

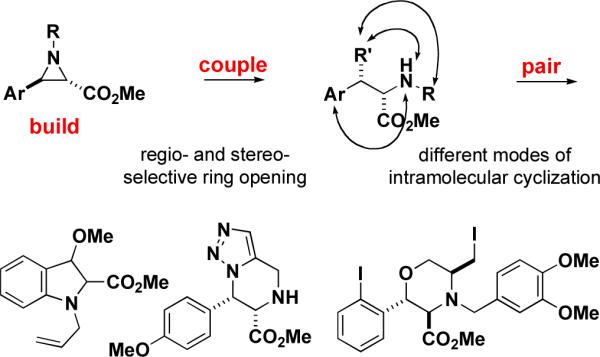
Abstract
The efficient synthesis of small molecules that collectively comprise optimal small-molecule screening collections is an important goal. With this in mind, we have used N-alkyl aziridines in a regio- and stereochemically controlled synthesis of polycyclic heterocycles based on nucleophilic ring opening and subsequent intramolecular cyclization.

Causal disease genes are being discovered with increasing frequency. The majority of the disease targets, which include transcription factors, regulatory RNAs, and signaling proteins without enzymatic activities, are believed to be difficult if not impossible (`undruggable') to modulate with small molecules. The underlying problem appears to be that the compounds comprising standard small-molecule screening collections used in probe and drug development lack the necessary structural elements.1 On the other hand, small molecules synthesized using diversity-oriented synthesis (DOS) have been discovered that target many of the most challenging targets, including ones viewed as `undruggable'.2
The `build/couple/pair' strategy in DOS addresses many of the goals of this type of synthetic chemistry.3,4,5 Here, we describe experiments aimed at using N-alkyl aziridines as building blocks in syntheses producing several alkaloid skeletons using a nascent build/couple/pair strategy.
N-alkyl aziridines were investigated for several reasons. First, the regio- and stereochemical outcomes of nucleophilic additions to aziridines are directed by the nature of the substituents bound to the ring. Theoretical studies indicate that there is a strong preference of nucleophiles for addition to a carbon neighboring π orbitals that can stabilize the transition state6, and such predictions have been confirmed in many experimental studies.7 We therefore expected that incorporating an aryl group as a substituent would promote nucleophilic addition at the adjacent carbon of the aziridine ring (Scheme 1). Second, many ring-opening reactions take place through an SN2 mechanism.8,9 The inversion of stereochemistry during the coupling step would allow for the control of the relative stereochemistry of the products. Third, N-alkyl aziridines were expected to form products with favorable hydrogen bonding potential and without sterically bulky activating groups. Although precedent indicates that the addition of nucleophiles to activated aziridines (substituted with electron-withdrawing groups at the nitrogen) is more facile than that to N-alkyl aziridines, known sulfonamidyl, acyl, or phosphoryl activating groups would alter the hydrogen bonding properties of the amine they mask and add significant steric bulk to the molecule. Fourth, the target aziridines can be prepared on multi-gram scale without protecting groups and with control over both their relative and absolute stereochemistry. A number of efficient methods of preparing aziridines have been reported10-14, and precedent suggests that these methods would be amenable to incorporation of a variety of builiding blocks. Also, the use of protecting groups in a parallel synthesis is undesirable; their addition and removal adds two steps onto an otherwise concise synthetic route, and the deprotection conditions may not be compatible with all combinations of skeletons and building blocks.
Scheme 1.
Conceptual depiction of aziridines as building blocks in diversity-oriented syntheses using the `build/couple/pair' strategy.
To test the usefulness of this aziridine-based plan, compounds 2, 3, and 4 were prepared as outlined in Scheme 2. Starting from p-methoxybenzaldehyde, dibromochalcone 1 was formed via Horner-Wadsworth-Emmons olefination with trimethyl acetophosphonate and subsequent bromination. Although 1 was isolated as a single diastereomer, both the cis and trans isomers of 2 resulted from the addition of propargylamine in DMF15, an observation that has, in previous studies, been taken as evidence that the addition takes place through an α-bromocinnamate formed in situ.16 Aziridines 3 and 4 were prepared in the same way from related building blocks and, in all cases, the cis and trans partners were separable by column chromatography. Regardless of the mechanism of their formation, this method allowed access to both diastereomers of a number of aziridines.
Scheme 2.

Preparation of azridines 2, 3, and 4. Reagents and conditions: (a) (MeO)2POCH2CO2Me, NaH, THF, 0 °C to rt; (b) Br2, CH2Cl2, rt, 78% (2 steps); (c) propargylamine, DMF, rt, 50%, 4:1 cis:trans.
We next explored a number of coupling and ring-opening reactions. We found that when aziridine trans-2 was treated with trimethylsilylazide in refluxing acetonitrile17,18, tetrahydrotriazolopyrazine 5 was isolated in 45% yield (Scheme 3). The reaction is presumed to proceed through a propargylamine azide intermediate that cyclizes spontaneously under thermal conditions to yield the triazole product. The stereochemistry of the substituents on the piperazine ring was determined to be syn by 1H nOe NMR experiments. Interestingly, under the same conditions, aziridine cis-2 yielded a crude reaction mixture that contained the same major product as that for the corresponding trans-aziridine, and, in fact, the same tetrahydrotriazolopiperazine product 5 was isolated in 24% yield following preparative TLC purification. Upon further analysis, both mixtures contained the same minor product, which, although not isolated, had a 1H NMR spectrum consistent with the anti-diastereomer of 5. To test whether this presumed epimerization occurred spontaneously under the reaction conditions, purified triazole 5 was subjected to refluxing acetonitrile for 48 h and analyzed. 1H NMR analysis indicated that no epimerization had taken place. Scrambling of the benzylic stereocenter was observed during Lewis acid-catalyzed nucleophilic addition to electron-rich aziridines (described below); a similar epimerization may have occurred in this case during the ring-opening step.
Scheme 3.
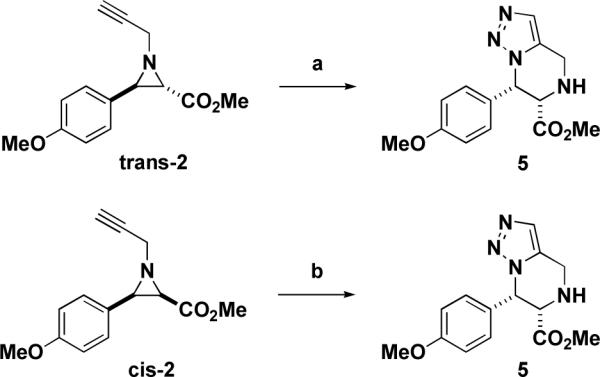
Synthesis of tetrahydrotriazolopyrazine 5. Reagents and conditions: (a) TMSN3, MeCN, reflux, 45%, >10:1 syn:anti; (b) TMSN3, MeCN, reflux, 24%, 3:1 syn:anti.
Next, we turned our attention to the coupling of the aziridines with different alcohol nucleophiles. Following a method reported by Vessiere19, we found that exposure of the unactivated aziridine cis-3 to BF3·OEt2 in methanol quickly formed amine 6 as a single diastereomer (Table 1, entry 1). The stereochemistry of all ring-opened products was inferred from the corresponding cyclized products. These conditions were applied to allyl aziridine 4 cis to form amine 7 (Table 1, entry 2) and also facilitated the ring opening of cis-3 when performed in allyl alcohol to form amine 8. (Table 1, entry 3) These conditions also induced the ring opening of trans aziridines effectively (data not shown).
Table 1.
BF3·OEt2-catalyzed ring-opening experiments.![]()
| entry | substrate | alcohol | product | yield |
|---|---|---|---|---|
| 1 | 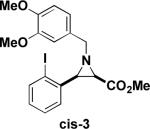 |
MeOH | 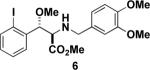 |
82% |
| 2 | 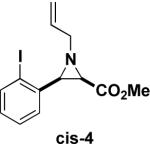 |
MeOH | 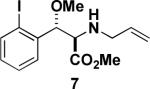 |
67% |
| 3 | 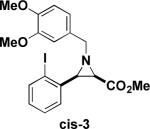 |
allyl alcohol | 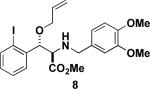 |
65% |
To determine whether these coupled products are amenable to an iodine-induced intramolecular cyclization, ring-opened allyl amine 8 was treated with iodine in THF. A separable 2:1 mixture of morpholine derivatives 9 and 10 was formed in a 30% yield (shown in Scheme 4). The relative stereochemistry of the major product was determined by 1H nOe NMR studies. The same result was achieved when the parent compound 8 was treated with N-iodosuccinimide in methylene chloride. Importantly, many of the products of this pathway contain reactive handles (aryliodide, methyl ester, primary alkyliodide) that can be modified subsequently to increase the building block diversity of products of this synthetic pathway.
Scheme 4.
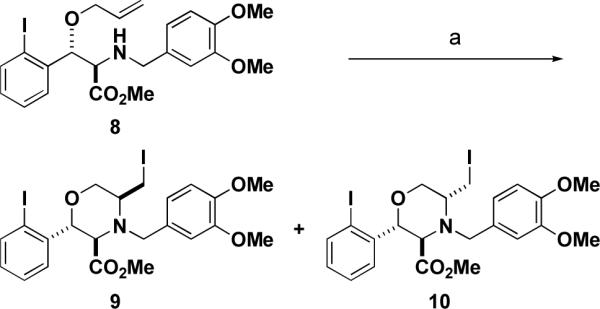
Intramolecular iodoamination to form substituted morpholines. Reagents and conditions: (a) I2, THF, rt, 37% yield, 2:1 9:10.
Aryl iodide-containing aziridines 3 and 4 were synthesized in order to exploit reported intramolecular arylamination methods. Using the ring-opened products 6, 7, and 11 (prepared as described above with trimethylsilylcyanide instead of trimethylsilylazide), copper- and palladium-catalyzed cyclization conditions were explored (Table 2). As illustrated in Table 2, entry 1, the desired dihydroindole 13 was isolated by column chromatography after heating aryl iodide 7 in DMF with a catalytic amount of copper(I) iodide, cesium carbonate, and a ligand reported by Buchwald and coworkers.20 However, the major product of the reaction was the corresponding indole elimination product 12, while the desired product 13 was isolated as the minor component and as a mixture of diastereomers (although the starting material was a single diastereomer). Experiments attempting a palladium-catalyzed cyclization, based on a method published by both Buchwald21 and Fukuyama22 (Table 2, entries 2 and 4), and copper-catalyzed reactions performed at lower temperatures (Table 2, entries 3 and 5) affected no reaction with amine 11, as determined by 1H NMR and/or LC/MS analysis. Experiments with 11 at higher temperature (Table 2, entry 6) led to the consumption of starting material and a complex but promising mixture, as did an experiment with 6 with a vast excess of catalyst (Table 2, entry 7).
Table 2.
Intramolecular arylamination experiments.![]()
| entry | substrate | conditions | result |
|---|---|---|---|
| 1 | 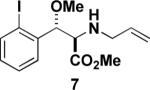 |
1.2 equiv. 2-acetylcyclohexanone, 30 mol% Cul, 2.0 equiv. Cs2CO3, DMF, 90 °C | 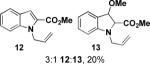 |
| 2 | 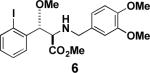 |
20 mol% Pd(PPh3)4, Et3N, reflux | no reaction |
| 3 | 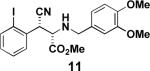 |
1.4 equiv. 2-acetylcyclohexanone, 40 mol% Cul, 2.0 equiv. Cs2CO3, DMF, 50 °C | no reaction |
| 4 | 11 | 5 mol% Pd2(dba)3, 20 mol% P(o-tol)3, 4 equiv KOt-Bu, toluene, rt | no reaction |
| 5 | 11 | 2.0 equiv Cul, 5 equiv Cs2CO3, benzene, rt | no reaction |
| 6 | 11 | 20 mol% 2-acetylcyclohexanone, 5 mol% Cul, 2 equiv. Cs2CO3, DMF, rt → 55 °C → 90 °C | starting material consumed, complex mixture formed |
| 7 | 6 | 4.4 equiv. 2-acetylcyclohexanone, 1.1 equiv Cul, 2.0 equiv Cs2CO3, DMF, 50 °C | starting material consumed, complex mixture formed |
Related intramolecular, metal-catalyzed reactions have been reported to occur in high yield at room temperature.21,22 None of the reported substrates involved amines substituted in the α-position with coordinating groups, however, which led to our suspicion that the amines used in the experiments summarized in Table 2 form a stable five-membered metal chelate that proceeded with the oxidative insertion reaction only at elevated temperature (Figure 1). If so, changing the carbomethoxy substituent to one that cannot form such a chelate would be expected to increase the efficiency of this reaction and would prevent the epimerization of the neighboring stereocenter.
Figure 1.
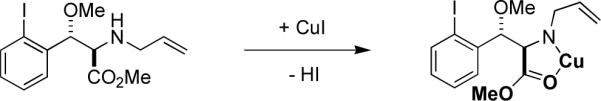
Proposed chelate to explain low reactivity of substrates in metal-mediated coupling reactions.
In summary, here we describe studies into the use of aziridines as components of a nascent build/couple/pair synthetic strategy. Aziridines were prepared with aryl substituents to direct ring opening with trimethylsilylazide and with different alcohols in the presence of BF3·OEt2. These products were substrates for a thermal [3 + 2] cycloaddition, an iodoamination, and an intramolecular arylamination, respectively. The intermediates and products synthesized in these studies will be submitted for inclusion in the Broad Institute screening collection. Although the yields of some of these reactions are modest, this work demonstrates that aziridines can be prepared with substituents that direct subsequent additions and allow for intramolecular cyclizations to form structurally diverse, rigid, and functionalized heterocycles. Incorporation of a variety functional groups and building blocks into this aziridine system should allow for a number of other pairing reactions to afford heterocycles having different ring sizes and properties.
Supplementary Material
Acknowledgement
The NIGMS-sponsored Center of Excellence in Chemical Methodology and Library Design (Broad Institute CMLD; P50 GM069721) funded this research. S.L.S. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Representative experimental details for the synthesis and the characterization data for key intermediates are provided. Supplementary data associated with this article can be found, in the online version, at doi:xxx.
References and notes
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat. Rev. Drug Disc. 2007;6:29. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber SL. Nature. 2009;457:153. doi: 10.1038/457153a. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 4.Burke MD, Schreiber SL. Angew. Chem., Int. Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen TE, Schreiber SL. Angew. Chem., Int. Ed. 2008;47:48. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen IMB. J. Phys. Chem. A. 1998;102:3193. [Google Scholar]
- 7.Park G, Kim S-C, Kang H-Y. Bull. Korean Chem. Soc. 2005;26:1339. [Google Scholar]
- 8.Pineschi M, Bertolini F, Crotti P, Macchia F. Org. Lett. 2006;8:2627. doi: 10.1021/ol060822m. [DOI] [PubMed] [Google Scholar]
- 9.Hu XE. Tetrahedron Lett. 2002;43:5315. [Google Scholar]
- 10.Williams AL, Johnston JN. J. Am. Chem. Soc. 2004;126:1612. doi: 10.1021/ja0385282. [DOI] [PubMed] [Google Scholar]
- 11.Unthank MG, Hussain N, Aggarwal VK. Angew. Chem., Int. Ed. 2006;45:7066. doi: 10.1002/anie.200602782. [DOI] [PubMed] [Google Scholar]
- 12.Evans DA, Faul MM, Bilodeau MT. J. Org. Chem. 1991;56:6744. [Google Scholar]
- 13.Li Z, Conser KR, Jacobsen EN. J. Am. Chem. Soc. 1993;115:5326. [Google Scholar]
- 14.Legters J, Thijs L, Zwanenburg B. Tetrahedron Lett. 1989;30:4881. [Google Scholar]
- 15.Davoli P, Spaggiari A, Ciamaroni E, Forni A, Torre G, Prati F. Heterocyc. 2004;63:2495. [Google Scholar]
- 16.Weber FG, Bandlow C. Z. Chem. 1973;13:467. [Google Scholar]
- 17.Chandrasekhar M, Sekar G, Singh VK. Tetrahedron Lett. 2000;41:10079. [Google Scholar]
- 18.Li Z, Fernandez M, Jacobsen EN. Org. Lett. 1999;1:1611. doi: 10.1021/ol990992h. [DOI] [PubMed] [Google Scholar]
- 19.Bodenan J, Chanet-Ray J, Vessiere R. Synthesis. 1992:288. [Google Scholar]
- 20.Shafir A, Buchwald SL. J. Am. Chem. Soc. 2006;128:8742. doi: 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]
- 21.Peat AJ, Buchwald SL. J. Am. Chem. Soc. 1996;118:1028. [Google Scholar]
- 22.Yamada K, Kubo T, Tokuyama H, Fukuyama T. Synlett. 2002:231. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.