

Abstract
Here we describe a Sleeping Beauty (SB) transposition system that utilizes a conditional SB transposase allele, which can be activated by Cre recombinase to drive the transposition of a mutagenic transposon in virtually any tissue and control the type of cancer produced. To demonstrate the potential of this system for modeling cancer in mice, we used it to screen for hepatocellular carcinoma (HCC) associated genes in mice by specifically limiting SB transposition to the liver. Among 8,060 non-redundant insertions subsequently cloned from 68 tumor nodules we identified 19 highly significant candidate disease loci, which encode genes like EGFR and MET that are known HCC genes and others like UBE2H that are not strongly implicated in HCC but represent potential new therapeutic targets for treating this neoplasm. With these improvements, transposon-based insertional mutagenesis now offers great potential for better understanding the cancer genome and for identifying new targets for therapeutic development.
Keywords: Hepatocellular carcinoma, Sleeping Beauty transposon, EGFR, Pyrosequencing
Introduction
Transposon-tagged mutagenesis has proven invaluable for functional genomic screens in organisms such as Drosophila melanogaster1, 2, but transposons such as SB that are capable of transposing in mouse cells have only recently been identified3. Due to SB's low transposition frequency in the mouse germline4-6, it was generally assumed that it would be impossible to mobilize SB at high enough frequencies in somatic cells to induce cancer. Two groups have shown this is incorrect by successfully mobilizing a mutagenic SB transposon in somatic cells at frequencies high enough to induce cancer in wild-type mice7 or accelerate the formation of tumors in p19Arf-deficient mice8.
In order to screen for cancer-associated genes in different types of cancer using SB, we sought to develop a conditional SB transposition system. For this, we decided to knock-in the SB transposase (SB11) carrying a floxed-stop (lsl) cassette into the mouse Rosa26 locus, which encodes a ubiquitously expressed nonessential gene9. Genes knocked-in to the Rosa26 locus are widely expressed and not subject to epigenetic silencing normally observed with transgenes9. Expression of the transposase knock-in (Rosa26-lsl-SB11), blocked due to the presence of the floxed-stop cassette, can subsequently be reactivated in any target tissue using a tissue-specific Cre recombinase to drive the transposition of the T2/onc mutagenic transposon7, 8 (see Supplementary Fig. 1a online).
HCC is the third leading cause of cancer-related death10 and an aggressive tumor has dismal prognosis since less than 30% of patients will be eligible for potential curative treatment at the time of diagnosis11. HCC is prevalent worldwide but differences in disease incidence rates reflect regional diversity mostly related to geographic distribution of viral hepatitis11. Gender also influences risk with males showing a 4:1 increase in prevalence over females, with existing preliminary molecular data that explains this gender discrepancy12. Mutations in the TRP53 gene are commonly found in HCC, suggesting its importance in liver tumorigenesis13, 14. In these experiments, we used a hepatocyte-specific Albumin-Cre (Alb-Cre) transgene to activate transposase expression specifically in the liver. As mutations in p53 are the most frequently described mutations in HCC, a conditional dominant negative p53 transgene15 was included (p53-lsl-R270H) (see Supplementary Fig. 1a online). Triple transgenic (Rosa26-lsl-SB11; T2/onc; Alb-Cre) and quadruple transgenic (Rosa26-lsl-SB11; T2/onc; Alb-Cre; p53-lsl-R270H) mice were generated and aged for liver tumorigenesis (see Supplementary Fig. 1b online).
In the present study, novel liver cancer-associated genes were identified using a conditional SB transposon forward insertional mutagenesis screen combined with a high-throughput sequencing technique. Information obtained from this screen will provide further insight to the genetic mechanisms associated with the disease and allow for possible development of therapeutic regimes.
Results
Hepatocyte-specific transposition and tumorigenesis
To demonstrate that transposase is activated exclusively in the liver, immunohistochemical (IHC) analyses was performed on mice carrying both Alb-Cre and Rosa26-lsl-SB11 transgenes using anti-SB transposase antibody (Fig. 1a). To confirm that transposition is occurring in the livers of experimental transgenic animals, excision PCR8 was also performed and evidence of excised amplicons was observed (see Supplementary Fig. 2a online). Experimental and control animals from both sexes were sacrificed initially at ∼100-days but no visible lesions were seen in any organs (data not shown). Preneoplastic liver nodules were first detected at ∼160-days in both male triple and quadruple transgenic animals. However, the quadruple transgenic animals displayed more numerous and larger nodules than triple transgenic animals (see Supplementary Fig. 2b online). For triple and quadruple transgenic control cohorts, double and triple transgenic mice carrying all possible combinations of the four transgenes were also generated and aged. No evidence of tumorigenesis was seen in control male littermates sacrificed at similar age (data not shown). From 101- to 223-days, 4 out of 6 (67%) quadruple transgenic male experimental animals had livers with macroscopic preneoplastic nodules (Fig. 1b) and a total of 67 nodules were isolated (see Supplementary Table 1 online). In contrast, 3 out of 7 (43%) triple transgenic male animals from 105- to 289-days had a total of 36 preneoplastic nodules isolated (see Supplementary Table 1 online). Excision PCR assays were positive in the livers of non-tumor producing experimental animals, indicating transposition events had occurred (see Supplementary Fig. 2a online).
Figure 1.
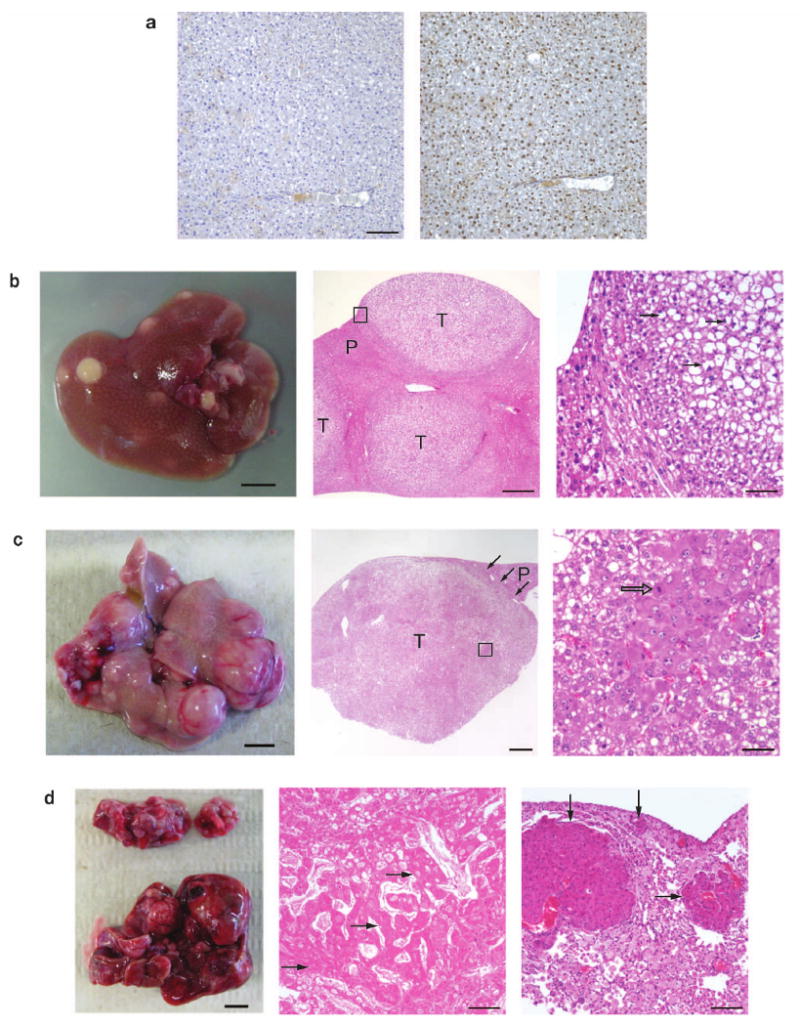
Accelerated tumorigenesis in p53-deficient livers compared with wild-type livers. (a) Albumin-Cre (Alb-Cre) efficiently deletes the floxed-stop (lsl) cassette within the Rosa26-lsl-SB11 transgene, allowing for SB transposase expression and subsequent somatic transposition events. Left panel, immunohistochemistry (IHC) of (Alb-Cre; Rosa26-lsl-SB11) double-transgenic liver section treated without the primary antibody (negative control). Right panel, IHC of serial liver section treated with the primary anti-SB transposase antibody. Sections were lightly stained with hematoxylin after IHC. Scale bar, 100 μm. (b) Quadruple male transgenic mouse liver at 159-days, displaying many preneoplastic nodules (left panel, scale bar, 0.5 cm). Middle (low magnification) and right (high magnification of boxed area from middle panel) panels show the tumor histology of large preneoplastic nodules using hematoxylin-eosin (HE) staining. These proliferative lesions were often compressing surrounding parenchyma and the cells within the preneoplastic foci and adenomas were frequently vacuolated, containing distinct lipid vacuoles or clear cytoplasm (arrows). Nuclei were equal to or smaller in size than those in the normal hepatic parenchyma, and occasionally contained mitotic figures indicative of cell division. Proliferative lesions were frequently bordered by hepatocytes with markedly enlarged nuclei that were occasionally karyomegalic. T, preneoplastic tumor nodule; P, parenchymal liver cells; scale bars for middle and right panels were 500 μm and 100 μm, respectively. (c) Triple-transgenic male experimental mouse liver at 330-days showing advanced tumor development. Note the many large irregular nodules showing a hypervascular phenotype (left panel, scale bar, 0.5 cm). Middle panel shows the HE histological section of one large neoplastic nodule typical of hepatocellular adenoma consisting of variably vacuolated hepatocytes filled with lipid. Three black arrows indicate border between adenoma and non-neoplastic hepatic parenchyma (P), which is slightly compressed. Right panel shows high magnification of boxed area in the middle panel. Note enlarged nuclei of hepatocytes with moderate variation in nuclear size, prominent nucleoli, and mitotic figure (open arrow). T, preneoplastic tumor nodule; P, parenchymal liver cells; scale bars for middle and right panels were 1000 μm and 50 μm, respectively. (d) Triple-transgenic male experimental mouse at 440-days with HCC (bottom, left panel) and lung metastasis (top, left panel). HE staining of the liver (middle panel) and lung (right panel), showing advanced HCC in the liver and its metastases into the lung. A partial HCC section composed of irregular trabeculae of neoplastic, diffusely necrotic hepatocytes (black arrows) that are multifocally vacuolated. Trabeculae are separated by dilated sinusoids containing variable amount of fibrin. The lung contains multiple variably sized metastatic nodules of HCC (black arrows) that markedly compress the pulmonary parenchyma. Pulmonary alveoli are filled with large numbers of foamy macrophages. Scale bar, 100 μm.
Detailed histopathological analyses revealed that the livers of triple and quadruple transgenic mice at ∼150-days contain frequent preneoplastic foci of cellular alteration with a few adenomas (Fig. 1b). One triple transgenic male mouse that was examined at 330-days displayed a liver with multiple large hypervascularized tumors, indicating hepatic adenoma (Fig. 1c). Two triple transgenic male mice examined at much later stages (440- and 460-days) displayed livers with HCC characteristics and more importantly, lung metastasis (Fig. 1d). One quadruple transgenic male mice examined at 432-days also displayed a liver with HCC characteristics and lung metastases (see Supplementary Table 1 online). Preneoplastic nodules from all triple and quadruple transgenic livers were positive for SB transposase (SB)-, Albumin (Alb)- and Ki67-immunostain using IHC (Fig. 2a), indicating that these nodules resulting from transposition events originated from hepatocytes and have increased rates of proliferation. The lung metastases were positive for SB-, Alb- and Ki67-immunostain using IHC, indicating that they had derived from the HCC (Fig. 2b). The majority of preneoplastic nodules expressed Alpha-fetoprotein (Afp), a biomarker for human HCC, as detected by RT-PCR (Fig. 3d), but only a small subset of nodules expressed enough Afp that it could be detected by IHC (data not shown). RT-PCR also demonstrated the expression of Osteopontin (Opn) in all preneoplastic nodules, a gene associated with HCC metastasis16 (Fig. 3d). Semi-quantitative RT-PCR shows upregulation of Opn and Afp expression as liver tumorigenesis progressed from adenoma to HCC (see Supplementary Fig. 2c online). IHC analyses for β-catenin levels demonstrate increasing levels of expression as tumorigenesis progressed from preneoplastic nodules to hepatic adenoma to HCC (see Supplementary Fig. 3 online). β-catenin gene mutations or increasing levels of its expression are also observed in human HCC17. Interestingly, triple (n=4) and quadruple (n=4) female experimental animals sacrificed from 178- to 342-days and 178- to 344-days, respectively, did not have any visible liver lesions (see Supplementary Table 1 online). However, two female triple transgenic animals (512- and 575-days) and one quadruple transgenic animal (432-days) did present livers with small preneoplastic nodules (see Supplementary Table 1 online). The low frequency and late latency of liver nodules in female experimental animals mirrors the strong gender bias in HCC tumor incidence seen in human patients. In addition, IHC analyses of non-tumor forming female liver sections with the proliferative marker Ki67 and Afp, were both positive (see Supplementary Fig. 4a online). Therefore, our conditional SB liver tumor model is useful in elucidating genetic mechanisms for HCC tumorigenesis, including lesions ranging from early hepatic adenomas to fully developed HCC (including metastatic HCC).
Figure 2.
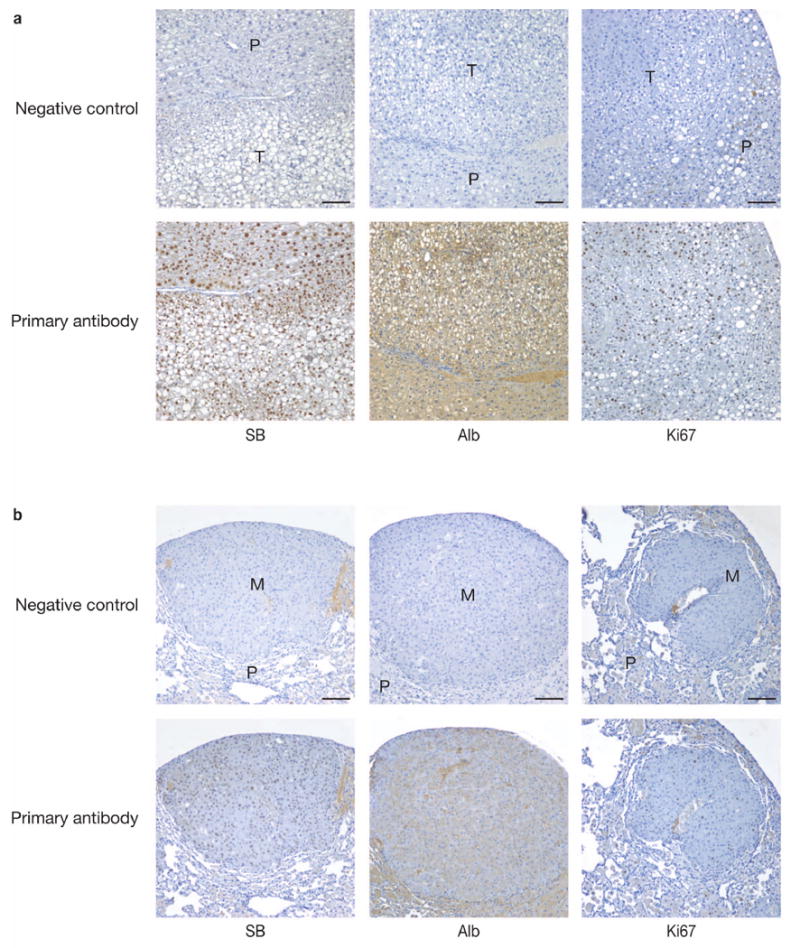
Immunohistochemical (IHC) analyses of liver preneoplastic nodules. (a) Paraffin-embedded liver tissue sections were stained with antibodies against SB transposase (SB), Albumin (Alb) and for the proliferative marker, Ki67. All liver tumor sections taken from either triple- or quadruple-transgenic experimental male animals were positive for SB, Alb and Ki67. Representative IHC liver sections from a 160-day old quadruple transgenic male mouse are shown. Top panels, negative control of liver sections not treated with the primary antibody; Bottom panels, IHC of serial liver sections treated with the indicated primary antibody; T, preneoplastic nodule; P, parenchymal cells; scale bars, 100 μm. (b) IHC analyses of the HCC-derived lung metastasis. Paraffin-embedded lung tissue sections were stained with antibodies against SB, Alb and Ki67. Top panels, negative control of lung sections not treated with the primary antibody; bottom panels, IHC of serial lung sections treated with the indicated primary antibody; P, parenchymal lung cells; M, metastasis from HCC; scale bars, 100 μm.
Figure 3.
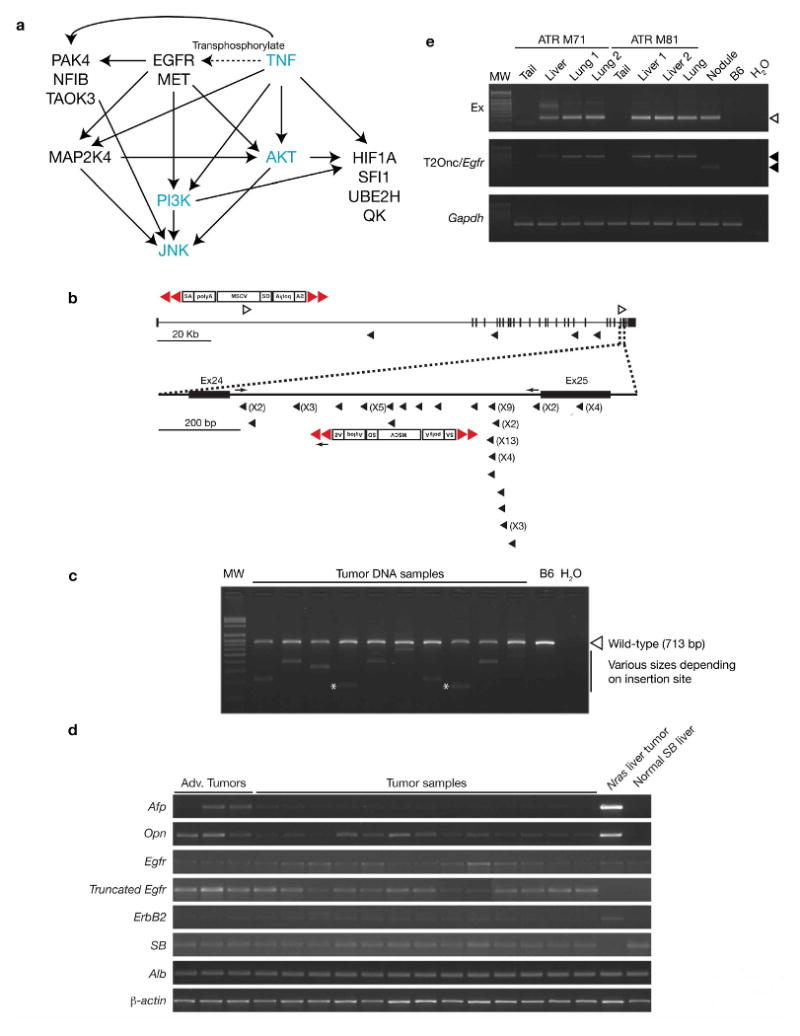
Frequent mutagenic transposon insertions into Epidermal growth factor receptor (Egfr) and Egfr interacting genes. (a) Ingenuity pathway analysis using 17 of the CIS genes obtained from the liver cancer screen. Out of the 17 genes entered, 10 genes were referenced and displayed in the network function pathways associated with post-translational modification, cancer and tumor morphology. The EGFR signaling pathway showing interactions with JNK, TNF and PI3K/AKT regulatory pathways is shown. CIS genes are in black and other genes in this network are in blue. (b) Diagrammatic representation of transposon insertions into intron 24 of Egfr. Schematic representation of the mutagenic transposon (T2/onc) is shown. Red triangles, inverted repeats/direct repeats (IR/DR) transposon flanking sequences; SA, splice acceptor; polyA, polyadenalytion signal; MSCV, LTR of the murine stem cell virus; SD, splice donor; open arrowhead, sense orientated insertion of the T2/onc relative to the Egfr gene; arrowhead anti-sense orientated insertion of the T2/onc relative to the Egfr gene; arrows, endogenous and vector primers used for the Egfr PCR genotyping shown in (c); numbers in parentheses, indicate the frequency of transposon insertions at each particular site from different liver preneoplastic nodules of experimental animals. (c) Confirming transposon insertions in intron 24 of Egfr. PCR genotyping was performed using genomic DNA isolated from individual tumor nodules. A subset of these samples was subjected to Egfr PCR genotyping using endogenous and vector primers. Resulting gel electrophoresis shows the endogenous Egfr (713 bp) band (open arrowhead), with the transposon-integrated band of varying sizes depending on the insertion site within intron 24. All amplicons corresponded with the pyrosequencing data except for 2 insertion sites (asterisks) that were missed by pyrosequencing. MW, 100-bp molecular standard; B6, C57BL/6 tail genomic DNA; H2O, double-distilled water used as a negative control. (d) RT-PCR analyses of tumor nodules using various markers. All neoplastic nodules were positive for SB transposase (SB) and Albumin (Alb) transcripts, indicating that transposition events are occurring and nodules were from a hepatocyte origin, respectively. A majority of tumor nodules were positive for Alpha-fetoprotein (Afp) transcripts, a clinical marker for HCC, and Osteopontin (Opn), which is over-expressed in various cancers including HCC. Nodules taken from a 330-day triple-transgenic mouse with advanced tumors (shown in Fig. 1c) were strongly positive for Afp and Opn. All tumor nodules tested were positive for endogenous Egfr and for truncated-Egfr transcripts. NRAS liver tumor, HCC control taken from a tumorigenic liver over-expressing NRAS G12V oncogene23; SB normal liver, normal liver taken from a SB transposase-expressing mouse; β-actin, control to show equal loading of mRNA used for RT-PCR. RT-negative controls were also performed for each sample and no visible bands were seen for any of the markers tested (data not shown). (e) Confirming for transposition events and transposon insertions in intron 24 of Egfr for HCCs and lung metastases. PCR genotyping was performed with genomic DNA isolated from the tails, livers and lung metastases of two triple male transgenic mice (ATR M71, 440-days and ATR M81, 460-days). Top panel shows the excision PCR assays (Ex) for transposition events in the lung metastases and HCCs (open arrowhead). No excision was detected in the tails of the triple male transgenic mice. Middle panel shows the PCR genotyping using only the endogenous Egfr forward and T/JB3 primers (T2/onc/Egfr) to confirm for transposon insertion in intron 24 of Egfr for the lung metastases and HCCs. Resulting gel electrophoresis demonstrates the transposon-integrated band (arrowhead) for both the lung metastases (lung) and HCCs (liver), but not in their tails. Gapdh, demonstrate equal genomic DNA template loading (100 ng) used for PCR reaction. Nodule, a different liver tumor nodule was used to compare between different transposon insertion site; MW, 100-bp molecular standard; B6, C57BL/6 tail genomic DNA; H2O, double-distilled water used as a negative control.
Sequencing for common insertion sites (CISs) from tumor samples
A flowchart for SB somatic cell mutagenesis and barcode-assisted integration site amplification procedure is presented as Supplementary Figure 1c online. Briefly, T2/onc integration sites from 68 preneoplastic nodules (3 from triple- and 65 from quadruple-transgenic animals) were cloned and sequenced using bar-coded primers and linker-mediated PCR followed by pyrosequencing18, which has made it possible to sequence tens-of-thousands of T2/onc integrations sites from a mixture of tumors in a single sequencing run (see Supplementary Methods online). Pyrosequencing of linker-mediated PCR products from these tumors generated over 140,000 individual sequences. Sequences containing less than 16 bp of genomic sequence were eliminated, leaving roughly 106,000 sequences. From these, 85,652 sequences were uniquely mapped at 95% identity to the mouse genome. As SB has a tendency to “local hop”, we excluded all insertions that mapped to the transposon donor chromosome (chromosome 15). This was done to ensure that CISs that could have occurred simply due to the bias of recovering insertion sites near the original donor concatemer due to the local hopping phenomenon were not reported6. We also eliminated insertions that did not map to the canonical TA insertion site required for SB integration19-21, giving us a total of 68,782 sequences. We then combined all insertions that mapped to the same TA dinucleotide and originated from the same neoplastic nodule, leaving a final tally of 8,060 non-redundant insertions. Next, we looked for regions in the genome (common insertions sites, CISs) that had more SB insertions than predicted by random chance since these CISs are most likely to harbor disease-related genes. Based on Monte Carlo criteria for statistical significance (see Supplementary Methods online) we defined CISs as regions in the genome with 6 insertions located within 130 kb of each other, 5 insertions within 65 kb or 4 insertions within 20 kb. Thirty CISs were identified according to these criteria in total. Of these CISs, 11 appear to represent “background” non-significant events due to false priming at a specific site in the genome since T2/onc insertions all begin at the same nucleotide, loci with no annotated genes, or were also present among CIS defined by control insertion site mapping experiments using 3-week old transgenic mouse tail DNA carrying both the T2/onc and Rosa26-SB11 transgenes (see Supplementary Methods online). The final CIS list is shown in Table 1 and 8,060 non-redundant insertions can be found as Supplementary Data online. Interestingly, significant overlap with this CIS list was seen in another set of liver tumors induced by a Villin-Cre transgene (manuscript in preparation), further attesting to the significance of these genes for HCC. Importantly, the specific insertion sites obtained from individual preneoplastic nodules at early tumorigenesis were found to be unique for each nodule, thus indicating that each nodule is a unique clone. Certain genes, such as Egfr, are reproducibly mutated by insertion mutations in nodules from the same mouse. However, these insertions are not in identical TA dinucleotides. We therefore conclude, that each preneoplastic nodule was derived from an independent event resulting from random transposon insertional mutagenesis events. In contrast, our lung metastasis analysis, described below, demonstrates clonal relationships can be detected between primary tumors and metastatic derivatives because identical T2/onc insertions occur in individual metastasis samples and a primary liver HCC tumor taken from the same mouse.
Table 1.
Common insertion sites for HCC-associated genes
| Gene | Chr | Position | Range | n | Nodules | Mouse |
|---|---|---|---|---|---|---|
| Egfr | 11 | 16765887-16872714 | 107 kb | 69 | 58 | 5 |
| Novel EST gene cluster | 9 | 3000138-3038047 | 38 kb | 20 | 17 | 5 |
| Sfi1 | 11 | 3046719-3136227 | 90 kb | 13 | 13 | 4 |
| Zbtb20 | 16 | 43349510-43460987 | 111 kb | 8 | 7 | 4 |
| ENSMUSESTG0000001569 | 10 | 52995507-53074648 | 79 kb | 7 | 5 | 2 |
| Nfib | 4 | 82058117-82133086 | 75 kb | 7 | 7 | 4 |
| Taok3 | 5 | 117614813-117701538 | 87 kb | 7 | 6 | 3 |
| Slc25a13 | 6 | 6047524-6159681 | 112 kb | 7 | 7 | 2 |
| Qk | 17 | 10379929-10457807 | 78 kb | 6 | 6 | 3 |
| Rnf13§ | 3 | 57552266-57663120 | 111 kb | 6 | 6 | 3 |
| Met | 6 | 17449763-17545224 | 95 kb | 6 | 6 | 3 |
| March1 | 8 | 68422058-68551102 | 129 kb | 6 | 5 | 2 |
| Psd3 | 8 | 70451840-70580359 | 129 kb | 6 | 6 | 3 |
| Map2k4 | 11 | 65524193-655861144 | 62 kb | 5 | 5 | 4 |
| Trpm7 | 2 | 126659349-126720778 | 61 kb | 5 | 5 | 3 |
| Ube2h§ | 6 | 30181012-30207531 | 27 kb | 5 | 4 | 3 |
| Vrk2§ | 11 | 26373044-26373912 | 869 bp | 4 | 3 | 3 |
| Hif1a | 12 | 75021346-75031073 | 10 kb | 4 | 4 | 2 |
| Pak4§ | 7 | 29367702-29371179 | 3 kb | 4 | 4 | 3 |
Chr, chromosome; Range, chromosomal position of transposon insertions; n, frequency of transposon insertions;
genes that did not have any transposon insertions from liver tumors generated with the Villin-Cre transgenic mice used in the gastrointestinal cancer study (manuscript in preparation). Nodules, number of preneoplastic nodules from which the CIS was determined from; Mouse, number of mice from which the nodules were isolated from. Position based on the Ensembl NCBI m37 April 2007 mouse assembly.
Ingenuity pathway analysis of CIS gene candidates
Next, we used Ingenuity Pathways Analysis (IPA) (Ingenuity® Systems, www.ingenuity.com) to obtain a better understanding of the possible pathways and interactions between CIS genes. Of the 17 CIS genes analyzed, the 3 most significant signaling/disease functional annotations are post-translational modification (p-value, 4.61E-09), cancer (p-value, 8.09E-06) and tumor morphology (p-value, 8.09E-06) (see Supplementary Table 2 online). The CIS list includes several genes that have been implicated in tumor formation and apoptosis of tumor cell lines: EGFR, HIF1A, MAP2K4, MET, PAK4, VRK2, TRPM7 and TAOK3. IPA identified two network pathways overrepresented by CIS genes. The first network includes two transcription factors (NFIB and HIF1A) and the second pathway involves genes that interact with TNF. The combined pathways from IPA are summarized in Figure 3a.
Frequent transposon insertions in Egfr
Interestingly, transposon insertions in the Epidermal growth factor receptor (Egfr) gene were detected in 85% of preneoplastic liver nodules isolated from experimental animals. These transposon insertions were most frequently detected in intron 24 of Egfr (Table 1 and Fig. 3b). The majority of insertions were in the antisense orientation, suggesting they are Egfr-truncating insertions. Three-primer PCR genotyping using endogenous Egfr and transposon primers performed with genomic DNA isolated from individual tumor nodules confirmed the presence of transposon vectors in this locus (Fig. 3c). RT-PCR also confirmed the presence of the predicted truncated Egfr transcript in these preneoplastic nodules (Fig. 3d).
Egfr insertions were also identified in preneoplastic nodules taken from a triple transgenic mouse, indicating that Egfr mutations also contribute to tumorigenesis in a non-predisposed genetic background. These insertions are predicted to result in the production of a truncated Egfr protein (about 984 amino acids) containing the majority of the kinase domain but lacking the carboxy-terminal domain. Indeed, this truncated Egfr was detected by Western blot analysis in the liver tumors of older experimental triple transgenic male mice (see Supplementary Fig. 4b online).
Lung metastases derived from hepatocellular carcinoma
Analysis of genomic DNA taken from metastases from two triple transgenic male mice also demonstrated transposon insertion in intron 24 of Egfr, indicating that they were derived from the HCCs (Fig. 3e). Thirty-two additional lung metastases nodules were isolated from a 432-day old quadruple transgenic male animal. Insertion sites from these metastasis nodules were compared to 3 individual HCC nodules taken from the same animal in order to identify a clonal relationship between primary liver tumors and metastases, and between metastases. One of the liver HCCs (HCC3) seem to share a common ancestor with a second HCC (HCC2) as both have identical Egfr gene insertions, which are distinct from the Egfr insertion in HCC1 (Fig. 4a). Most of the metastases share 4 additional insertions with HCC2 indicating that the metastases share a common ancestor with HCC2. Three additional insertion mutations were found in most of the metastases (Fig. 4a). From the phylogenetic tree generated from the insertion sites (see Supplementary Methods online and Fig. 4b), primary liver tumor HCC2 and all lung metastases have the closest common ancestor suggesting that the lung metastases actually are derived from liver tumor HCC2. This preliminary data suggests that SB-induced tumorigenesis allows one to derive clonal relationships between primary and metastatic derivatives, and to discover metastases-specific insertion mutations that may drive this biological process.
Figure 4.
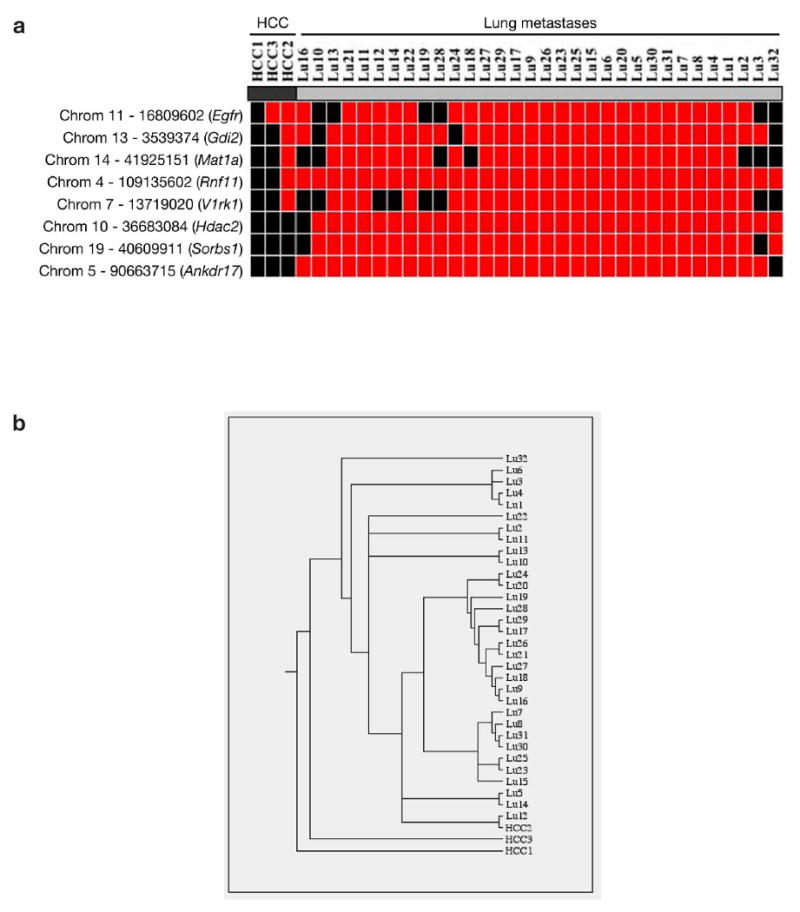
Using SB-induced tumorigenesis to derive clonal relationships between primary and metastatic derivatives. (a) Heat-map showing the clonal relationship between lung metastases and HCC samples. Heat-map was generated by mapping insertion sites from 32 lung metastases nodules and 3 HCC nodules taken from the same experimental mouse (ATRP M232). Importantly, 3 additional insertion mutations were common in most of the metastases, indicating potential genes involved with the metastatic process. Red, insertion detected at the indicated locus; Black, no insertion detected at the indicated locus. (b) Phylogenic tree generated from the insertion sites of the lung metastases and HCCs.
Comparison with human hepatocellular carcinoma samples
Representative oligonucleotide microarray analysis (ROMA) of 100 human HCCs showed that 17 human homologues of our CIS genes were also affected by either gain or loss in copy numbers in HCC (see Supplementary Table 3 online). The effect of transposon insertions on CIS gene expression was also predicted and included in Supplementary Table 3 and Supplementary Methods online. When comparing with human HCC samples (n=100), genes with distinct copy number gains identified in human HCC samples and were CIS genes in our mouse model include EGFR, SLC25A13, MET and UBE2H. Genes with distinct copy number losses in human HCC samples and were CIS genes included MARCH1, PSD3, MAP2K4 and NFIB. In addition, we analyzed another cohort of 132 human samples spanning the whole spectrum of human hepatocarcinogenesis: normal liver (n=10), cirrhotic liver (n=13), low-grade dysplastic nodules (n=10), high-grade dysplastic nodules (n=8) and HCC (n=91). Fifteen of the CIS genes were analyzed by combined single nucleotide polymorphism and gene expression-arrays. The most appealing candidates for clinical correlations were selected based on recurrent gene copy number changes and correlated gene expression changes compared to control samples (see Supplementary Methods online). Out of the 15 genes, only 3 genes fulfilled these criteria: MAP2K4, QKI and UBE2H. MAP2K4 and QKI have losses of DNA copy numbers with significant decrease in mRNA levels, whereas UBE2H has DNA copy number gains with significant increase in mRNA levels (see Supplementary Fig. 5a online). Associations between MAP2K4, QKI and UBE2H expression and clinico-pathological variables were analyzed in 82 HCV-related HCC patients treated with liver resection (see Supplementary Methods online). Although these genes didn't display a significant difference in outcome measured by tumor recurrence or survival due to the small sample population, high expression levels of UBE2H displayed a non-significant trend towards lower survival rates (p=0.09) compared with low expression levels (see Supplementary Fig. 5c online). Tyrosine kinase receptors EGFR and MET, both located on chromosome 7, recently shown that copy number gains of this chromosome are a frequent event in HCC, and characterized as a molecular class of HCC patients22 (see Supplementary Fig. 5b online).
Functional validation of several CIS genes
Since Ubiquitin-conjugating enzyme E2H (UBE2H) is a candidate HCC oncogene, its oncogenic potential was tested using a cell proliferation assay. AML12 cells (adult mouse hepatocyte cell line transgenic for the human TGF-α gene) stably transfected with an Ube2h expression vector have a higher proliferative rate than normal untransfected cells or AML12 cells transfected with an empty vector (see Supplementary Fig. 6 online).
To test whether the truncated form of EGFR could contribute to neoplastic growth in vivo, the Fumaryl acetoacetate hydrolase (Fah)-deficient mouse model was utilized as previously described23. Two vectors were generated: One that co-expresses Fah and Luciferase (pT2/FAHIL); and the other, a truncated form of EGFR (exon 1 to exon 24) (pT2/PGK-Truncated EGFR) (Fig. 5a). The vectors were administered to Fah-deficient mice that express the SB11 transposase knocked into the Rosa26 locus (Fah/SB11) by tail vein hydrodynamic injection24. Upon withdrawal of NTBC, the mice underwent liver repopulation, as evidenced by stable weight gain and increasing Luciferase expression (Fig. 5b). One experimental mouse injected with pT2/FAHIL and pT2/PGK-Truncated EGFR was sacrificed 43-days post-injection and several patches of liver hyperplastic nodules were visible (Fig. 5c). These nodules were shown by RT-PCR to express Fah and the truncated form of EGFR (Fig. 5d). These hyperplastic liver nodules were confirmed by IHC to co-express both Fah and EGFR (Fig. 5f,g). Importantly, adjacent normal appearing liver tissue was negative for both transcripts (Fig. 5d).
Figure 5.
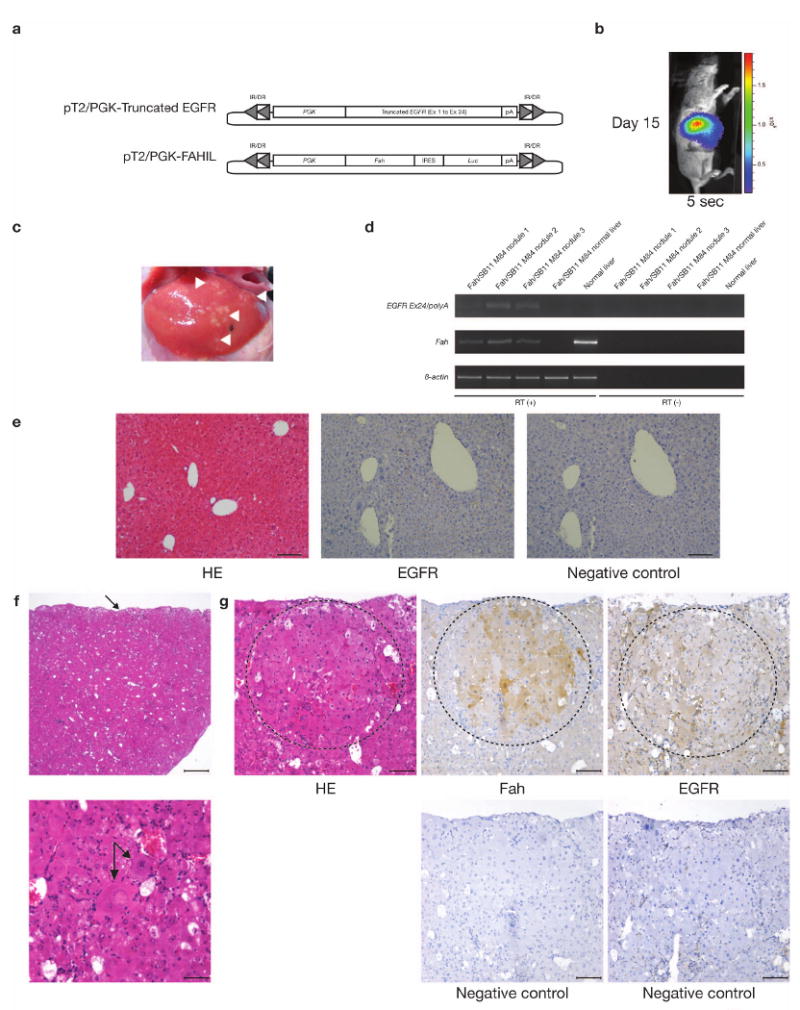
Validating the oncogenic potential of truncated EGFR using the Fumarylacetoacetate hydrolase (Fah)-deficient mouse model. (a) Vectors used for tail vein hydrodynamic injection. pT2/PGK-Truncated EGFR, truncated EGFR cDNA (exon 1 to exon 24) placed under the control of the Phosphoglycerate kinase (PGK) promoter and flanked by SB inverted repeat/direct repeat (IR/DR) recognition sequences essential for transposition. pT2/PGK-FAHIL, Fumaryl acetoacetate hydrolase (Fah) cDNA placed under the control of the PGK promoter fused with an IRES-Luciferase (Luc) reporter gene, flanked by SB IR/DRs. (b) Luciferase activity in Fah/SB11 M84 taken 15-days post-injected with pT2/PGK-Truncated EGFR and pT2/PGK-FAHIL. Exposure time was 5-seconds. (c) Validating the tumorigenic potential of truncated EGFR using the Fah-deficient mouse model. At 43-days post-hydrodynamic injection, Fah/SB11 M84 was killed and the abdominal cavity opened, revealing many patches of small hyperplastic liver nodules (arrowheads). These nodules were carefully removed for RNA extraction and subsequent RT-PCR analyses. Adjacent normal liver tissue was also extracted for comparison. (d) RT-PCR analyses of the liver nodules and adjacent normal tissue. Liver hyperplastic nodules were expressing both Fah and the truncated form of EGFR, while the adjacent normal tissue was negative for both transcripts. RT (+), first strand cDNA synthesis with reverse transcriptase added; RT (−), first strand cDNA synthesis without reverse transcriptase. (e) Normal histology of Fah-deficient liver (hematoxylin-eosin stain, HE) and lack of EGFR detectable by immunohistochemical (IHC) staining. EGFR, treated with EGFR primary antibody; Negative control, serial section not treated with the indicated primary antibody. Scale bars, 100 μm. (f) Histology of liver hyperplastic nodules induced by truncated form of EGFR. Top HE panel, capsular surface of the liver was irregularly nodular (arrow) but overall hepatic architecture was preserved with regularly spaced central veins and portal tracts. Scale bar, 500 μm. Bottom HE panel, a portion of hepatic lobule containing variably sized hepatocytes with 2 cytomegalic and karyomegalic hepatocytes in the center one of which is binucleated (arrows). Occasional hepatocytes have vacuolated cytoplasm. Hepatic cords are not evident due to cellular crowding. Scale bar, 50 μm. (g) Hyperplastic nodule (enclosed within dashed circular line) within hepatic parenchyma consisted of closely packed sheets of variably sized hepatocytes including a karyomegalic cell. Note mild compression of surrounding hepatic parenchyma. There was low degree of inflammation represented by scattered neutrophils and lymphocytes and mild extramedullary hematopoiesis. IHC analyses of serial liver sections treated with the indicated primary antibody confirmed the co-expression of Fah and EGFR in liver nodules. Most of the hepatocytes within hyperplastic nodule (enclosed within dashed circular line) expressed Fah. Hepatocytes within hyperplastic nodule (enclosed within dashed circular line) and within surrounding parenchyma stained weakly for EGFR. EGFR staining is also prominent in the cytoplasmic membranes of cells bordering sinusoids. Negative control, serial sections not treated with the indicated primary antibody. Scale bars, 100 μm.
Discussion
The recent development of target-based therapeutics for treating cancer has sparked a worldwide effort to identify all of the genes and signaling pathways that cause this disease. Transposon-based insertional mutagenesis represents a powerful method for identifying cancer genes but until now it has been impossible to control transposition in a manner that would allow different types of cancer to be modeled. Using a conditional SB transposase allele and a hepatocyte-specific Cre recombinase, we were successful in screening for HCC-associated genes in mice. As expected, quadruple transgenic mice displayed more numerous and larger tumor nodules than triple transgenic animals, as a result of the p53 predisposed genetic background. Importantly, our conditional SB liver tumor model is useful in elucidating genetic mechanisms for all stages of HCC tumorigenesis, from early hepatic adenoma to fully developed HCC and including metastases.
Using pyrosequencing technology, it was possible to amplify and sequence tens-of-thousands of SB insertion sites from a mixture of tumors in a single sequencing run, thereby greatly facilitating the use of transposons for cancer gene identification. Among 8,060 non-redundant insertions subsequently cloned from 68 tumor nodules, 19 highly significant common insertion sites (CISs) were identified. The CIS list contains several genes that have been implicated in tumor formation and apoptosis of tumor cell lines: EGFR, HIF1A, MAP2K4, MET, PAK4, VRK2, TRPM7 and TAOK3. Ingenuity pathway analysis (IPA) identified two network pathways overrepresented by these CIS genes. The first network includes two transcription factors, NFIB and HIF1A, which are capable of transducing phosphorylation-signaling cascades from EGFR. HIF1A has also been suggested to play a role in tumor vascularization25. The second pathway involves genes that interact with TNF. TNF can induce tyrosine phosphorylation and internalization of EGFR, playing a critical role in NF-KB activation26. NF-KB, in turn, plays an important role in regulating apoptosis during liver tumor formation27.
Transposon insertions in the Egfr gene that truncate the carboxy-terminus were common in SB-induced liver tumors. Deletions of the carboxy-terminal domain of EGFR (966-1006) have been shown to result in higher autokinase activity and in transforming ability in vitro and in vivo28. Internal deletions in the carboxy-terminus of EGFR have also been detected in naturally occurring EGFR mutants displaying tumorigenic properties28-30, probably resulting in constitutively active forms of the protein due to the destabilization of the inactive EGFR monomeric complex31. It has been suggested that truncated Egfr can form a heterodimer with ErbB2 and transphosphorylate the tyrosine sites32. Tyrosine phosphorylated ErbB2 could then lead to the activation of other signaling pathways by other mechanisms and may play a role in HCC tumorigenesis33. EGFR over-expression has also been demonstrated in several human cancers such as breast, gut and HCC34-37. EGFR is over-expressed in 15-40% of HCC although there are data suggesting activation of EGF signaling in close to 50% of HCCs37, 38. Extra copies of the EGFR gene were seen in 17 out of 38 (45%) HCC tumors but increased expression did not correlate with the increase in EGFR copy number34. Recent findings suggest that EGF signaling could be even related to HCC development based on significant differences in EGF genotype prevalence according to risk of developing HCC39. In addition, target therapy against EGFR using erlotinib have shown interesting preliminary results in phase II clinical trials in HCC40, 41.
Genes with distinct copy number gains identified in human HCC samples and that were CIS genes in our mouse model include EGFR, SLC25A13, MET and UBE2H. EGFR and MET are known proto-oncogenes, while SLC25A13 and UBE2H may have novel oncogenic activities in HCC. MET encodes the tyrosine kinase receptor for Hepatocyte growth factor and is overexpressed in HCC42. While our algorithm predicted a Met gene disruption in our SB-induced tumors (see Supplementary Table 3 online), we suspect that these insertions actually activate Met as an oncogene since 5 out of 6 could produce a kinase domain containing truncated protein or activate the gene by enhancer insertion. It is also possible that loss of function of Met makes a positive contribution to tumor development since Met knockout mice have been reported to be more sensitive to liver tumor development43. Genes with distinct copy number losses in human HCC samples and that were CIS genes included MARCH1, PSD3, MAP2K4 and NFIB. MAP2K4, has been identified as a putative tumor-suppressor gene in human solid tumors of breast, prostate and pancreas, and may have a similar function in the liver44-46. PSD3 and MARCH1 have not been shown to be involved in cancer but based on data presented here, may have potential tumor suppressor activity in HCC. Interestingly, NFIB is a transcription factor that is known to be up-regulated in hepatitis-induced HCC47. Another interesting finding is that a large number of the CIS genes have human homologues that map to chromosome 7 which has copy number amplifications in more than 15% of human HCC cases48, 49. In addition, another cohort of 132 human samples spanning the whole spectrum of human hepatocarcinogenesis was compared with 15 of the CIS genes by combined SNP- and gene expression-arrays. Preliminary results indicate a non-significant trend to higher tumor recurrence and poorer survival rates associated with higher expression levels of UBE2H. From our validation experiments and human comparative studies, there is evidence to suggest a novel role of UBE2H in liver tumorigenesis. Furthermore, validation experiments confirmed the contribution of truncated EFGR to neoplastic growth in vivo.
Recently, a molecular classification of HCC has been proposed based on gene copy number alteration and expression profiling22. Based on hierarchical clustering of gene expression data, Chiang et al. identified five classes: CTNNB1, proliferation, IFN-related, novel class defined by polysomy of chromosome 7 and an unannotated class. We did not recover recurrent insertions in Ctnnb1 or several of the other known human HCC genes. We did observe an increase in β-catenin protein expression however. Nevertheless, we may be modeling one or more of the non-CTNNB1 subclasses of HCC, a prediction we intend to verify using mRNA microarray profiling of SB-induced HCC. Nevertheless, based on our comparison of CIS genes to gene copy number and expression changes in human HCC, it appears that three of the genes on our list are strong candidates for drivers of HCC: UBE2H, QKI, and MAP2K4. Moreover, several of the other CIS genes have been studied specifically in the context of human HCC and are likely to play a role in the development of this disease. These genes include MET, EGFR, and HIF1A. Taken together, this indicates that the SB screen yields a high fraction of relevant events in human HCC.
These studies, combined with others showing that conditional transposon-based insertional mutagenesis can be used to model solid tumors in other organ sites such as brain and gastrointestinal tract (unpublished results), define a powerful new method for dissecting the cancer genome and for developing better treatments for cancer. In conclusion, future directions include using this technology for further validation of both HCC- and metastasis-associated genes generated by this study.
Methods
Generation of transgenic animals
Albumin-Cre (Alb-Cre) transgenic animals were purchased from Jackson Laboratory, USA50. They were initially bred with T2/onc homozygotes to obtain doubly transgenic animals carrying both Alb-Cre and T2/onc. The T2/onc transgenic line with the donor concatemer on chromosome 15 generated as previously described8, was used in this study. Simultaneously, transgenic animals heterozygous for Rosa26-lsl-SB11 and p53-lsl-R270H (purchased from NCI, Frederick Mouse Repository) were interbred to obtain doubly transgenic animals. The two doubly transgenic lines were finally interbred to generate the required triple (Alb-Cre/T2/onc/Rosa26-lsl-SB11), quadruple (Alb-Cre/T2/onc/Rosa26-lsl-SB11/p53-lsl-R270H) and control animals of various transgene combinations. The genetic background of these animals was mixed, allowing for a diverse genetic population analyses.
PCR genotyping
Identification of the various genotypes from both adult transgenic animal and pups were performed as follows: Firstly, genomic DNA was isolated from tail clippings using standard proteinase K treatment, phenol-chloroform extraction and ethanol precipitation. Genomic DNA was then dissolved in sterile TE [10mM tris-HCl (pH7.5), 1mM EDTA (pH 8)] and quantified using a Nanodrop spectrophotometer. PCR genotyping was performed using 100 ng of diluted genomic DNA as template. PCR primers used for Alb-Cre were forward 5′-CACACTGAAATGCTCAAATGGGAGA-3′ and reverse 5′-GGCAAATTTTGGTGTACGGTCAGTA-3′ (amplicon 456 bp); T2/onc forward 5′-CGCTTCTCGCTTCTGTTCGC-3′ and reverse 5′-CCACCCCCAGCATTCTAGTT-3′ (amplicon 264 bp); Rosa26-lsl-SB11 were Rosa26 wild-type forward 5′-CTGTTTTGGAGGCAGGAA-3′, Rosa26 wild-type reverse 5′-CCCCAGATGACTACCTATCCTCCC-3′, SB reverse 5′-CTAAAAGGCCTATCACAAAC-3′ (Rosa26 wild-type and Rosa26-lsl-SB11 amplicons are 420 bp and 266 bp, respectively); p53-lsl-R270H were p53 wild-type forward 5′-TTACACATCCAGCCTCTGTGG-3′, p53 wild-type reverse 5′-CTTGGAGACATAGCCACACTG-3′, p53-lsl-R270H conditional forward 5′-AGCTAGCCACCATGGCTTGAGTAAGTCTGCA-3′ (p53 wild-type and p53-lsl-R270H conditional allele amplicons are 170 bp and 270 bp, respectively). PCR conditions for Taq polymerase (CLP) were used according to the manufacturer's instructions with an initial denaturing step of 94°C for 5 min; 35-cycles of denaturing at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min; followed by a final extension at 72°C for 7 min. PCR products were separated on a 2% agarose gel and genotype determined by the absence or presence of expected amplicons.
Liver tumor analysis
The whole liver was carefully removed from the sacrificed animal, washed and placed in cold phosphate buffered saline (PBS). The number of surface liver tumor nodules was counted for all liver lobes. All reasonably sized tumor nodules (>2 mm in diameter) were carefully removed from the liver lobes using fine forceps and placed in fresh cold PBS. These separated nodules were then halved using a sterile razor blade and split into samples for DNA and RNA extraction. Tissue samples for RNA were stored at -80°C in RNAlater (Sigma) to prevent RNase contamination and degradation. Histological sections were only taken for larger tumor nodules (>2 mm in diameter), in addition to the samples for DNA and RNA extraction. DNA extraction was done as previously described in the PCR genotyping section. Extraction of RNA was done using the Trizol reagent (Invitrogen) using protocols described by the manufacturer. Formalin fixed-paraffin embedded sections from various tissues were sectioned at 5 microns using a standard microtome (Leica), mounted and heat-fixed onto glass slides. Tissue section slides were either processed and stained with hematoxylin-eosin (HE) using standard protocols, or used for immunohistochemistry (IHC) as described in the next section.
Immunohistochemistry (IHC)
Formalin fixed-paraffin embedded sections from various tissues were sectioned at 5 microns, mounted and heat-fixed onto glass slides to be used for IHC analyses. Briefly, the glass section slides were dewaxed and rehydrated through a gradual decrease in ethanol concentration. The antigen epitopes on the tissue sections were then unmasked using a commercially available unmasking solution (Vector Laboratories) according to the manufacturer's instructions. The tissue section slides were then treated with 3% hydrogen peroxide to remove any endogenous peroxidases. Blocking was performed at 4°C using a M.O.M. mouse immunoglobulin-blocking reagent (Vector Laboratories) in a humidified chamber for several hours. The sections were then incubated overnight at 4°C in a humidified chamber using various primary antibodies: SB transposase (1:100) (R&D Systems), Alb (1:200) (abcam), Afp (1:100) (GeneTex), Ki67 (1:200) (Novocastra), β-catenin (1:500) (BD) and Fah (1:250) (AbboMax). After primary incubation, sections were washed thoroughly in PBS before incubating with horseradish peroxidase-secondary antibody raised against the primary antibody initially used. After thorough washes with PBS, the sections were treated with freshly prepared DAB substrate (Vector Laboratories) and allowed for adequate signal to develop before stopping the reaction in water. For EGFR IHC, EGFr Kit (Clone 31G7) (Zymed Laboratories, Invitrogen) was used according to the manufacturer's instructions, except for the following modification: An additional overnight blocking step using the M.O.M. mouse immunoglobulin-blocking reagent (Vector Laboratories) was incorporated after proteinase K treatment in order to reduce background staining. Finally, sections were then lightly counter-stained with hematoxylin, dehydrated through gradual increase in ethanol concentration, cleared in Citrosol and mounted in Permount (Fisher).
Pyrosequencing
Protocol for amplicon sequencing using the GS20 Flex pyrosequencing machine was as previously described by Roche. Briefly, 100 ng of genomic DNA isolated from individual tumors was digested with either BfaI or NlaIII, for left or right transposon IR/DR, respectively. A small volume of this enzyme digest was used for splinkerette linker attachment using the appropriate linker. To make the BfaI linker, the following oligonucleotide sequences were annealed together using standard protocols, top strand 5′-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC-3′ and bottom strand 5′- TAGTCCCTTAAGCGGAG-3′. As for the NlaIII linker, the following oligonucleotide sequences were annealed together using standard protocols, top strand 5′-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGACCATG-3′ and bottom strand 5′- GTCCCTTAAGCGGAGCC-3′. Linker ligations were performed overnight at 16°C using T4 DNA ligase (NEB). The ligation reaction was cleaned using MinElute 96 well plates (Qiagen) in a vacuum manifold and resuspended in 40 μl of sterile double-distilled water (DDW). This resuspended solution was then digested with either BamHI or XhoI, for left or right transposon IR/DR, respectively. A small volume was then used for primary PCR using the following primers: Left IR/DR primer (BfaI), 5′-CTGGAATTTTCCAAGCTGTTTAAAGGCACAGTCAAC-3′; right IR/DR primer (NlaIII), 5′-GCTTGTGGAAGGCTACTCGAAATGTTTGACCC-3′ and common splinkerette primer was used for both IR/DRs, 5′-GTAATACGACTCACTATAGGGC-3′. PCR conditions for Taq polymerase (CLP) were used according to the manufacturer's instructions of an initial denaturing step of 94°C for 5 min; 30-cycles of denature at 94°C for 30 sec, annealing at 60°C for 30 sec and extension at 72°C for 1.5 min; followed by a final extension at 72°C for 5 min. One microliter of the diluted first PCR product sample (1:75) was used as template for the secondary PCR under the following conditions: Nested versions of the above primers carrying the required fusion sequences for GS20 Flex pyrosequencing (Fusion A and Fusion B), as well as a unique 10 bp barcode recognition sequence for each tumor sample. Primers were designed as such that the nested transposon primer have the Fusion A and barcode attached (Fusion A – barcode – nested primer) and the nested linker primer has the Fusion B sequence attached (linker nested – Fusion B). PCR conditions for Taq polymerase (Roche FastStart High Fidelity) were used according to the manufacturer's instructions of an initial denaturing step of 94°C for 5 min; 35-cycles of denature at 94°C for 30 sec, annealing at 60°C for 30 sec and extension at 72°C for 1.5 min; followed by a final extension at 72°C for 5 min. After the secondary PCR, the reaction was purified using MinElute 96 well plates (Qiagen) in a vacuum manifold and resuspended in 30 μl of sterile TE. The amount of DNA in each PCR sample was quantified using the QuantIT picogreen kit (Invitrogen) and the samples were diluted to a final concentration of 2 × 105 molecules/μl for pyrosequencing.
Selection criteria for common insertion sites (CISs)
Egfr PCR genotyping
PCR genotyping was used to confirm the presence of the T2/onc transposon insertion in intron 24 of the Egfr gene. Briefly, genomic DNA was isolated from individual tumor nodules using protocols already described in the PCR genotyping section. PCR genotyping was performed using 100 ng of diluted genomic DNA as template. PCR primers used for Egfr intron 24 were forward, 5′-TACATGGTCAAAATCTCTCCAATAGGTC-3′ and reverse, 5′-ATTAGAAAGGGCAACGAAGCTTGC-3′, with an expected amplicon of 713 bp. A third primer specific for the IR/DR-R (T/JB3) of the T2/onc transposon vector was also included, 5′-AGGGAATTTTTACTAGGATTAAATGTCAGG-3′. PCR conditions were as described previously in the PCR genotyping section. The amplicon sizes varied depending on the position of the T2/onc transposon vector insertion site. When the T2/onc/Egfr amplicon is expected to overlap the endogenous Egfr product, a PCR genotyping using only the T/JB3 and Egfr intron 24 forward primers is used instead with the same PCR conditions.
RT-PCR
Extraction of RNA from tumor nodules was done using the Trizol reagent (Invitrogen) using protocols described by the manufacturer. First strand cDNA synthesis was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche) as described by the manufacturer using 1 μg total RNA as template. Both reactions using with (RT+) and without (RT−) the reverse transcriptase were performed for all the samples. Subsequent PCR was performed using 1 μl of the cDNA as template with various primer pairs. Primer sequences for Alpha-fetoprotein (Afp) were forward 5′-CCTGTGAACTCTGGTATCAG-3′ and reverse 5′-GCTCACACCAAAGCGTCAAC-3′ (amplicon 410 bp); Osteopontin (Opn) forward 5′-CTTTCACTCCAATCGTCCCTAC-3′ and reverse 5′-GCTCTCTTTGGAATGCTCAAGT-3′ (amplicon 305 bp); Sleeping Beauty (SB) transposase forward 5′-ATGGGAAAATCAAAAGAAATCAGCC-3′ and reverse 5′-CGCACCAAAGTACGTTCATCTCTA-3′ (amplicon 221 bp); Albumin (Alb) forward 5′-CCCCACTAGCCTCTGGCAAAAT-3′ and reverse 5′-CTTAAACCGATGGGCGATCTCACT-3′ (amplicon 127 bp); Epidermal growth factor receptor (Egfr) forward 5′-GATAGATGCTGATAGCCGCCCAAAG-3′ and reverse 5′-TCATGCTCCAATAAACTCACTGCTT-3′(amplicon 772 bp); truncated-Egfr forward (same forward primer used for Egfr) and reverse (specific for the T2/onc SV40-polyA) 5′-TGCTTTATTTGTGAAATTTGTGATGCTATTG-3′ (amplicon 320 bp); Receptor tyrosine-protein kinase erbB2 (ErbB2) forward 5′-CCCAGATCTCCACTGGCTCC-3′ and reverse 5′-TTCAGGGTTCTCCACAGCACC-3′ (amplicon 376 bp); β-actin forward 5′-GTGACGAGGCCCAGAGCAAGAG-3′ and reverse 5′-AGGGGCCGGACTCATCGTACTC-3′ (amplicon 938 bp); Neomycin (Neo) forward 5′-ATGATTGAACAAGATGGATTGCACG-3′ and reverse 5′-AAGGTGAGATGACAGGAGATCCTG-3′ (amplicon 321 bp); Ubiquitin-conjugating enzyme E2H (Ube2h) forward 5′-CTGAGCGGACCCCACGGGAC-3′ and reverse 5′-CAGCAACTGGGGCAGGAAGG-3′ (amplicon 505 bp); Fumarylacetoacetate hydrolase (Fah) forward 5′-ATGAGCTTTATTCCAGTGGCC-3′ and reverse 5′-ACCACAATGGAGGAAGCTCG-3′ (amplicon 503 bp); truncated EGFR forward 5′-GACCCCCAGCGCTACCTTGTCATTCAG-3′ and reverse (specific for the rabbit β-globin polyA) 5′-GCCACACCAGCCACCACCTTCTG-3′ (amplicon 140 bp). PCR conditions are similar to PCR genotyping described previously except 25 to 30 cycles were performed to avoid amplicon saturation.
Representational oligonucleotide microarray analysis (ROMA)
Microarray analysis was performed on human HCC samples as previously described51.
Cell proliferation assay
AML12 (CRL-2254) was obtained from America Type Culture Collection (ATCC) and maintained according to the recommended culture conditions. An expression vector for Ube2h (MC200579) mouse cDNA was obtained from Origene. The empty vector (pcDNA) purchased from Invitrogen, was used as a negative control. Cell transfections were performed using Lipofectamine LTX (Invitrogen) with PLUS (Invitrogen) according to the manufacturer's recommendation. Transfected cell lines were grown in media containing neomycin (0.5 mg/ml) for 2 weeks to select for stable cell populations. Stable cell populations for each expression vector were obtained from 3 individual transfections. Cell proliferation rate of the stable cell populations was determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer's protocols.
Hydrodynamic injection
Hydrodynamic injections were performed as previously described23. Briefly, Fumarylacetoacetate hydrolase (Fah)-null mice carrying the Rosa26-SB11 transgene were generated. Truncated-EGFR (exon 1 to exon 24) was PCR amplified from pBabe-Puro-LTR-EGFR (a kind gift from Dr Heidi Gruelich, Dana Farber Cancer Institute) using the following primers: Exon 1 forward 5′-ATGCGACCCTCCGGGACGGC-3′ and exon 24 reverse 5′-CTGAATGACAAGGTAGCGCTGGGGGTC-3′ was placed under the control of a Phosphoglycerate kinase (PGK) promoter and cloned into the pT2 vector containing the SB flanking IR/DR recognition sequences to obtain pT2/PGK-Truncated EGFR. Two other constructs were also prepared: pT2/PGK-FAHIL, vector containing the Fah and Luciferase gene under the control of the PGK promoter52. Twenty micrograms of each construct was hydrodynamically injected into 6-week old Fah-null/Rosa26-SB11 male mice (Fah/SB11) using previously established conditions24. These mice are normally maintained with 7.5 μg/ml 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) drinking-water but replaced with normal drinking water after hydrodynamic injection of transposon vectors. These experimental animals were observed for weight changes and Luciferase activity as previously described23.
Supplementary Material
Supplementary Figure 1 Determining common transposon integration sites from the liver cancer mouse model. (a) Transgenes used to generate the liver cancer mouse model. T2/onc, mutagenic transposon vector that can cause mis-expression of proto-oncogenes or disrupt tumor suppressor genes. IR/DR, inverted repeat/direct repeat transposon flanking sequences; SA, splice acceptor; pA, polyadenalytion signal; MSCV 5′ LTR, 5′-LTR of the murine stem cell virus; SD, splice donor. Rosa26-lsl-SB11, SB transposase (SB11) carrying a floxed-stop (lsl) cassette engineered into the mouse Rosa26 locus. p53-lsl-R270H, conditional dominant negative p53 transgene (asterisk indicates location of p53 R270H mutation). Alb-Cre, Albumin promoter driving Cre recombinase for the removal of floxed-stop cassettes and activation of both SB transposase and dominant negative p53 R270H protein in hepatocytes. (b) Breeding strategy for generating experimental animals. Transgenic mice carrying the 4 individual transgenes were bred to obtain doubly transgenic animals. These doubly transgenic mice were subsequently bred to obtain either triple (non-predisposed genetic background) or quadruple (predisposed genetic background) experimental animal. Control animals carrying various combinations of transgenes were also generated but not shown in the diagram. (c) Flowchart for high-throughput barcode-assisted amplification procedure used to obtain transposon common insertion sites.
Supplementary Figure 2 Analyses of preneoplastic liver nodules isolated from experimental animals. (a) Excision PCR analyses demonstrating evidence of transposon (T2/onc) excision in livers taken from non-tumor producing experimental and control livers. TF, triple-transgenic female livers; QF, quadruple-transgenic female livers; TM, triple-transgenic male livers; QM, quadruple-transgenic male livers (red and black, tumor- and non-tumor producing, respectively); ARP, transgenic animal containing Alb-Cre, Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; RP, transgenic animal containing Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; ORP, transgenic animal containing the T2/onc, Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; Tumor, genomic DNA isolated from a liver neoplastic nodule; H2O, double-distilled water negative control; D, indicates the age of the animal in days; Donor, 2.4 kb PCR amplicon; Excision, 233 bp PCR amplicon; MW, 100-bp molecular standard; Gapdh, demonstrate equal genomic DNA template loading (100 ng) used for PCR reaction. (b) Tumorigenic livers extracted from 160-day old triple- (left) and quadruple- (right) experimental male transgenic littermates showing accelerated tumor formation in the latter. Arrowheads, denote smaller preneoplastic nodules in the triple-transgenic littermate; scale bar, 0.5 cm. (c) Semi-quantitative analysis of RT-PCR products from Figure 3d. The ImageJ software was used to quantify the band intensity of the RT-PCR amplicons for Afp, Opn, Egfr and ErbB2. Arbitrary units shown relative to β-actin levels. Advanced tumors, n=3; Early tumors, n=13; HCC, n=1; Normal liver, n=1. Values are the mean ± SD.
Supplementary Figure 3 Immunohistochemical (IHC) analyses of liver nodules at various stages of tumorigenesis showing positive reaction to β-catenin. Normal liver, normal C57BL/6 liver; ATRP M81, preneoplastic nodules from a 156-day quadruple transgenic male mouse; ATRP M121, preneoplastic nodules from a 178-day quadruple transgenic male mouse; ATR M81, preneoplastic nodules from a 460-day triple transgenic male mouse; NRAS liver tumor, HCC control taken from a tumorigenic liver over-expressing NRAS G12V oncogene. Negative control, IHC of liver sections not treated with the primary antibody; Primary antibody, IHC of serial liver sections treated with the indicated primary antibody; scale bars, 100 μm.
Supplementary Figure 4 Analyses of liver samples from female experimental animals and Western blot analyses for truncated Egfr. (a) Immunohistochemical (IHC) analyses of a 344-day old non-tumor producing quadruple-transgenic female liver showing positive reaction to Afp and Ki67 (arrowheads). These sections were also IHC positive for SB and Alb (data not shown). Negative control, IHC of liver sections not treated with the primary antibody; Primary antibody, IHC of serial liver sections treated with the indicated primary antibody; scale bars, 100 μm. (b) Western blot analysis for the truncated Egfr protein. Top panel, Using a phospho-Egf receptor (Tyr845) antibody, the truncated Egfr was detected at around the 150 kDa size (open arrowhead) and the wild-type Egfr can be weakly seen in some of the samples at around the 170 kDA size (arrowhead). Quadruple experimental animals: ATRP M81 preneoplastic liver sample, 156-days; ATRP F6 non-tumor producing liver, 279-days; ATRP F67 non-tumor producing liver, 344-days. Triple experimental animals: ATRP M175 preneoplastic liver sample, 375-days; ATRP M51 individual preneoplastic liver samples, 330-days; ATR M71 HCC sample and lung metastasis, 440-days. Truncated Egfr was detected in majority of samples, faintly in the lung metastasis but not in B6 and ATR M71 liver samples. Bottom panel, GAPDH monoclonal antibody was used to demonstrate protein loading for the Western blot. Lung, lung metastasis; B6, protein isolated from C57BL/6 liver.
Supplementary Figure 5 Integrative genomic analysis of CIS candidate genes in human hepatitis C virus (HCV)-related hepatocellular carcinoma (HCC). (a) Gene expression of 3 candidate genes in human HCC: MAP2K4, QKI and UBE2H. (b) Gene expression and its correlation with DNA copy numbers of EGFR and MET in human HCC. Left panels (a, b), changes in gene expression in the whole spectrum of human HCC. Normal, normal liver; Cirrhosis, cirrhotic tissue; LGDN, low-grade dysplastic nodules; HGDN, high-grade dysplastic nodules; HCC, hepatocellular carcinoma. Right panels (a, b), correlation between DNA copy numbers and gene expression for each candidate gene (log2 expression values). (c) Association between UBE2H expression and overall survival of HCV-induced HCC patients. Non-significant trend towards poorer survival associated with high UBE2H expression (red line) compared with low expression levels (black line) in HCV-induced HCC patients.
Supplementary Figure 6 Validating the effect of over-expressing Ube2h in AML12 cell line by cell proliferation assay. (a) RT-PCR of AML12 cells stably transfected with the Ube2h expression vector. Three different cell populations of Ube2h transfected cells from separate transfection experiments were generated. Representative RT-PCR of 2 transfected cell populations are presented. AML12, normal untransfected AML12 cells; RT (+), first strand cDNA synthesis with reverse transcriptase added; RT (−), first strand cDNA synthesis without reverse transcriptase. Presence of Neomycin (Neo) resistance gene was confirmed in Ube2h stably transfected cell populations. (b) Semi-quantitative RT-PCR using ImageJ was used to confirm the over-expression levels of Ube2h in transfected cells. Values are relative to β-actin levels and standardized to untransfected AML12. (c) Representative graph showing the proliferative effect of over-expressing Ube2h in AML12 cells. Cells were initially seeded at 10,000 cells and cell proliferation rate determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay at the indicated time points. Ube2h, AML12 transfected with the Ube2h expression vector; Empty, AML12 transfected with the pcDNA empty vector. Values are the representative mean ± SD of experiments done in triplicate, *p<0.01.
Supplementary Table 1 Frequency of liver tumor nodules in experimental mice
Supplementary Table 2 Ingenuity pathway analysis of CIS gene list
Supplementary Table 3 Comparison between CIS genes with human HCC array CGH data analysis
Acknowledgments
The authors wish to thank Christine E. Nelson, Stefanie S. Breitbarth, Michelle K. Gleason and Geoff Hart for their excellent technical support; Jason B. Bell for performing the hydrodynamic injections. We are also grateful to the Minnesota Supercomputing Institute for providing extensive computational resources (hardware and systems administration support) used to carry out the sequence analysis.
A.V. is supported by a Sheila Sherlock fellowship from the European Association for the Study of the Liver. L.S.C. is supported by a 1K01CA122183-01 grant from the National Cancer Institute. N.G.C., N.A.J. and L.T. are supported by the Department of Health and Human Services, National Institutes of Health and the National Cancer Institute. J.M.L is supported by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK076986-01), Spanish National Institute of Health (SAF-2007-61898) and Samuel Waxman Cancer Research Foundation. D.A.L. is supported by U01 CA84221 and R01 CA113636 grants from the National Cancer Institute.
Footnotes
Note: Supplementary information is available on the Nature Biotechnology website.
References
- 1.Spradling AC, et al. Gene disruptions using P transposable elements: an integral component of the Drosophila genome project. Proc Natl Acad Sci U S A. 1995;92:10824–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellen HJ, et al. P-element-mediated enhancer detection: a versatile method to study development in Drosophila. Genes Dev. 1989;3:1288–1300. doi: 10.1101/gad.3.9.1288. [DOI] [PubMed] [Google Scholar]
- 3.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 4.Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 5.Horie K, et al. Efficient chromosomal transposition of a Tc1/mariner- like transposon Sleeping Beauty in mice. Proc Natl Acad Sci U S A. 2001;98:9191–9196. doi: 10.1073/pnas.161071798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keng VW, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 7.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 8.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 9.Zambrowicz BP, et al. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 11.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 12.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 13.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 14.Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer Biol. 2000;10:185–200. doi: 10.1006/scbi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 15.de Vries A, et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci U S A. 2002;99:2948–2953. doi: 10.1073/pnas.052713099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohde F, et al. Expression of osteopontin, a target gene of de-regulated Wnt signaling, predicts survival in colon cancer. Int J Cancer. 2007;121:1717–1723. doi: 10.1002/ijc.22868. [DOI] [PubMed] [Google Scholar]
- 17.Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res. 2001;61:2085–2091. [PubMed] [Google Scholar]
- 18.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Luenen HG, Colloms SD, Plasterk RH. The mechanism of transposition of Tc3 in C. elegans. Cell. 1994;79:293–301. doi: 10.1016/0092-8674(94)90198-8. [DOI] [PubMed] [Google Scholar]
- 20.Vos JC, De Baere I, Plasterk RH. Transposase is the only nematode protein required for in vitro transposition of Tc1. Genes Dev. 1996;10:755–761. doi: 10.1101/gad.10.6.755. [DOI] [PubMed] [Google Scholar]
- 21.Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. Embo J. 1996;15:5470–5479. [PMC free article] [PubMed] [Google Scholar]
- 22.Chiang DY, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wangensteen KJ, et al. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology. 2008;47:1714–1724. doi: 10.1002/hep.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell JB, et al. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc. 2007;2:3153–3165. doi: 10.1038/nprot.2007.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee TK, et al. Regulation of angiogenesis by Id-1 through hypoxia-inducible factor-1alpha-mediated vascular endothelial growth factor up-regulation in hepatocellular carcinoma. Clin Cancer Res. 2006;12:6910–6919. doi: 10.1158/1078-0432.CCR-06-0489. [DOI] [PubMed] [Google Scholar]
- 26.Hirota K, Murata M, Itoh T, Yodoi J, Fukuda K. Redox-sensitive transactivation of epidermal growth factor receptor by tumor necrosis factor confers the NF-kappa B activation. J Biol Chem. 2001;276:25953–25958. doi: 10.1074/jbc.M011021200. [DOI] [PubMed] [Google Scholar]
- 27.Arsura M, et al. Transient activation of NF-kappaB through a TAK1/IKK kinase pathway by TGF-beta1 inhibits AP-1/SMAD signaling and apoptosis: implications in liver tumor formation. Oncogene. 2003;22:412–425. doi: 10.1038/sj.onc.1206132. [DOI] [PubMed] [Google Scholar]
- 28.Chang CM, et al. A minor tyrosine phosphorylation site located within the CAIN domain plays a critical role in regulating tissue-specific transformation by erbB kinase. J Virol. 1995;69:1172–1180. doi: 10.1128/jvi.69.2.1172-1180.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boerner JL, Danielsen A, Maihle NJ. Ligand-independent oncogenic signaling by the epidermal growth factor receptor: v-ErbB as a paradigm. Exp Cell Res. 2003;284:111–121. doi: 10.1016/s0014-4827(02)00096-4. [DOI] [PubMed] [Google Scholar]
- 30.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 31.Landau M, Fleishman SJ, Ben-Tal N. A putative mechanism for downregulation of the catalytic activity of the EGF receptor via direct contact between its kinase and C-terminal domains. Structure. 2004;12:2265–2275. doi: 10.1016/j.str.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Sasaoka T, et al. Involvement of ErbB2 in the signaling pathway leading to cell cycle progression from a truncated epidermal growth factor receptor lacking the C-terminal autophosphorylation sites. J Biol Chem. 1996;271:8338–8344. doi: 10.1074/jbc.271.14.8338. [DOI] [PubMed] [Google Scholar]
- 33.Bekaii-Saab T, Williams N, Plass C, Calero MV, Eng C. A novel mutation in the tyrosine kinase domain of ERBB2 in hepatocellular carcinoma. BMC Cancer. 2006;6:278. doi: 10.1186/1471-2407-6-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckley AF, Burgart LJ, Sahai V, Kakar S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am J Clin Pathol. 2008;129:245–251. doi: 10.1309/WF10QAAED3PP93BH. [DOI] [PubMed] [Google Scholar]
- 35.Rajkumar T, Gullick WJ. The type I growth factor receptors in human breast cancer. Breast Cancer Res Treat. 1994;29:3–9. doi: 10.1007/BF00666177. [DOI] [PubMed] [Google Scholar]
- 36.Lopes LF, Bacchi CE. EGFR and gastrointestinal stromal tumor: an immunohistochemical and FISH study of 82 cases. Mod Pathol. 2007;20:990–994. doi: 10.1038/modpathol.3800932. [DOI] [PubMed] [Google Scholar]
- 37.Villanueva A, et al. Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 39.Tanabe KK, et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. Jama. 2008;299:53–60. doi: 10.1001/jama.2007.65. [DOI] [PubMed] [Google Scholar]
- 40.Philip PA, et al. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;23:6657–6663. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 41.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salvi A, et al. In vitro c-met inhibition by antisense RNA and plasmid-based RNAi down-modulates migration and invasion of hepatocellular carcinoma cells. Int J Oncol. 2007;31:451–460. [PubMed] [Google Scholar]
- 43.Takami T, et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67:9844–9851. doi: 10.1158/0008-5472.CAN-07-1905. [DOI] [PubMed] [Google Scholar]
- 44.Su GH, Song JJ, Repasky EA, Schutte M, Kern SE. Mutation rate of MAP2K4/MKK4 in breast carcinoma. Hum Mutat. 2002;19:81. doi: 10.1002/humu.9002. [DOI] [PubMed] [Google Scholar]
- 45.Xin W, et al. MAP2K4/MKK4 expression in pancreatic cancer: genetic validation of immunohistochemistry and relationship to disease course. Clin Cancer Res. 2004;10:8516–8520. doi: 10.1158/1078-0432.CCR-04-0885. [DOI] [PubMed] [Google Scholar]
- 46.Nakayama K, et al. Homozygous deletion of MKK4 in ovarian serous carcinoma. Cancer Biol Ther. 2006;5:630–634. doi: 10.4161/cbt.5.6.2675. [DOI] [PubMed] [Google Scholar]
- 47.Iizuka N, et al. Differential gene expression in distinct virologic types of hepatocellular carcinoma: association with liver cirrhosis. Oncogene. 2003;22:3007–3014. doi: 10.1038/sj.onc.1206401. [DOI] [PubMed] [Google Scholar]
- 48.Moinzadeh P, Breuhahn K, Stutzer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade--results of an explorative CGH meta-analysis. Br J Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zimmermann U, et al. Chromosomal aberrations in hepatocellular carcinomas: relationship with pathological features. Hepatology. 1997;26:1492–1498. doi: 10.1053/jhep.1997.v26.pm0009397989. [DOI] [PubMed] [Google Scholar]
- 50.Postic C, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 51.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilber A, et al. Messenger RNA as a source of transposase for sleeping beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol Ther. 2007;15:1280–1287. doi: 10.1038/sj.mt.6300160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Determining common transposon integration sites from the liver cancer mouse model. (a) Transgenes used to generate the liver cancer mouse model. T2/onc, mutagenic transposon vector that can cause mis-expression of proto-oncogenes or disrupt tumor suppressor genes. IR/DR, inverted repeat/direct repeat transposon flanking sequences; SA, splice acceptor; pA, polyadenalytion signal; MSCV 5′ LTR, 5′-LTR of the murine stem cell virus; SD, splice donor. Rosa26-lsl-SB11, SB transposase (SB11) carrying a floxed-stop (lsl) cassette engineered into the mouse Rosa26 locus. p53-lsl-R270H, conditional dominant negative p53 transgene (asterisk indicates location of p53 R270H mutation). Alb-Cre, Albumin promoter driving Cre recombinase for the removal of floxed-stop cassettes and activation of both SB transposase and dominant negative p53 R270H protein in hepatocytes. (b) Breeding strategy for generating experimental animals. Transgenic mice carrying the 4 individual transgenes were bred to obtain doubly transgenic animals. These doubly transgenic mice were subsequently bred to obtain either triple (non-predisposed genetic background) or quadruple (predisposed genetic background) experimental animal. Control animals carrying various combinations of transgenes were also generated but not shown in the diagram. (c) Flowchart for high-throughput barcode-assisted amplification procedure used to obtain transposon common insertion sites.
Supplementary Figure 2 Analyses of preneoplastic liver nodules isolated from experimental animals. (a) Excision PCR analyses demonstrating evidence of transposon (T2/onc) excision in livers taken from non-tumor producing experimental and control livers. TF, triple-transgenic female livers; QF, quadruple-transgenic female livers; TM, triple-transgenic male livers; QM, quadruple-transgenic male livers (red and black, tumor- and non-tumor producing, respectively); ARP, transgenic animal containing Alb-Cre, Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; RP, transgenic animal containing Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; ORP, transgenic animal containing the T2/onc, Rosa26-lsl-SB11 and p53-lsl-R270H transgenes; Tumor, genomic DNA isolated from a liver neoplastic nodule; H2O, double-distilled water negative control; D, indicates the age of the animal in days; Donor, 2.4 kb PCR amplicon; Excision, 233 bp PCR amplicon; MW, 100-bp molecular standard; Gapdh, demonstrate equal genomic DNA template loading (100 ng) used for PCR reaction. (b) Tumorigenic livers extracted from 160-day old triple- (left) and quadruple- (right) experimental male transgenic littermates showing accelerated tumor formation in the latter. Arrowheads, denote smaller preneoplastic nodules in the triple-transgenic littermate; scale bar, 0.5 cm. (c) Semi-quantitative analysis of RT-PCR products from Figure 3d. The ImageJ software was used to quantify the band intensity of the RT-PCR amplicons for Afp, Opn, Egfr and ErbB2. Arbitrary units shown relative to β-actin levels. Advanced tumors, n=3; Early tumors, n=13; HCC, n=1; Normal liver, n=1. Values are the mean ± SD.
Supplementary Figure 3 Immunohistochemical (IHC) analyses of liver nodules at various stages of tumorigenesis showing positive reaction to β-catenin. Normal liver, normal C57BL/6 liver; ATRP M81, preneoplastic nodules from a 156-day quadruple transgenic male mouse; ATRP M121, preneoplastic nodules from a 178-day quadruple transgenic male mouse; ATR M81, preneoplastic nodules from a 460-day triple transgenic male mouse; NRAS liver tumor, HCC control taken from a tumorigenic liver over-expressing NRAS G12V oncogene. Negative control, IHC of liver sections not treated with the primary antibody; Primary antibody, IHC of serial liver sections treated with the indicated primary antibody; scale bars, 100 μm.
Supplementary Figure 4 Analyses of liver samples from female experimental animals and Western blot analyses for truncated Egfr. (a) Immunohistochemical (IHC) analyses of a 344-day old non-tumor producing quadruple-transgenic female liver showing positive reaction to Afp and Ki67 (arrowheads). These sections were also IHC positive for SB and Alb (data not shown). Negative control, IHC of liver sections not treated with the primary antibody; Primary antibody, IHC of serial liver sections treated with the indicated primary antibody; scale bars, 100 μm. (b) Western blot analysis for the truncated Egfr protein. Top panel, Using a phospho-Egf receptor (Tyr845) antibody, the truncated Egfr was detected at around the 150 kDa size (open arrowhead) and the wild-type Egfr can be weakly seen in some of the samples at around the 170 kDA size (arrowhead). Quadruple experimental animals: ATRP M81 preneoplastic liver sample, 156-days; ATRP F6 non-tumor producing liver, 279-days; ATRP F67 non-tumor producing liver, 344-days. Triple experimental animals: ATRP M175 preneoplastic liver sample, 375-days; ATRP M51 individual preneoplastic liver samples, 330-days; ATR M71 HCC sample and lung metastasis, 440-days. Truncated Egfr was detected in majority of samples, faintly in the lung metastasis but not in B6 and ATR M71 liver samples. Bottom panel, GAPDH monoclonal antibody was used to demonstrate protein loading for the Western blot. Lung, lung metastasis; B6, protein isolated from C57BL/6 liver.
Supplementary Figure 5 Integrative genomic analysis of CIS candidate genes in human hepatitis C virus (HCV)-related hepatocellular carcinoma (HCC). (a) Gene expression of 3 candidate genes in human HCC: MAP2K4, QKI and UBE2H. (b) Gene expression and its correlation with DNA copy numbers of EGFR and MET in human HCC. Left panels (a, b), changes in gene expression in the whole spectrum of human HCC. Normal, normal liver; Cirrhosis, cirrhotic tissue; LGDN, low-grade dysplastic nodules; HGDN, high-grade dysplastic nodules; HCC, hepatocellular carcinoma. Right panels (a, b), correlation between DNA copy numbers and gene expression for each candidate gene (log2 expression values). (c) Association between UBE2H expression and overall survival of HCV-induced HCC patients. Non-significant trend towards poorer survival associated with high UBE2H expression (red line) compared with low expression levels (black line) in HCV-induced HCC patients.
Supplementary Figure 6 Validating the effect of over-expressing Ube2h in AML12 cell line by cell proliferation assay. (a) RT-PCR of AML12 cells stably transfected with the Ube2h expression vector. Three different cell populations of Ube2h transfected cells from separate transfection experiments were generated. Representative RT-PCR of 2 transfected cell populations are presented. AML12, normal untransfected AML12 cells; RT (+), first strand cDNA synthesis with reverse transcriptase added; RT (−), first strand cDNA synthesis without reverse transcriptase. Presence of Neomycin (Neo) resistance gene was confirmed in Ube2h stably transfected cell populations. (b) Semi-quantitative RT-PCR using ImageJ was used to confirm the over-expression levels of Ube2h in transfected cells. Values are relative to β-actin levels and standardized to untransfected AML12. (c) Representative graph showing the proliferative effect of over-expressing Ube2h in AML12 cells. Cells were initially seeded at 10,000 cells and cell proliferation rate determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay at the indicated time points. Ube2h, AML12 transfected with the Ube2h expression vector; Empty, AML12 transfected with the pcDNA empty vector. Values are the representative mean ± SD of experiments done in triplicate, *p<0.01.
Supplementary Table 1 Frequency of liver tumor nodules in experimental mice
Supplementary Table 2 Ingenuity pathway analysis of CIS gene list
Supplementary Table 3 Comparison between CIS genes with human HCC array CGH data analysis