ value of about 0.6 on V2/Kp. The reaction of three p-substituted benzyl alcohols showed only small substituent effects on V1/Et, but a ρ value of −0.6 can be estimated for the effects on V1/Kb. These results are qualitatively similar to those with yeast ADH.
The interpretation of the results from the studies with the p-substituted aromatic substrates is complicated because of the involvement of the catalytic zinc that binds the oxygen of the substrate and the histidine that participates in acid/base catalysis through a hydrogen bond relay system. The enzyme can modulate the development of charge in the transition state. By analogy with the homologous horse liver ADH, we can suggest that the alcohol binds to the catalytic zinc, becomes deprotonated by the catalytic histidine, and is stabilized as the alkoxide by a low barrier hydrogen bond to the hydroxyl group of Thr-45 [39,46,47]. Rapid reaction kinetics suggest that the zinc-OH2 loses a proton to form zinc-OH− before alcohol displaces the water [48]. Subsequently, the hydride ion is transferred from the alkoxide to the oxidized nicotinamide ring and the zinc-bound aldehyde is formed. The inverse solvent isotope effects for the hydride transfer step (2-fold faster in D2O than H2O) with ethanol or benzyl alcohol and a proton inventory study with ethanol suggest that the transition state has a charge on the oxygen of −0.3 [40,46]. Thus, the transition state may more closely resemble aldehyde than alcoholate, with respect to the charge on the oxygen, but the charge at C-7 in the transition state may be different.
The new results for reactions of p-substituted substrates with SceADH2 are qualitatively similar to the previous ones, but differ somewhat in the magnitudes of the effects and are more simply interpreted. The new results show that there are good linear correlations for both V2/Et and V2/Kp with σ+, producing ρ values of 1.9 and 1.7 with p-substituted benzaldehydes, respectively (Fig 2). In contrast to the previous results [12], it appears that there is no significant effect of the p-substituents on binding of the benzaldehydes. Note that V2/Kp describes the bimolecular reaction of the enzyme-NADH complex with benzaldehyde to form the enzyme-NAD+ complex and free benzyl alcohol, whereas V2/Et describes the conversion of the ternary enzyme-NADH-benzaldehyde complex to form free enzyme, NAD+ and benzyl alcohol. When hydride transfer is the rate-limiting step and the kinetic mechanism is rapid equilibrium random [12,14], the difference in the ρ values for V2/Kp and V2/Et would correspond to the ρ value for the binding of aldehyde. However, fitting of log(1/Kp) against σ+ gave a ρ value of −0.2 ± 0.2, not a statistically significant fit. The ρ values of 1.7–1.9 appear to be larger than the ρ value of 1.5 for the equilibrium constant, which would be consistent with a substituent effect on the transition state. This would imply that the transition state develops some negative charge relative to aldehyde (as the hydride ion attacks), which is stabilized by electron withdrawing groups (promoting the reduction). The ρ values of 2.3–2.6 for reactions of benzaldehydes with NaBH4 and HCN also suggest development of some negative charge [12], but as noted above, enzyme catalysis may modulate the charge and have different ρ values.
For the oxidation of the benzyl alcohols, the substituents slightly, but not significantly, affect V1/Et and V1/Kb, and there does not appear to be an effect on binding of the alcohol (slope of Hammet plot = −0.13 ± 0.38). In contrast, the previous results with yeast ADH found that binding of alcohol was favored by hydrophobic effects [14].
Because it appears that hydride transfer is the rate limiting step for alcohol oxidation or aldehyde reduction, and there are only small effects of the substituents on binding, we can simplify the analysis of the effects by considering only the V/Km parameters, which report on the energetics of the conversion of free substrate to the transition state for the forward and reverse reactions: E-NAD+ + alcohol == E-NADH + aldehyde. This reaction is described by a rearranged Haldane equation: KeqKia/Kiq = (V1/Kb)/(V2/Kp). This relationship requires that ρ(Keq) = ρ(V1/Kb) − ρ(V2/Kp), and the present results fit within experimental error: (−1.5 ≈ 0.1 – 1.7), when the reaction is written for alcohol oxidation. The conclusion from this analysis is that the major effect of the substituents is due to the free energies of the ground states of the substrates rather than the transition state energies. Electron withdrawing substituents promote aldehyde reduction because they favor the thermodynamics of the reaction. The activation energies for alcohol oxidation may be slightly affected by the p-substituents, whereas the activation energies for aldehyde reduction are significantly affected by the substitutents. The structure-activity studies do not fully define the transition state, but the lack of significant substituent effects on alcohol oxidation is consistent with a transition state that electronically resembles alcohol. Further studies of isotope effects and a comprehensive analysis of the tunneling are required.
The structure-activity results with SceADH2 are consistent with the previous studies with commercial ADH, and the same general interpretation applies, but how can we account for the quantitative differences between the previous and present studies? Different enzyme preparations were used, and it is possible that the commercial enzyme was a mixture of isoenzymes, although the ADH2 in that preparation might have contributed most of the activity. Different buffers and pH values were used, and the lower pH used in this study would facilitate the reduction of aldehydes by promoting protonation of the alcoholate. Experimental errors giving a 2-fold difference on kinetic constants would yield differences of 0.3 on log plots, which is similar to the average differences between fitted and experimental values in Figs. 1 and 2. In order to increase the confidence in the kinetic analyses in this study, the range of compounds was extended to the -CN and -CF3 compounds, which are much more electron-withdrawing. Using a narrower range of compounds gives different ρ values. It is also possible that hydride transfer is not completely rate limiting for all substrates with SceADH2, but the extremely low turnover numbers and catalytic efficiencies compared to good substrates is most consistent with a rapid equilibrium random mechanism. Despite the differences in the studies, it is gratifying that the major features of the structure-activity relationships are the same for the yeast ADHs.
Acknowledgments
This work was supported by NIH Grants AA06223 and GM078446 and NSF Grant MCB 91-18657. We thank Drs. Axel J. Ganzhorn, David W. Green, and Jeffrey S. Kavanaugh for preliminary observations that led to this work and Mark D. Hermes for some results. We thank Kristine B. Berst and The University of Iowa Macromolecular Structure Facility for the mass spectral analysis of the peptides from ScbADH1. We thank Dr. Amnon Kohen for helpful discussions. We thank Drs. E. T. Young and B. D. Hall for the host yeast strains and the plasmids containing the genes for SceADH1 and SceADH2.
Abbreviations
- ADH
alcohol dehydrogenase
- SceADH1
ADH I from Saccharomyces cerevisiae
- SceADH2 and SceADH3
ADH isoenzymes II and III from S. cerevisiae
- ScbADH1
ADH I from Saccharomyces carlsbergensis
- comADH
commercial yeast ADH
- V1/Et
turnover number for alcohol oxidation
- V1/Kb
V1/EtKb, catalytic efficiency for alcohol oxidation
- V2/Et and V2/Kp
V2/EtKp, turnover number and catalytic efficiency for aldehyde reduction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sund H, Theorell H. Alcohol Dehydrogenases. The Enzymes. 1963;7:25–83. [Google Scholar]
- 2.Brändén CI, Jörnvall H, Eklund H, Furugren B. Alcohol Dehydrogenases. The Enzymes, 3rd Ed. 1975;11:103–190. [Google Scholar]
- 3.Sun HW, Plapp BV. Progressive sequence alignment and molecular evolution of the Zn-containing alcohol dehydrogenase family. J Mol Evol. 1992;34:522–535. doi: 10.1007/BF00160465. [DOI] [PubMed] [Google Scholar]
- 4.Green DW, Sun HW, Plapp BV. Inversion of the substrate specificity of yeast alcohol dehydrogenase. J Biol Chem. 1993;268:7792–7798. [PubMed] [Google Scholar]
- 5.Dickinson FM, Dalziel K. The specificities and configurations of ternary complexes of yeast and liver alcohol dehydrogenases. Biochem J. 1967;104:165–172. doi: 10.1042/bj1040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jörnvall H. The primary structure of yeast alcohol dehydrogenase. Eur J Biochem. 1977;72:425–442. doi: 10.1111/j.1432-1033.1977.tb11267.x. [DOI] [PubMed] [Google Scholar]
- 7.Young T, Williamson V, Taguchi A, Smith M, Sledziewski A, Russell D, Osterman J, Denis C, Cox D, Beier D. The alcohol dehydrogenase genes of the yeast, Saccharomyces cerevisiae: isolation, structure, and regulation. In: Hollaender A, DeMoss RD, Kaplan S, Konisky J, Savage D, Wolfe RS, editors. Genetic Engineering of Microorganisms for Chemicals. Plenum Publishing; New York: 1982. pp. 335–361. [DOI] [PubMed] [Google Scholar]
- 8.Bennetzen JL, Hall BD. The primary structure of the Saccharomyces cerevisiae gene for alcohol dehydrogenase. J Biol Chem. 1982;257:3018–3025. [PubMed] [Google Scholar]
- 9.Russell DW, Smith M, Williamson VM, Young ET. Nucleotide sequence of the yeast alcohol dehydrogenase II gene. J Biol Chem. 1983;258:2674–2682. [PubMed] [Google Scholar]
- 10.Young ET, Pilgrim D. Isolation and DNA sequence of ADH3, a nuclear gene encoding the mitochondrial isozyme of alcohol dehydrogenase in Saccharomyces cerevisiae. Mol Cell Biol. 1985;5:3024–3034. doi: 10.1128/mcb.5.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganzhorn AJ, Green DW, Hershey AD, Gould RM, Plapp BV. Kinetic characterization of yeast alcohol dehydrogenases. Amino acid residue 294 and substrate specificity. J Biol Chem. 1987;262:3754–3761. [PubMed] [Google Scholar]
- 12.Klinman JP. The mechanism of enzyme-catalyzed reduced nicotinamide adenine dinucleotide-dependent reductions. Substituent and isotope effects in the yeast alcohol dehydrogenase reaction. J Biol Chem. 1972;247:7977–7987. [PubMed] [Google Scholar]
- 13.Klinman JP. Acid-base catalysis in the yeast alcohol dehydrogenase reaction. J Biol Chem. 1975;250:2569–2573. [PubMed] [Google Scholar]
- 14.Klinman JP. Isotope effects and structure-reactivity correlations in the yeast alcohol dehydrogenase reaction. A study of the enzyme-catalyzed oxidation of aromatic alcohols. Biochemistry. 1976;15:2018–2026. doi: 10.1021/bi00654a032. [DOI] [PubMed] [Google Scholar]
- 15.Welsh KM, Creighton DJ, Klinman JP. Transition-state structure in the yeast alcohol dehydrogenase reaction: the magnitude of solvent and alpha-secondary hydrogen isotope effects. Biochemistry. 1980;19:2005–2016. doi: 10.1021/bi00551a001. [DOI] [PubMed] [Google Scholar]
- 16.Cha Y, Murray CJ, Klinman JP. Hydrogen tunneling in enzyme reactions. Science. 1989;243:1325–1330. doi: 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- 17.Wills C, Jörnvall H. The two major isozymes of yeast alcohol dehydrogenase. Eur J Biochem. 1979;99:323–331. doi: 10.1111/j.1432-1033.1979.tb13260.x. [DOI] [PubMed] [Google Scholar]
- 18.Rainbow C. Brewer’s Yeast. In: Rose AH, Harrison JS, editors. The Yeasts. Academic Press; New York: 1970. p. 148. [Google Scholar]
- 19.Hammond JR. The Yeasts. Academic Press; New York: 1993. Brewer’s Yeast; pp. 7–67. [Google Scholar]
- 20.Børsting C, Hummel R, Schultz ER, Rose TM, Pedersen MB, Knudsen J, Kristiansen K. Saccharomyces carlsbergensis contains two functional genes encoding the Acyl-CoA binding protein, One similar to the ACB1 gene from S. cerevisiae and one identical to the ACB1 gene from S. monacensis. Yeast. 1997;13:1409–1421. doi: 10.1002/(SICI)1097-0061(199712)13:15<1409::AID-YEA188>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 21.Dunn B, Sherlock G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008;18:1610–1623. doi: 10.1101/gr.076075.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherman F, Fink GR, Hicks JB. Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1986. [Google Scholar]
- 23.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 24.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gould RM, Plapp BV. Substitution of arginine for histidine-47 in the coenzyme binding site of yeast alcohol dehydrogenase I. Biochemistry. 1990;29:5463–5468. doi: 10.1021/bi00475a009. [DOI] [PubMed] [Google Scholar]
- 26.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Theorell H, Yonetani T. Liver alcohol dehydrogenase-DPN-pyrazole complex: A model of a ternary intermediate in the enzyme reaction. Biochem Z. 1963;338:537–553. [PubMed] [Google Scholar]
- 28.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 29.Shearer GL, Kim K, Lee KM, Wang CK, Plapp BV. Alternative pathways and reactions of benzyl alcohol and benzaldehyde with horse liver alcohol dehydrogenase. Biochemistry. 1993;32:11186–11194. doi: 10.1021/bi00092a031. [DOI] [PubMed] [Google Scholar]
- 30.Ganzhorn AJ, Plapp BV. Carboxyl groups near the active site zinc contribute to catalysis in yeast alcohol dehydrogenase. J Biol Chem. 1988;263:5446–5454. [PubMed] [Google Scholar]
- 31.Fan F, Lorenzen JA, Plapp BV. An aspartate residue in yeast alcohol dehydrogenase I determines the specificity for coenzyme. Biochemistry. 1991;30:6397–6401. doi: 10.1021/bi00240a008. [DOI] [PubMed] [Google Scholar]
- 32.Wratten CC, Cleland WW. Product inhibition studies on yeast and liver alcohol dehydrogenases. Biochemistry. 1963;2:935–941. doi: 10.1021/bi00905a007. [DOI] [PubMed] [Google Scholar]
- 33.Dickinson FM, Monger GP. A study of the kinetics and mechanism of yeast alcohol dehydrogenase with a variety of substrates. Biochem J. 1973;131:261–270. doi: 10.1042/bj1310261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickenson CJ, Dickinson FM. A study of the oxidation of butan-1-ol and propan-2-ol by nicotinamide-adenine dinucleotide catalysed by yeast alcohol dehydrogenase. Biochem J. 1975;147:541–547. doi: 10.1042/bj1470541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook PF, Cleland WW. pH variation of isotope effects in enzyme-catalyzed reactions. 1. Isotope- and pH-dependent steps the same. Biochemistry. 1981;20:1797–1805. doi: 10.1021/bi00510a014. [DOI] [PubMed] [Google Scholar]
- 36.Dickinson FM, Dalziel K. Substrate specificity and stereospecificity of alcohol dehydrogenases. Nature. 1967;214:31–33. doi: 10.1038/214031a0. [DOI] [PubMed] [Google Scholar]
- 37.Hammett LP. Physical Organic Chemistry. McGraw-Hill; New York: 1970. [Google Scholar]
- 38.Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. Wiley; New York: 1979. pp. 1–63. [DOI] [PubMed] [Google Scholar]
- 39.Ramaswamy S, Eklund H, Plapp BV. Structures of horse liver alcohol dehydrogenase complexed with NAD+ and substituted benzyl alcohols. Biochemistry. 1994;33:5230–5237. doi: 10.1021/bi00183a028. [DOI] [PubMed] [Google Scholar]
- 40.Sekhar VC, Plapp BV. Rate constants for a mechanism including intermediates in the interconversion of ternary complexes by horse liver alcohol dehydrogenase. Biochemistry. 1990;29:4289–4295. doi: 10.1021/bi00470a005. [DOI] [PubMed] [Google Scholar]
- 41.Lee KM, Dahlhauser KF, Plapp BV. Reactivity of horse liver alcohol dehydrogenase with 3-methylcyclohexanols. Biochemistry. 1988;27:3528–3532. doi: 10.1021/bi00409a060. [DOI] [PubMed] [Google Scholar]
- 42.Klinman JP. Probes of mechanism and transition-state structure in the alcohol dehydrogenase reaction. CRC Crit Rev Biochem. 1981;10:39–78. doi: 10.3109/10409238109114635. [DOI] [PubMed] [Google Scholar]
- 43.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 44.Pu J, Ma S, Garcia-Viloca M, Gao J, Truhlar DG, Kohen A. Nonperfect synchronization of reaction center rehybridization in the transition state of the hydride transfer catalyzed by dihydrofolate reductase. J Am Chem Soc. 2005;127:14879–14886. doi: 10.1021/ja054170t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dworschack RT, Plapp BV. pH, isotope, and substituent effects on the interconversion of aromatic substrates catalyzed by hydroxybutyrimidylated liver alcohol dehydrogenase. Biochemistry. 1977;16:2716–2725. doi: 10.1021/bi00631a020. [DOI] [PubMed] [Google Scholar]
- 46.Ramaswamy S, Park DH, Plapp BV. Substitutions in a flexible loop of horse liver alcohol dehydrogenase hinder the conformational change and unmask hydrogen transfer. Biochemistry. 1999;38:13951–13959. doi: 10.1021/bi991731i. [DOI] [PubMed] [Google Scholar]
- 47.Plapp BV. Catalysis by Alcohol Dehydrogenases. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. CRC Press, Taylor and Francis; Boca Raton: 2006. pp. 811–835. [Google Scholar]
- 48.Kovaleva EG, Plapp BV. Deprotonation of the horse liver alcohol dehydrogenase-NAD+ complex controls formation of the ternary complexes. Biochemistry. 2005;44:12797–12808. doi: 10.1021/bi050865v. [DOI] [PubMed] [Google Scholar]
- 49.Plapp BV. Enhancement of the activity of horse liver alcohol dehydrogenase by modification of amino groups at the active sites. J Biol Chem. 1970;245:1727–1735. [PubMed] [Google Scholar]
- 50.Bäcklin KI. The equilibrium constant of the system ethanol, aldehyde, DPN+, DPNH and H+ Acta Chem Scand. 1958;12:1279–1285. [Google Scholar]