

Abstract
A new approach that improves the efficiency and specificity of Polymerase Chain Reaction (PCR) has been developed. Heat sensitive 3’-protected derivatives of 2’-deoxyribonucleoside 5’-triphosphates (dNTPs) have been synthesized and used as substitutes for natural dTTP, dCTP, dATP and dGTP in PCR. Since 3’-protected dNTPs are either non-substrates or terminating substrates for Taq DNA polymerase they do not support primer extension/elongation at low stringency conditions during PCR sample preparation when PCR artifacts such as primer dimers and mis-priming products can form. At initial heat-denaturing step and during PCR sequence the 3’-protecting group is cleaved releasing 3’-unprotected dNTP that is a natural substrate for DNA polymerase. As a result, the primer extension/elongation proceeds only at elevated temperature of PCR, when the interaction of primers and template is highly stringent and specific. Several 3’-protecting groups covering a wide range of deprotection kinetics have been tested. The 3’-O-tetrahydrofuranyl derivatives of dNTPs have demonstrated the best properties leading to a drastically reduced accumulation of PCR artifacts such as “primer dimers” and “mis-priming” products. Overall, PCR with 3’-THF protected dNTPs demonstrated substantially improved performance and was more efficient and specific compared to PCR with standard dNTPs.
During the last two decades the polymerase chain reaction (PCR) became one of the most powerful techniques in biomedical and biological studies and applications due to its ability to copy and amplify efficiently and selectively nucleic acid regions of interest in the presence of large quantities of other nucleic acid sequences1-4. Although PCR is recognized as a highly accurate and specific technique, it is not free from artifacts, such as primer dimers and mis-priming products5,6. These unwanted products are largely formed under low stringency hybridization conditions that exist prior to PCR during sample preparation at ambient temperature and during the initial heat-denaturing ramp of the PCR process. Once formed, these off-target products can compete with amplification of the desired DNA target and significantly decrease the efficiency of PCR. While undesired products are commonly encountered in PCR, these non-specificity issues can be particularly challenging for high-sensitivity analytical PCR systems required in forensics7,8, detection of blood-borne infectious agents9,10, microbial pathogens11, genetic disorders12, and defective or cancerous genes13. Non-specific amplification artifacts such as mis-priming products or primer dimers are a big problem in quantitative real time PCR with SYBR Green detection, because they cause the background to appear earlier and interfere with quantification of the results.
A common solution for the reduction of unwanted PCR side products is a general approach collectively referred to as Hot Start PCR. This approach relies on the prevention of primer extension/elongation until the initial heat denaturing step brings the PCR mixture to a high temperature where the stringency of the primer/target hybridization is optimal. At high temperature specificity is controlled thermodynamically by increased preference for more stable perfectly matched primer/nucleic acid complexes compared to weaker, partially matched, primer/primer and primer/nucleic acid complexes.
The original Hot Start PCR approach is performed by a temporary withdrawal of critical component(s) of the PCR mixture, such as DNA polymerase, primers or magnesium ion from the solution thus preventing the primer extension reaction from happening14,15. The critical PCR component is added to the tube only after the whole mixture reaches high stringency conditions during the initial heat denaturing step. Hot Start PCR activation can be also achieved by placing a temporary physical barrier, such as a wax or sequester, between critical PCR components6, 16-19. The barrier melts or dissolves at high temperature of the PCR cycling allowing the critical components to mix with the rest of the solution resulting in improved PCR performance. However, these technical solutions are laborious and prone to mechanical errors especially for high throughput applications.
An advanced Hot Start PCR approach includes modified DNA polymerases that are made inactive at room temperature when a PCR mixture is typically prepared. A suppression of enzymatic activity of DNA polymerase is achieved either by temporary chemical modification of some critical aminoacid of the polymerase20, 21, by creating mutant enzyme22, or through a tight complex of DNA polymerase with a specific antibody23, protein24 or oligonucleotide aptamer25, 26 that prevents normal function of the enzyme. During the initial heat denaturing step, the enzyme is released from chemical or physical suppressors resulting in a functionally active enzyme molecule. The use of Hot Start DNA polymerases capable of thermal activation leads to vastly improved PCR performance, while substantially reduccing the labor and probability of technical mistakes. However, there are known thermostable DNA polymerases that are not available as Hot Start version. Conversion of DNA polymerase into a Hot Start form is a costly and lengthy development project.
Finally, there is an approach that relies on modified primers. These primers possess either unique structural feature or chemical modification that make them non-extendable or poorly extendable by DNA polymerase at low temperature27-37. At the elevated temperature of PCR, however, the primers loose structural constrains or convert to unmodified primers either chemically or enzymatically. Since unmodified primers become active and extendable after the initial denaturing step (Hot Start), they support PCR amplification. The effectiveness of modified primers in preventing of accumulation of PCR artifacts and their influence on PCR performance is dependent on either the efficiency of the structural constraint or chemical modification that suppresses the primer extension at low stringency conditions, and then, after Hot Start, the ability of primers to fully recover the extension properties. Precise effect of structural or chemical modification of the primer on PCR is hard to predict without performing experiments. Also, the modification, structural or chemical, has to be incorporated into each individual primer thus making this approach less attractive for applications where multiple modified primers are required.
Here we describe a novel and general solution to Hot Start PCR that is free of the shortcomings of other Hot Start approaches. The key element of the approach is substitution of natural 2’-deoxyribonucleoside 5’-triphosphates (dNTPs) in a PCR mixture with dNTP derivatives containing a thermolabile 3’-protecting group. In this work we describe several types of 3’-protected dNTPs and proof-of-principal experiments demonstrating that use of these compounds reduces the formation and accumulation of the off-target artifacts that interfere with the efficiency and specificity of PCR and leads to a substantial improvement in PCR performance. Preliminary results on the synthesis and use of 3’-protected dNTPs in PCR have been published recently38,39. Although modified dNTPs were used to improve PCR40-43, they were never previously proposed as a tool for Hot Start PCR applications. The report provides initial PCR results that should be of general interest to users of PCR, and serve as a prelude for our continued investigations aimed at demonstrating utility of 3’-protected dNTPs in heat-triggered PCR applications.
EXPERIMENTAL SECTION
Chemical and biochemicals
The chemicals used in this study were commercially obtained through Acros, Fisher Scientific, Sigma-Aldrich, or ChemGenes. Primers were obtained through TriLink BioTechnologies, Inc. Synthesis of 3’-protected 2’-deoxynucleoside 5’-triphosphates is described in Supporting Information section. Bacteriophage lambda genomic DNA was purchased from Roche Applied Science, HIV-1 recombinant genomic DNA was a component of the Gene Amplimer kit purchased from Applied Biosystems, and Human genomic DNA was purchased from Promega. Taq DNA polymerase (recombinant), 10 × PCR buffer and 50 mM MgCl2 were purchased from Invitrogen. The set of standard dNTP was purchased from New England Biolabs.
Analytical Instruments
HPLC was accomplished on a Beckman System Gold Nouveau Model 126 with Model 168 photodiode array detector. NMR spectra were recorded on Bruker Model AX 500 spectrometer (NuMega, San Diego, CA, USA). Electrospray mass spectrometry analyses were recorded on Finnigan MAT LCQ mass spectrometer.
Study of kinetics of conversion of 3’-protected derivatives of dTTP to 3’-unprotected dTTP in PCR buffer (Figures 1, 2; Table)
Figure. 1.
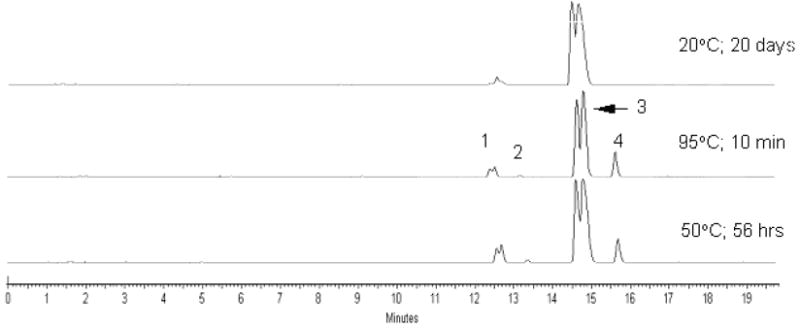
Anion exchange HPLC analyses of the 3’-THF dTTP after incubation in PCR buffer at different temperatures. Peak identification: (1) 3’-THF dTDP; (2) dTDP; (3) 3’-THF dTTP; (4) dTTP. Samples were analyzed by AX-HPLC on Dionex DNA Pac P-100 analytical column (4 × 250 mm) using a gradient of 1M LiCl in 25 mM Tris-base, pH 10.0 from 0 to 50% over 40 min, 1 mL/min.
Figure 2.
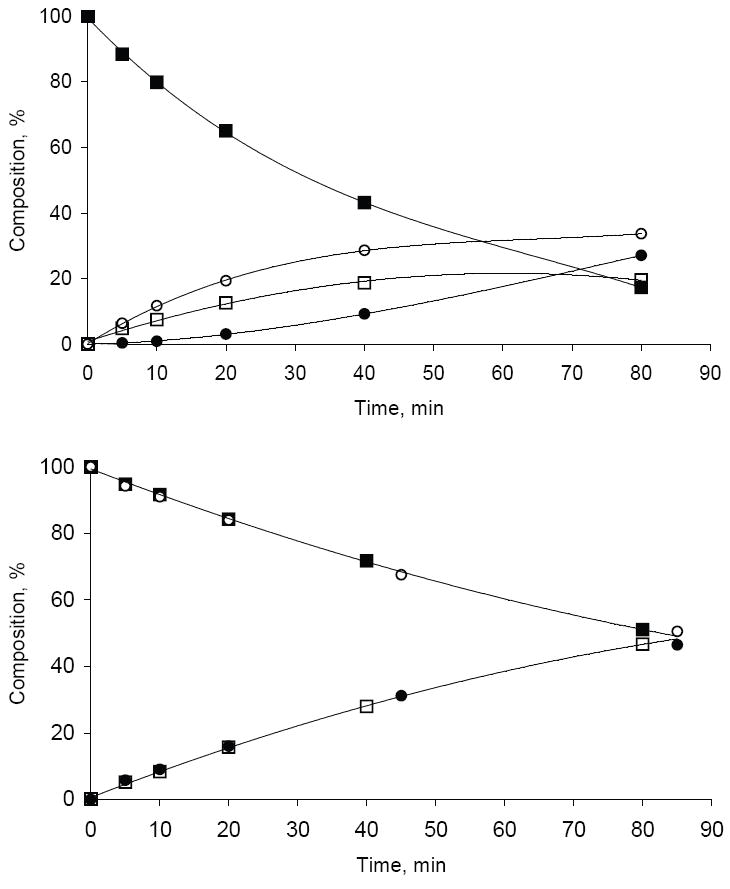
(A) Kinetic curves of conversion of 3’-THF dTTP in PCR buffer at 95°C: 3’-THF dTTP (■), dTDP (●), dTTP (○), 3’-THF dTDP (□). (B) Superimposed kinetic curves of conversion of 5’-triphosphate group to 5’-diphosphate group for 3’-THF dTTP and dTTP in PCR buffer at 95°C. For 3’-THF dTTP: sum of 3’-THF dTTP and dTTP (■), sum of 3’-THF dTDP and dTDP (□); for dTTP: dTTP(○), dTDP (●).
Table.
Estimated concentration of dTTP (μM; ±10%) generated from 3’-protected dTTP (200 μM initial concentration) in PCR buffer during PCR cycle sequence.
| Cycle number | THPa | MTHP | THF | Ac | Mac | Pac | H |
|---|---|---|---|---|---|---|---|
| Ob | 4 | 12 | 14 | 14 | 99 | 139 | 197 |
| 10 | 16 | 34 | 36 | 28 | 126 | 163 | 188 |
| 20 | 22 | 47 | 56 | 39 | 127 | 155 | 181 |
| 30 | 33 | 62 | 67 | 47 | 122 | 148 | 172 |
| 40 | 36 | 71 | 76 | 55 | 115 | 140 | 165 |
| τ1/2, minc | 250 | 100 | 80 | 200 | 8 | 6 | n/a |
3’-protected dTTP derivatives: 3’-THP, 3’-MTHP, 3’-THF, 3’-Ac, 3’-Mac, 3’-Pac; H: dTTP control
after initial 5 min denaturing step at 95°C
half-reaction time for deprotection of 3’-derivatives of dTTP at 95°C in PCR buffer estimated from kinetic curves (Figure S-1)
For kinetics studied at 95°C the aliquots of 0.2 mM solution of 3’-protected dTTP in 50 μL of 1 × PCR buffer (50 mM KCl, 2.5 mM MgCl2, 10 mM Tris-HCl, pH 8.4 at 25 °C) were placed in thin-walled 200 μL PCR tubes. One control tube was placed at −20°C immediately while other tubes were placed in a PCR thermal cycler running at 95°C. At a specified time, the tube was removed from thermal cycler and placed at −20°C. Each sample was analyzed by reverse phase HPLC on DeltaPak C18 analytical column (3.9 × 300 mm) using a gradient of acetonitrile in 100 mM TEAA, pH 7.2 over 40 min, 1 mL/min. On the resultant chromatogram, the HPLC peaks were integrated at 266 nm, half-reaction times were estimated from kinetic curves (see, Supporting information section) and presented in Table (estimated accuracy ± 10%). Similar approach was used to evaluate a stability of 3’-THF dTTP at room temperature and 50°C (Figure 1).
For kinetics studied in PCR conditions the experiment was performed as described above for 95°C experiment with the exception that the samples were incubated in PCR thermal cycler running the following PCR sequence: 95°C for 5 min (initial denaturation step), followed by 40 PCR cycles at 95°C for 40 sec, 56°C for 30 sec, 72°C for 120 sec. The tubes were removed after initial denaturation step before cycle 1, and after cycles 10, 20, 30, and 40. Samples were analyzed as described above and calculated concentrations of dTTP are presented in Table.
Endpoint PCR experiments with 3’-THF derivatives
PCR protocol used for experiments described in Figures 3 and 4 was set up by combining following components in a single, thin walled 200 μL tube. All reactions contained 1X PCR buffer (50 mM KCl, 2.5 mM MgCl2, 10 mM Tris-HCl, pH 8.4 at 25°C), 0.2 mM dNTPs and 0.5 U Taq DNA Polymerase in a 50 μL reaction volume covered by a drop of mineral oil (optional). HIV-1 reactions included 0.5 μM forward d(5′-GAATTGGGTGTCAACATAGCAGAAT-3′) and reverse d(5′-AATACTATGGTCCACACAACTATTGCT-3′) primers and 10 copies of HIV-1 recombinant DNA (as standardized from the Gene Amplimer kit), with 50 ng of human genomic DNA as a carrier. Reactions that amplified a 1.9 kbp region of the Lambda phage DNA included 0.5 μM each forward d(5′-AAGGAGCTGGCTGACATTTTCG-3′) and reverse d(5′-CGGGATATCGACATTTCTGCACC-3′) primers, 10000 copies of Lambda DNA and 50 ng of Human genomic DNA as a competing foreign DNA. The dNTP mixtures employed for these studies were either a mixture of four standard dNTPs, or a mixture where dTTP was substituted with a 3’-protected dTTP derivative. The enzyme was the last component added to each PCR tube just before placing the tube in the PCR instrument.
Figure 3.
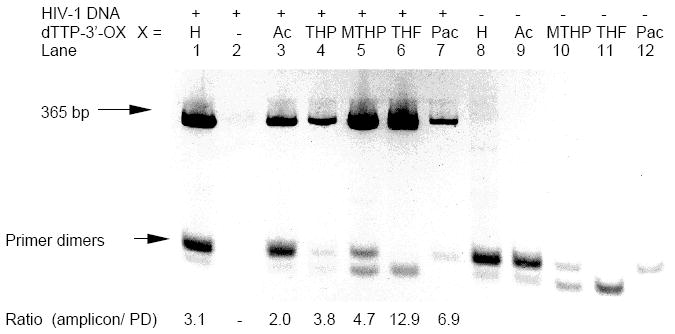
The 4% agarose gel analyses of PCR amplification of HIV-1 DNA template at 10 copies. Each lane contained dATP+dCTP+dGTP (200 μM each). To each reaction 3’-protected dTTP was added at 200 μM final concentration. Lanes 1 and 8: dTTP; lane 2: control (no dTTP or 3’-protected dTTP); lanes 3 and 9: 3’-Ac dTTP; lane 4: 3’-THP dTTP; lanes 5 and 10: 3’-MTHP dTTP; lanes 6 and 11: 3’-THF dTTP; lanes 7 and 12: 3’-Pac dTTP. Lanes 1-7: with HIV-1 DNA + HG DNA; lanes 8 -12: HG DNA only. At bottom: ratio of amplicon to primer dimers estimated by integration of UV-bands.
Figure 4.
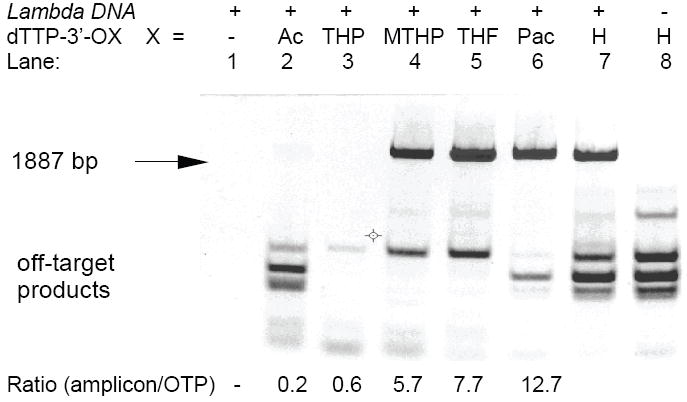
2% agarose gel analysis of PCR mixtures with Lambda phage DNA at 10,000 copies. Each lane contained dATP+dCTP+dGTP (200 μM each). To each reaction 3’-protected dTTP was added at 200 μM final concentration. Lane 1: control (no dTTP or 3’-protected dTTP); lane 2: 3’-Ac dTTP; lane 3: 3’-THP dTTP; lane 4: 3’-MTHP dTTP; lane 5: 3’-THF dTTP; lane 6: 3’-Pac dTTP; lanes 7 and 8: dTTP. Lanes 1-7: with Lambda DNA + HG DNA; lane 8: HG DNA only. At bottom: ratio of amplicon to off-target products estimated by integration of UV-bands.
The endpoint PCR protocol for the experiment shown in Figure 5 was set up as follows. A stock solution in 1 X PCR buffer that contained both forward and reverse HIV-1 primers, HIV-1 recombinant DNA with human genomic DNA as a carrier and Taq DNA Polymerase was prepared. After ~5 min incubation at room temperature the 45 μL aliquot from the stock solution was added to a thin walled 200 μL tube containing 5μL of dNTP/3’-THF dNTP mixture in 1 X PCR buffer. Before to be placed into the PCR instrument, the reaction tubes were additionally incubated at room temperature for ~5 min. The final reaction mixture contained both primers at 1 μM, dNTP or 3’-THF dNTP at 200 μM each, 10 copies of HIV-1 template, 50 ng of human genomic DNA and 1.25 U of Taq DNA polymerase. The dNTP mixtures employed for these studies were either a mixture of four standard dNTPs, or a mixture of 3’-protected and standard dNTPs where one, two, three or all four dNTPs were 3’-THF dNTPs.
Figure 5.
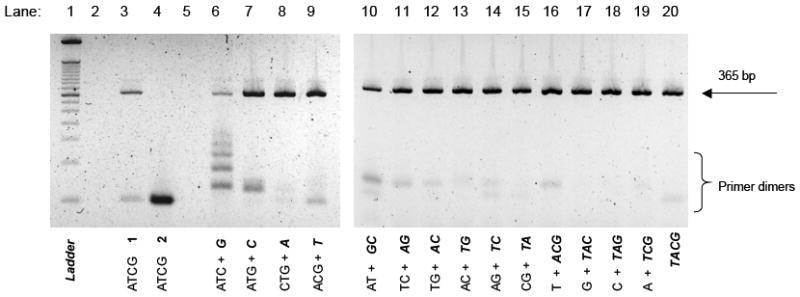
The 4% agarose gel analyses of PCR amplification of HIV-1 DNA 365 bp target at 10 copies. Each mixture contains standard dNTPs and/or 3’-THF dNTPs, 200 μM each. A, C, T, G and A, C, T, G stand for dATP, dCTP, dTTP, dGTP, and 3’-THF dATP, 3’-THF dCTP, 3’-THF dTTP, 3’-THF dGTP, respectively. Lane 1: 50 bp DNA ladder; lanes 2 and 5: empty; lane 3: standard ATCG (control 1, enzyme added as the last component); lane 4: standard ATCG (control 2, dNTP mixture was added as the last component); lane 6: ATC + G; lane 7: AGT + C; lane 8: GCT + A; lane 9: ACG + T; lane 10: AT + GC; lane 11: CT + GA; lane 12: GT + AC; lane 13: AC + TG; lane 14: AG + TC; lane 15: GC + AT; lane 16: T + GCA; lane 17: G + TCA; lane 18: C + GTA; lane 19: A + GCT; lane 20: TACG.
All PCR experiments were performed on a Perkin Elmer GeneAmp® 2400 thermal cycler (ramping rate ~ 1°C/sec) and were repeated at least four times. The thermal cycling conditions were 95°C for 5 min initial denaturation step, followed by 40 PCR cycles at 95°C for 40 sec, 56°C for 30 sec, 72°C for 120 sec, and final extension at 72°C for 7 min. An aliquot of 20 μL of each PCR sample was loaded on 2% or 4% agarose E-gel cartridge (Invitrogen) and run for 25 min according to the manufacturer’s suggested protocol. An image of the gel was acquired using UV transillumination with an Alpha Innotech Corporation Multi Image Light Cabinet with CCD Camera and quantified using AlphaEaseFC™ software. Accuracy of integration was estimated as ± 20%.
RESULTS AND DISCUSSION
3’-protected dNTPs
We prepared derivatives of four natural dNTPs that contained a thermally labile 3’-protecting group and tested these compounds as substitutes for natural dNTPs in PCR. The choice of an adequate 3’-protecting group for dNTPs was a challenge due to the complexity of the PCR system and variability of parameters that potentially can influence the outcome of the amplification reaction. The ideal 3’-protecting group should be stable in a PCR buffer at ambient temperature during preparation and manipulation of the PCR sample, but labile at high temperatures (56-95 °C) required for the PCR process (Scheme). The 3’-protected dNTPs are either non-substrates or terminating substrates for thermostable DNA polymerases, such as Thermus aquaticus (Taq) DNA polymerase. Regardless of the mechanism of action of 3’-protected dNTPs38,39,44 further primer extension and/or elongation is halted at ambient temperature where the primer-template hybridization is not stringent. The PCR amplification can start only after heat-triggered activation when the 3’-protecting group is removed to produce a corresponding unprotected dNTP with a free 3’-hydroxyl group that is the normal substrate for Taq DNA polymerase. At high temperatures of PCR the conditions for specificity of primer-template hybridization are optimal, therefore the level of undesired products such as primer-dimers and/or mis-priming products is substantially reduced.
Scheme.
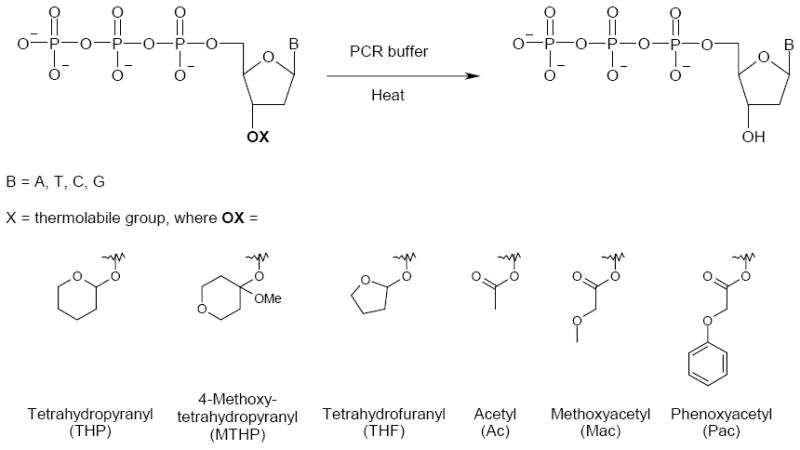
Proposed heat-triggered conversion of 3’-protected dNTP to 3’-OH dNTP in PCR buffer and examined 3’-protecting groups.
We chose to test several 3’-protecting groups that differed significantly in deprotection properties45. For initial testing we prepared 3’-protected ether and ester derivatives of 2’-deoxythymidine: 3’-O-tetrahydropyranyl (3’-THP), 3’-O-(4-methoxy)tetrahydropyranyl (3’-MTHP), 3’-O-tetrahydrofuranyl (3’-THF), 3’-O-acetyl (3’-Ac), 3’-O-methoxyacetyl (3’-Mac) and 3’-O-phenoxyacetyl (3’-Pac) (Scheme). When we determined later that the 3’-O-tetrahydrofuranyl group showed the best PCR properties we also synthesized 3’-O-tetrahydrofuranyl derivatives of 2’-deoxyadenosine, 2’-deoxycytidine and 2’-deoxyguanosine. The 5’-triphosphorylation of all 3’-protected 2’-deoxynucleoside derivatives was accomplished by the procedure described by Ludwig and Eckstein46. The 3’-protected dNTPs were isolated in 25-40% average yield after consecutive anion-exchange and reverse phase chromatography purification and conversion to a sodium salt form. For details of syntheses see Supporting Information section.
The long-term storage stability of 3’-THF, 3’-MTHP and 3’-THP dNTPs in frozen water buffer was evaluated. According to our preliminary results all compounds can be stored as frozen solutions in water at −20°C for at least one year without a noticeable decrease of their performance in PCR. In addition, all acid sensitive 3’-ether derivatives were stable at 4°C in aqueous solution at pH >8.0 for at least three months, while the base sensitive 3’-Pac and 3’-Mac ester derivatives were unstable if kept at pH>6.0.
Studies on the thermal conversion of 3’-protected dTTP derivatives to dTTP
The rate of cleavage of a 3’-protecting group and formation of unprotected dTTP was determined for all synthesized 3’-derivatives of dTTP at 95°C in PCR buffer (Table, Figure S-1). The time points were analyzed by reverse-phase or anion-exchange HPLC that allowed separation of dTTP with or without a 3’-protecting group. It also resolved two diastereomeric forms of 3’-THP and 3’-THF derivatives (Figure 1). The cleavage rate of 3’-protecting groups was found to fall in the following order: THP~Ac >MTHP~THF>Mac~Pac. At 95°C in PCR buffer (pH 8.4 at 25°C) the loss of 3’- ester protecting group from least stable 3’-Pac and 3’-Mac dTTP derivatives was fast (t1/2 ~ 6-8 min) and was anticipated to be complete before the end of a typical PCR experiment. On the other hand, the most stable 3’-protecting groups such as 3’-Ac and 3’-THP (t1/2 ~ 3-4 hrs) were expected to mainly survive during PCR process and therefore would provide a limited level of unprotected dNTPs available for target amplification. The 3’-THF and 3’-MTHP protecting groups with intermediate deprotecting rates (t1/2 ~ 1.5 hrs) were considered as most suitable for PCR application.
Since common PCR buffers contain Tris-HCl, it brings an important issue of dependence of pH of the PCR buffer on its temperature47. This property may lead to a positive or negative effect on the overall rate of conversion of 3’-protected dNTP to the unprotected dNTP during PCR. The 3’-ether derivatives (THP, MTHP and THF) are stable in PCR buffer (pH~8) when the samples are prepared at room temperature. When a PCR process starts and temperature rises to 95°C, the pH of the Tris-HCl buffered solution drops by approximately two units to pH~6 where the stability of a 3’-ether function is much lower45. It should result in pH-driven ~100-fold acceleration of 3’-deprotection rate. In addition, a temperature-driven (going from ~20°C to 95°C) acceleration of the reaction rate adds another ~1000-fold to the kinetics of 3’-deprotection. In our estimation, the combined temperature-and-pH effect results in ~105 and ~5×102 faster deprotection kinetics at 95°C compared to the rate of cleavage of 3’-ether group at 20°C and 50°C, respectively (Figure 1). In comparison, two opposite effects determine the overall change of 3’-deprotection rate of base-labile 3’-ester derivatives at elevated temperature. While the temperature-driven (from 20°C to 95°C) rate acceleration should increase the kinetics of 3’-deprotection by ~1000-fold, the pH-driven stabilization of the 3’-ester protecting group due to a less basic conditions (from pH ~8 at 20°C to pH~6 at 95°C) should slow this kinetics down by ~100-fold. The combined effect gives ~10-fold overall rate acceleration. In this context 3’-ether protecting groups are preferable for Tris-HCl containing PCR buffers because they stabilize 3’-protected dNTPs at low temperatures during sample preparation but enhance the 3’-deprotection kinetics at high temperatures of PCR.
Thermal conversion of the 5’-triphosphate chain of dNTP to 5’-diphosphate one is a major reaction that is characteristic for all natural dNTPs, especially below pH 8.0 48. Our separate experiments showed that at standard PCR conditions the 3’-THF group does not have any effect on the rate of conversion of 5’-triphosphate group to 5’-diphosphate group (Figure 2A and 2B). Due to continued conversion of 3’-THF dTTP to 3’-THF dTDP the effective concentration of dTTP generated from 3’-THF dTTP is leveled-off at ~ 30% after 40 min of incubation at 95°C (Figure 2A). Thermal conversion of a 5’-triphosphate group to a 5’-diphosphate group in PCR buffer was observed for all of the above 3’ derivatives of dTTP tested. Formation of a small amount of 2’-deoxythymidine 5’-mono- and 5’-tetraphosphate was also detected.
In another set of experiments the rate of accumulation of dTTP from 3’-protected dTTP was determined during a typical PCR cycle sequence. The data points were collected after initial denaturation step before the first PCR cycle and after 10, 20, 30 and 40 PCR cycles. During the initial 5 min denaturation step, prior to PCR cycle 1, and after 10, 20, 30 and 40 cycles a variable amount of dTTP was generated depending on the nature of 3’-protecting group. Estimated concentration of dTTP was ranging from few μM to more that 100 μM. The 3’-Pac and 3’-Mac derivatives generate highest level of dTTP after first 10 cycles. After reaching the maximum the level of dTTP decreases due to continues conversion of dTTP to dTDP. More stable 3’-Ac, 3’-THP, 3’-MTHP and 3’-THF derivatives generate lower level of dTTP with a maximum concentration at cycle 40. Although the absolute rates of accumulation of dTTP in PCR cycling experiment are specific to PCR temperature/time profile, PCR buffer and to technical parameters of the PCR instrument, they correlate qualitatively with the rates determined at 95°C for isothermal conditions (Table).
PCR experiments with 3’-protected dTTPs and end-point detection in model HIV-1 and Lambda DNA systems
Five 3’-protected derivatives of dTTP were evaluated as substitutes for dTTP in the PCR amplification of a 365-bp HIV-1 recombinant DNA and ~1.9-kbp Lambda DNA targets in the presence of Human Genomic DNA (HG DNA) as a competing foreign DNA (Figure 3 and 4). With standard dNTPs the HIV-1 system is specifically prone to accumulation of primer dimers 6. In the model Lambda DNA (+HG DNA) system with standard dNTPs, several bands of relatively long mis-priming amplification products can be seen on the gel. Although the nature of these mis-priming products was not investigated, apparently they originated from added HG DNA (Figure 4, lane 8).
The results of PCR experiments with 3’-protected dTTP and HIV-1 DNA template showed a strong suppression of primer dimers, especially for the 3’-MTHP and 3’-THF derivatives (Figure 3). Interestingly, despite the fact that 3’-Ac dTTP and 3’-THP dTTP were shown to be relatively stable in PCR buffer (Figure 1), the limited amount of 3’-OH dTTP generated by heat activation during PCR was sufficient to support the amplification of a relatively short 365-bp HIV-1 target with moderate efficiency. In Lambda DNA system the use of 3’-MTHP, 3’-THF and 3’-Pac derivatives of dTTP as substitutes for standard dTTP led to an efficient accumulation of 1887-bp amplicon and substantial decrease of the mis-priming products. The 3’-Ac dTTP and 3’-THP dTTP derivatives appeared to be inefficient in amplification of the 1887-bp fragment (Figure. 4).
The optimal rate of conversion of 3’-protected dNTPs to unprotected dNTPs in PCR buffer at high temperature is a key requirement for successful PCR amplification. The 2’-deoxythymidine derivatives with 3’-protecting groups such as 3’-Pac and 3’-Mac, that are not stable in PCR buffer, are not easy to handle and can only be used with considerable care. They start to convert to unprotected dTTP in PCR buffer even before the PCR cycling begins thus reducing the positive effect. To avoid the unwanted cleavage of 3’-protecting group, PCR mixtures were prepared on ice and the 3’-Pac dTTP was added at the last moment. After brief mixing the tubes were placed immediately in the pre-heated PCR instrument. This procedure ensured an optimal performance of 3’-Pac dTTP derivative in PCR (Figure 3 and 4). However, the use of 3’-Pac and 3’-Mac ester derivatives of dNTPs in PCR applications appeared to be impractical especially in experiments where multiple samples have to be prepared.
A different problem is associated with 3’-derivatives of dTTP that are very stable (3’-Ac and 3’-THP). Since the rate of conversion of 3’-Ac and 3’-THP derivatives to 3’-OH dTTP is slow it was anticipated that they would not efficiently support PCR amplification especially at the later PCR cycles with a high demand for unprotected dTTP. To our surprise, both of the above mentioned 3’-derivatives of dTTP supported amplification of a relatively short 365-bp HIV-1 target with efficiency and specificity comparable to that for a standard dTTP (Figure 4). A different result was seen for a longer ~1.9-kbp Lambda DNA target (Figure 5). Under conditions used in our standard PCR experiment both 3’-Ac and 3’-THP derivatives of dTTP failed to support amplification of this longer target. Presumably there was either insufficient number of unprotected dTTP molecules generated during PCR and/or the rate of enzymatic polymerization became a limiting factor due to a low concentration of the unprotected dTTP available for incorporation. In either case the expected end result would be an incomplete elongation/extension of the primer leading to an accumulation of aborted amplicons incapable to efficiently support the amplification process.
The 3’-THF and 3’-MTHP derivatives of dNTPs with intermediate rates of thermal conversion to unprotected dNTPs generated sufficient amount of dNTPs to efficiently support PCR amplification of short and long targets and were sufficiently stable during preparation of the PCR mixtures thus preventing a premature loss of the 3’-protecting group. Overall, when 3’-THF and 3’-MTHP derivatives of dTTPs were used as substitutes for standard dTTP in both the HIV-1 and Lambda DNA systems examined, a robust accumulation of desired amplicons was observed together with a strong suppression of undesired off-target products (Figures 3 and 4).
The above experiments have demonstrated that substitution of only one 3’-THF dTTP, 3’-MTHP dTTP or 3’-Pac dTTP for a standard dTTP significantly reduced the level of off-target products in both HIV-1 and Lambda DNA systems. The UV-integration of the agarose gel images revealed a higher ratio of amplicon to off-target products in lanes where these 3’-protected dTTP derivatives were used as compared to the lane with control dTTP (Figure 3 and 4). The yield of desired target amplification product correlated qualitatively with the rate of conversion of the 3’-protected dTTP derivatives to unprotected dTTP in the PCR buffer at 95°C.
PCR experiments with 3’-THF dNTPs in HIV-1 system
Based on the results obtained for 3’-protected dTTP derivatives we have chosen the 3’-THF protecting group as a leading modification for further investigation. Although the 3’-Pac derivative of dTTP was also efficient in PCR experiments, we did not pursue this modification for dATP, dCTP and dGTP due to it’s stability issues (see above).
To further evaluate a benefit of 3’-THF dNTP derivatives in PCR the conditions of PCR mixture preparation were set up to increase a potential of formation of primer dimers. The PCR stock mixture containing all components except dNTPs/3’-THF dNTPs was prepared and kept for at least 5 min at room temperature to promote a complex formation between forward and reverse primers and Taq DNA polymerase. The hypothesis is that the pre-formed oligonucleotide-enzyme complexes are ready to immediately incorporate standard (unprotected) dNTPs into primers and form primer dimers as soon as all components are mixed together in PCR tube.
We investigated PCR performance in the HIV-1 system with one, two, three and four substitutions 3’-THF dNTP for dNTP (Figure 5). When stock solution of primers, template and enzyme was added to a mixture of standard dNTPs (lane 4, control 2) a strong band of primer dimer was detected on the gel with only traces of 365-bp amplicon. In comparison, when using standard dNTP mixture in a typical set up protocol (enzyme was added last to the tube) both bands of amplicon and primer dimer were detected (lane 3, control 1). When one 3’-THF dNTP was substituted for respective dNTP in PCR mixture a substantial improvement in amplification of 365-bp amplicon was evident with 3’-THF dATP and 3’-THF dTTP derivatives showing the best performance. The effect of combined action of two, three or four 3’-THF derivatives was much stronger resulting in drastic reduction of primer dimers and improved performance and specificity of PCR (lanes 10-20). At this time we cannot explain apparent and reproducible differences in the efficiency of suppression of primer dimers observed for 3’-THF derivatives of dCTP, dTTP, dATP, dGTP and their combinations. The differences may be a consequence of specific nucleotide sequences of the template and/or primers at their 3’-terminal area and/or the nature of the first dNTP required for primer extension. On the other hand the observed variation may be a reflection of sub-optimal experimental conditions (such as choice of heat activation time, PCR cycle parameters, buffer composition and pH). Although there were variations in average amplicon and primer dimer yields from experiment to experiment, the general trend for improved PCR performance with multiple 3’-THF substitutions was clear and reproducible in each individual experiment. For each PCR mixture containing three or four 3’-THF dNTPs a nearly complete elimination of primer dimer was observed even for set up conditions promoting formation of primer dimers. Further work on the optimization of PCR cycling parameters and/or further purification of 3’-THF-dNTPs may lead to further improvement of PCR performance. Enzymatic “mop-up” may help to remove traces of dNTP contamination in 3’-THF dNTP49.
During the typical initial heat-denaturing step (2-10 min at 95°C) of the PCR experiment only a fraction of molecules of 3’-derivative of dNTP is converted to 3’-unprotected dNTP. For our leading 3’-THF dNTP derivatives we estimate that a 5 min heat activation step at 95°C generates approximately 1.6 × 1016 molecules of 3’-unprotected dNTP. This amount would be sufficient to support an amplification of a 1000-bp long DNA fragment for at least 30-35 PCR cycles. An additional amount of 3’-unprotected dNTPs is generated during the following PCR cycles. Our estimates indicate that at any PCR cycle the mixture contains at least 104-1012 fold excess of unmodified dTTPs over amplified material through the cycle 35 and probably beyond that cycle (see, Supporting Information section). Even when slow deprotecting 3’-THP and 3’-Ac derivatives of dTTP are used, the 3’-deprotection rate appeared to be adequate to generate enough dTTP molecules to support detectable amplification of short target (365-bp) in HIV-1 system at conditions used.
The enzymatic kinetics could be another limiting factor when 3’-protected dNTPs are used in PCR. The lower limit of working concentration of unprotected dNTPs that can support current PCR temperature/time profile was not estimated in this work. Robust accumulation of amplicon, especially long ones, may fail when the concentration of unprotected dNTPs becomes a limiting factor as evident in the case of 3’-Ac and 3’-THP dTTP derivatives and 1.9 kbp Lambda DNA amplicon (Figure 4).
The overall PCR time/temperature profile used in this work is well suited for the evaluation of PCR properties of 3’-protected dNTPs. For comparison purposes we have chosen an initial denaturing time of 5 min and a relatively long denaturing time of 40 sec at 95°C in each cycle which are typical for Hot Start PCR applications with Hot Start DNA polymerases or modified primers 20, 26-28, 35. The annealing temperatures for HIV-1 and Lambda DNA primers were not optimized but were selected ~6-8°C below their predicted optimal annealing temperature to increase a possibility of formation of partially mismatched complexes. It allowed for evaluation and comparison of the performance of 3’-protected dNTPs and natural dNTPs at suboptimal PCR conditions. Although PCR cycling parameters used in this work may not be applicable for every PCR experiment that employs standard primers, enzymes and dNTPs, our preliminary studies indicate that substitution of 3’-THF dNTPs for natural dNTPs does not require modification or adjustment of any PCR temperature/time profile except a 2-5 min increase of initial denaturing time at 95°C. However, the 3’-THF dNTPs may be not an optimal choice in rapid cycling protocols where denaturing time of the cycle is less than 10 sec due to a relatively slow deprotecting kinetics.
Inhibitory properties of 3’-protected dNTPs
We did not investigate specifically if the 3’-protected dNTPs or the products of their degradation, such as 3’-protected and deprotected 2’-deoxynucleoside mono-, di- and tetraphosphates, have any substantial inhibitory effect on PCR amplification. However, at least in the case of 3’-THF dNTP derivatives the accumulation of above nucleotide by-products in the PCR mixture apparently does not compromise the overall performance of PCR. In addition, degradation products generated from the cleaved of 3’-THF group do not seem to have an adverse effect on PCR performance indicating the absence of any critical modification by these products of DNA polymerase and/or primer, or template, or amplicons, or dNTPs. Overall, while the absence of inhibition has not been proved directly, it is clear that efficient target amplification could not happen if substantial inhibitors of Taq DNA polymerase were present in PCR mixture.
CONCLUSIONS
A novel, general and efficient approach to Hot Start PCR was developed. It was demonstrated for two model systems that 3’-THF derivatives of dNTPs fully support PCR amplification and provide a substantial reduction in accumulation of primer dimers and/or mis-priming products. Although there were some variations observed in the effectiveness of different combinations of 3’-THF dNTPs, the overall tendency was that efficiency of target accumulation and suppression the primer dimers correlated with increased number of 3’-THF dNTPs used in the PCR. Further evaluation of performance of various 3’-protected dNTPs in PCR applications such as quantitative PCR, fast cycling real time PCR, reverse-transcription PCR, multiplex PCR with end-point and real time detection is in progress.
Acknowledgments
This work was supported by grant from National Institute of Health (Grant 1R43GM079836 – 01). We are grateful to Richard Hogrefe, Gerald Zon and Natasha Paul for critical reading of the manuscript, Haoqiang Huang, Joyclyn Yee, Stephanie Perry and Jonathan Shum for helpful discussions.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at htty://pubs.acs.org
Supplementary Material
References
- 1.Mullis KB. US4683202 U.S. Patent. 1987
- 2.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 3.Kolmodin LA, Williams JF. Methods Mol Biol. 1997;67:3–15. doi: 10.1385/0-89603-483-6:3. [DOI] [PubMed] [Google Scholar]
- 4.Arnheim N, Erlich H. Annu Rev Biochem. 1992;61:131–156. doi: 10.1146/annurev.bi.61.070192.001023. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Cui X, Arnheim N. Proc Natl Acad Sci USA. 1990;87:4580–4584. doi: 10.1073/pnas.87.12.4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chou Q, Russel M, Birch DE, Raymond J, Bloch W. Nucleic Acids Res. 1992;20:17–23. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budowle B, Schutzer SE, Einslen A, Kelley LC, Walsh AC, Smith JA, Marrone BL, Robertson J, Campos J. Science. 2003;301:1852–1853. doi: 10.1126/science.1090083. [DOI] [PubMed] [Google Scholar]
- 8.Sato Y, Hayakawa M, Nakajima T, Motani H, Kiuchi M. Leg Med (Tokyo) 2003;5(Suppl 1):S191–S193. doi: 10.1016/s1344-6223(02)00108-6. [DOI] [PubMed] [Google Scholar]
- 9.Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Clin Microbiol Rev. 2000;13:559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saldanha J, Minor P. J Med Virol. 1994;43:72–76. doi: 10.1002/jmv.1890430114. [DOI] [PubMed] [Google Scholar]
- 11.Sachse K, Frey J. Methods in Molecular Biology. 2003;216:352. [PubMed] [Google Scholar]
- 12.Traeger-Synodinos J. Molecular Aspects of Medicine. 2006;27:176–191. doi: 10.1016/j.mam.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Dahiya R, Deng G, Selph C, Carroll P, Presti J., Jr Biochem Mol Biol Int. 1998;44:407–415. doi: 10.1080/15216549800201422. [DOI] [PubMed] [Google Scholar]
- 14.D’Aquila RT, Bechtel LJ, Videler JA, Eron JJ, Gorczyca P, Kaplan JC. Nucleic Acids Res. 1991;19:3749. doi: 10.1093/nar/19.13.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullis KB. PCR Methods Appl. 1991;1:1–4. doi: 10.1101/gr.1.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Kaijalainen S, Karhunen PJ, Lalu K, Lindstrom K. Nucleic Acids Res. 1993;21:2959–2960. doi: 10.1093/nar/21.12.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riol H, Lévesque G, Murthy RV. Anal Biochem. 1994;221:210–212. doi: 10.1006/abio.1994.1402. [DOI] [PubMed] [Google Scholar]
- 18.Horton RM, Hoppe B, Conti-Tronconi BM. BioTechniques. 1994;16:42–43. [PubMed] [Google Scholar]
- 19.Barnes WM, Rowlyk KR. Molecular and Cellular Probes. 2002;16:167–171. doi: 10.1006/mcpr.2002.0407. [DOI] [PubMed] [Google Scholar]
- 20.Birch DE, Kolmodin L, Laird WJ, McKinney N, Wong J, Young KKY, Zangenberg GA, Zoccoli MA. Nature. 1996;381:445–446. [Google Scholar]
- 21.Moretti T, Koons B, Budowle B. BioTechniques. 1998;25:716–722. [PubMed] [Google Scholar]
- 22.Kermekchiev MB, Tzekov A, Barnes WM. Nucleic Acids Res. 2003;31:6139–6147. doi: 10.1093/nar/gkg813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kellogg DE, Rybalkin I, Chen S, Mukhamedova N, Vlasik T, Siebert PD, Chenchik A. Biotechniques. 1994;16:1134–1137. [PubMed] [Google Scholar]
- 24.Shigemori Y, Mikawa T, Shibata T, Oishi M. Nucleic Acids Res. 2005;33:e126. doi: 10.1093/nar/gni111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang C, Jayasena SD. J Mol Biol. 1996;264:268–278. doi: 10.1006/jmbi.1996.0640. [DOI] [PubMed] [Google Scholar]
- 26.Mizuguchi H, Nakatsuji M, Fujiwara S, Takagi M, Imanaka T. J Biochem (Tokyo) 1999;126:762–768. doi: 10.1093/oxfordjournals.jbchem.a022514. [DOI] [PubMed] [Google Scholar]
- 27.Lebedev AV, Paul N, Yee J, Timoshchuk VA, Shum J, Miyagi K, Kellum J, Hogrefe RI, Zon G. Nucleic Acids Res. 2008;36:e131. doi: 10.1093/nar/gkn575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brownie J, Shawcross S, Theaker J, Whitcombe D, Ferrie R, Newton C, Little S. Nucl Acids Res. 1997;25:3235–3241. doi: 10.1093/nar/25.16.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Will SG. US 6001611 U.S. Patent. 1999
- 30.Ailenberg M, Silverman M. Biotechniques. 2000;29:1018–1024. doi: 10.2144/00295st03. [DOI] [PubMed] [Google Scholar]
- 31.Kaboev OK, Luchkina LA, Tret’iakov AN, Bahrmand AR. Nucleic Acids Res. 2000;28:e94. doi: 10.1093/nar/28.21.e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullman EF, Lishanski A, Kurn N. US 6482590 U.S. Patent. 2002
- 33.Bonner AG. US2003162199 U.S. Patent. 2003
- 34.Laird WJ, Niemiec JT. US 2003044817 U.S. Patent. 2003
- 35.Kong D, Shen H, Huang Y, Mi H. Biotechnology Letters. 2004;26:277–280. doi: 10.1023/b:bile.0000015425.33690.88. [DOI] [PubMed] [Google Scholar]
- 36.Ankenbauer W, Heindl D, Laue F, Huber N. US 7122355 U.S. Patent. 2006
- 37.Zon G, Lebedev AV. US 20070281308 U.S. Patent. 2007
- 38.Koukhareva I, Haoqiang H, Yee J, Paul N, Hogrefe RI, Lebedev AV. Collection Symposium Series. 2008;10:259–263. doi: 10.1093/nass/nrn131. [DOI] [PubMed] [Google Scholar]
- 39.Koukhareva I, Haoqiang H, Yee J, Shum J, Paul N, Hogrefe RI, Lebedev AV. Nucleic Acids Symposium Series. 2008;52:259–260. doi: 10.1093/nass/nrn131. [DOI] [PubMed] [Google Scholar]
- 40.Wong KK, McClelland M. Nucleic Acids Res. 1991;19:1081–1085. doi: 10.1093/nar/19.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seela F, Röling A. Nucleic Acids Res. 1992;20:55–61. doi: 10.1093/nar/20.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dierick H, Stul M, De Kelver W, Marynen P, Cassiman J-J. Nucleic Acids Res. 1993;21:4427–4428. doi: 10.1093/nar/21.18.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kutyavin IV. Biochemistry. 2008;47:13666–13673. doi: 10.1021/bi8017784. [DOI] [PubMed] [Google Scholar]
- 44.Metzker ML, Raghavachari R, Richards S, Jacutin SE, Civitello A, Burgess K, Gibbs RA. Nucleic Acids Res. 1994;22:4259–4267. doi: 10.1093/nar/22.20.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green TW, Wuts PGM. Protective Groups in Organic Synthesis. Wiley-Interscience; New York: 1999. p. 779. [Google Scholar]
- 46.Ludwig J, Eckstein F. J Org Chem. 1989;54:631–635. [Google Scholar]
- 47.Durst RA, Staples BR. Clinical Chemistry. 1972;18:206–208. [PubMed] [Google Scholar]
- 48.Ihlenfeldt HG, Schmidt A, Muhlegger K, Leitenberger V. US6916616 US Patent. 2005
- 49.Metzker ML, Raghavachari R, Burgess K, Gibbs RA. BioTechniques. 1998;25:814–817. doi: 10.2144/98255st01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.