

Abstract
OBJECTIVE
The E23K variant in the Kir6.2 subunit of the ATP-sensitive K+ channel (KATP channel) is associated with increased risk of type 2 diabetes. The present study was undertaken to increase our understanding of the mechanisms responsible. To avoid confounding effects of hyperglycemia, insulin secretion and action were studied in subjects with the variant who had normal glucose tolerance.
RESEARCH DESIGN AND METHODS
Nine subjects with the E23K genotype K/K and nine matched subjects with the E/E genotype underwent 5-h oral glucose tolerance tests (OGTTs), graded glucose infusion, and hyperinsulinemic-euglycemic clamp with stable-isotope–labeled tracer infusions to assess insulin secretion, action, and clearance. A total of 461 volunteers consecutively genotyped for the E23K variant also underwent OGTTs. Functional studies of the wild-type and E23K variant potassium channels were conducted.
RESULTS
Insulin secretory responses to oral and intravenous glucose were reduced by ∼40% in glucose-tolerant subjects homozygous for E23K. Normal glucose tolerance with reduced insulin secretion suggests a change in insulin sensitivity. The hyperinsulinemic-euglycemic clamp revealed that hepatic insulin sensitivity is ∼40% greater in subjects with the E23K variant, and these subjects demonstrate increased insulin sensitivity after oral glucose. The reconstituted E23K channels confirm reduced sensitivity to inhibitory ATP and increase in open probability, a direct molecular explanation for reduced insulin secretion.
CONCLUSIONS
The E23K variant leads to overactivity of the KATP channel, resulting in reduced insulin secretion. Initially, insulin sensitivity is enhanced, thereby maintaining normal glucose tolerance. Presumably, over time, as insulin secretion falls further or insulin resistance develops, glucose levels rise resulting in type 2 diabetes.
The ATP-sensitive K+ channel (KATP channel) plays a central role in glucose-stimulated insulin secretion (GSIS). It is comprised of two subunits, an inward rectifier potassium channel (Kir6.2) and a sulfonylurea receptor (SUR1). An increase in the cytosolic ATP-to-ADP ratio in the pancreatic β-cell inhibits KATP channel activity and stimulates insulin secretion, whereas a lowering of the ATP-to-ADP ratio restores channel activity and suppresses insulin release. Mutations in the genes encoding Kir6.2 (KCNJ11) and SUR1 (ABCC8) are associated with neonatal diabetes mellitus (NDM) and hyperinsulinism (1,2). A common amino acid polymorphism in Kir6.2, Glu23Lys (E23K), is associated with susceptibility to type 2 diabetes (3). However, the mechanistic basis of this association and the phenotypic consequences in vivo remain controversial. An early study (4) observed no effects of the E23K variant on whole-cell K+ currents. Conversely, more detailed studies report overactivity of E23K channels that is variably attributed to a decrease in channel sensitivity to inhibitory ATP (5) or an increase in ATP sensitivity with an enhanced activation by free fatty acids (6). In both cases, net overactivity of the KATP channel is predicted to suppress GSIS.
Although initial studies in humans showed no changes in insulin secretion, subsequent studies (7–9) showed association of the E23K variant with reduced insulin secretion. If these studies are performed on diabetic subjects, reduced insulin secretion may result from the adverse effects of hyperglycemia on β-cell function. The present study was performed in order to provide quantitative indexes of insulin secretion and define dose-response relationships between glucose and insulin secretion rate (ISR) at various glucose concentrations in subjects with the E23K genotype and normal glucose levels. The contribution of alterations in insulin clearance to insulin levels as a function of the E23K genotype was also examined.
It is unclear whether the KATP channel regulates insulin sensitivity. Although an increase in insulin sensitivity is reported in animals with loss of KATP channels (10), similar studies have not been performed with gain of channel activity. A study in human subjects (11) suggested that the E23K genotype underlies an increase in insulin sensitivity, whereas insulin sensitivity was not altered in other studies (8,12–14). The present study was undertaken to address these gaps in our understanding of the physiological mechanisms underlying susceptibility to type 2 diabetes in subjects with the E23K variant. Since hyperglycemia can cause defects in insulin secretion, we focused on subjects with normal glucose tolerance. This allowed us to define the changes in insulin secretion and action that antedate diabetes onset and are due to the effects of the E23K variant per se. To resolve questions regarding the underlying mechanisms, we performed comprehensive assessments of the effect of E23K on KATP channel activity.
RESEARCH DESIGN AND METHODS
Nondiabetic subjects, aged <65 years in good health and with stable weight for 6 months, were recruited using advertisements. The studies were approved by the Human Research Protective Office.
Study 1 (intensive metabolic studies).
Nine subjects with the K/K genotype and nine age-, sex-, and BMI-matched subjects with the E/E genotype participated in three separate protocols designed to test insulin secretion, action, and clearance. All subjects were unrelated and had no family history of type 2 diabetes. Insulin secretion was assessed using the 5-h oral glucose tolerance test (OGTT) and graded glucose infusion (GGI). Insulin sensitivity was assessed during the hyperinsulinemic-euglycemic clamp and OGTT. Insulin clearance was assessed during OGTT and GGI.
Study 2 (cross-sectional study).
A total of 461 volunteers who responded to advertisements were genotyped for the E23K variant and underwent 5-h OGTTs for assessments of insulin secretion, action, and clearance.
Study protocols.
Subjects adhered to their regular diet and refrained from exercise for 3 days before the studies.
5-h OGTT.
Participants ingested a 75-g glucose load. Blood samples were collected at −15, 0, 10, 20, 30, 60, 90, 120, 150, 180, 240, and 300 min after ingestion to determine plasma glucose, insulin, and C-peptide concentrations. GGI involved the intravenous administration of glucose at increasing rates (1, 2, 3, 4, 6, and 8 mg · kg−1 · min−1) for 40 min each (15,16). This protocol raises the plasma glucose concentration from basal to hyperglycemic levels and defines the dose-response relationships between glucose and insulin secretion.
Hyperinsulinemic-euglycemic clamp with stable-isotope–labeled tracer infusion.
Studies (17,18) were performed after an overnight fast as described. A primed (22.5 μmol/kg)-constant (0.25 μmol · min−1 · kg−1) infusion of [6,6-2H2]glucose was started at 0700 h. After 3.5 h of tracer infusion, a hyperinsulinemic-euglycemic clamp was started and continued for 3.5 h. Insulin was infused at 40 mU/m2 per min. Dextrose (20%) with [6,6-2H2]glucose (∼2.5%) was infused to maintain plasma glucose concentration at ∼5.6 mmol/l. The infusion of [2H2]glucose was decreased by 75% of basal during the clamp for the expected decline in hepatic glucose production. Body composition was assessed using the same dual-energy X-ray absorptiometry instrument in all subjects (Delphi 4500-W; Hologic, Waltham, MA).
Analyses.
The areas under the curve (AUCs) were calculated using the trapezoid method (19). ISR was calculated using parameters for C-peptide kinetics and volume of distribution (20). Static, dynamic, and overall responsivity indexes were calculated as reported (21). The GGI allowed the dose-response relationship between ISR and glucose over the physiological range to be determined. ISRs and glucose concentrations represented the average of the values between 10 and 40 min at each infusion rate. Mean ISR for each glucose infusion rate was plotted against the mean glucose concentration. The slope of the line relating these two variables provided a measure of the sensitivity of the β-cell to glucose. Peripheral insulin sensitivity was calculated from the hyperinsulinemic-euglycemic clamp (17,18,22). Basal endogenous glucose rate of appearance was calculated using Steele's equations (20). Hepatic insulin sensitivity was assessed as the inverse of the product of the basal endogenous glucose production rate (μmol · kg fat-free mass−1 · min−1) and the fasting insulin concentration (pmol/l) × 1,000 (23,24). Whole-body insulin sensitivity was estimated using the oral glucose minimal model (25,26). Composite insulin sensitivity index was calculated using the method of Matsuda (23). Insulin clearance rate was calculated by dividing the 1) ISR AUC by the insulin AUC and the 2) C-peptide AUC by the insulin AUC (15,27).
Biochemical measurements.
Plasma glucose was measured using a glucose analyzer (Yellow Springs Instruments, Yellow Springs, OH). Plasma insulin and C-peptide were measured using radioimmunoassays (Linco Research, St.Louis, MO).
Genotyping.
DNA was prepared from peripheral blood lymphocytes and the KCNJ11 E23K polymorphism (dbSNP rs5219) typed using a TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA).
Statistical analyses.
Group differences were compared using the Student's t test for unpaired samples for continuous variables and the Fisher's exact test for categorical variables. Where appropriate, we used ANOVA and Tukey's test for post hoc analyses for continuous variables and χ2 test for categorical variables. ANCOVA were used to adjust for age, race, and BMI. SPSS version 15.0 (SPSS, Chicago, IL) was used for all analyses. A P value <0.05 was considered to be statistically significant. Results are reported as means ± SE.
In vitro studies of the E23K variant.
Recombinant KATP channel Kir6.2 (mouse) + SUR1 (hamster) was transiently expressed in COSm6 cells as described (28). Of the missense variants reported in humans, the Kir6.2 clone contained no variants, while the SUR1 clone contained the A1369S substitution. Patch-clamp experiments were performed as described (28). Bath and pipette solutions (K-INT) contained (in mmol/l): 150 KCl, 10 HEPES, and 1 EGTA (pH 7.4). For experiments with MgATP, the free Mg2+ concentration was kept at 2 mmol/l. The ATP dose response was quantified by fitting the raw data with a Hill equation:
where Irel is the current relative to that in the absence of ATP, {ATP} is the ATP concentration, K1/2,ATP is the half-maximal inhibitory ATP concentration, and H is the Hill coefficient, which was allowed to vary. Dose-response curves for tolbutamide inhibition were described by the product of two Hill components (28), where A represents the high-affinity and B the low-affinity components:
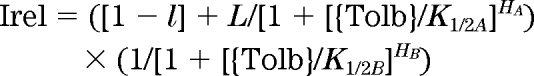 |
where {Tolb} is the tolbutamide concentration, K1/2A and K1/2B are the half-maximal inhibitory tolbutamide concentrations, HA and HB are the Hill coefficients, and l is the fraction of current blocked with high affinity.
86Rb+ efflux experiments were performed as described (28). Transfected cells were preincubated with or without metabolic inhibitors (2.5 μg/ml oligomycin plus 1 mmol/l 2-deoxy-d-glucose). At selected time points, the solution was aspirated and replaced with fresh solution and the aspirated solution was counted in a scintillation counter. The rate constant of ATP-sensitive K+–specific (86Rb+) efflux (k2) was obtained by fitting the data with a single exponential equation:
where the rate constant for nonspecific efflux (k1) was obtained from untransfected cells.
Two approaches were used to estimate Po,zero, the initial open probability (in zero ATP), of membrane patches containing multiple channels. (PIP2; method I) PIP2 was added to the patch until the current reached a saturating level (IPIP2). This was assumed to represent a maximum Po,zero of ∼0.97 (29). The fold increase in current was calculated (fold increase = IPIP2/Iinitial) and the Po,zero was estimated from the following equation:
(NA; method II) Mean Po,zero was estimated from stationary fluctuation analysis of macroscopic currents (30,31). Mean patch current (I) and variance (α2) in the absence of ATP were obtained by subtraction of the mean current and variance in 5 mmol/l ATP (i.e., all channels closed), respectively. Single-channel current (i) was assumed to be −3.75 pA, corresponding to single-channel conductance of 75 pS. Po,zero was estimated from the following equation:
Channel density was calculated as I(Po,max × i) from maximum patch current in zero ATP (I) and assuming a single-channel current (i) = 3.75 pA at −50 mV.
RESULTS
Insulin secretion in subjects with E23K variant.
We initially examined insulin secretion in subjects homozygous for the E23K variant, since they might exhibit the greatest difference if E23K genotype affected insulin secretion. The two groups were well matched for age, sex, and BMI (Table 1). Both groups had normal glucose concentrations in the fasting state and following oral and intravenous glucose administration (Table 1) (Fig. 1). The respective AUCs for glucose between E/E and K/K subjects during the OGTT (17.6 ± 0.1 vs. 18.7 ± 0.1 × 104 min · mmol−1 · l−1) and GGI (17.6 ± 0.1 vs. 17.6 ± 0.1 × 104 min · mmol−1 · l−1) were not different (Fig. 1). By contrast, the AUC for insulin in the OGTT (52.6 ± 5.7 vs. 71.0 ± 7.0 × 103 min · pmol−1 · l−1) and GGI (24.5 ± 2.1 vs. 36.1 ± 4.8 × 103 min · pmol−1 · l−1) was ∼40% lower (P < 0.05) in the K/K group. The AUC for C-peptide was also reduced (P < 0.05) in the K/K subjects in both the OGTT (30.2 ± 2.7 vs. 58.9 ± 15.1 × 104 min · pmol−1 · l−1) and GGI (26.4 ± 2.1 vs. 36.9 ± 3.3 × 104 min · pmol−1 · l−1). Static, dynamic, and overall β-cell response to glucose were also ∼40% lower (P < 0.05) in the K/K group (Fig. 2A) based on the OGTT and GGI. In addition, the dose-response curve relating glucose and ISR during the GGI was shifted downward and to the right in the K/K group (Fig. 2B). Accordingly, the ISR AUC (17.2 ± 1.2 vs. 26.1 ± 0.5 × 103 pmol/l) and mean ISR (70.6 ± 4.9 vs. 106.7 ± 9.9 pmol · l−1 · min−1) were lower (P < 0.05) in the K/K group. No significant differences were observed in insulin clearance rates in this study (P > 0.05).
TABLE 1.
Characteristics of study participants (study 1)
| E/E | K/K | P value | |
|---|---|---|---|
| n | 9 | 9 | |
| Women (%) | 6 (67) | 6 (67) | 1.0 |
| Caucasian (%) | 9 (100) | 9 (100) | 1.0 |
| Age (years) | 45.9 ± 2.8 | 46.6 ± 3.9 | 1.0 |
| Weight (kg) | 75.5 ± 5.7 | 72.9 ± 4.2 | 0.73 |
| Height (cm) | 166.1 ± 2.5 | 164.2 ± 3.9 | 0.69 |
| BMI (kg/m2) | 27.8 ± 1.3 | 26.4 ± 1.6 | 0.53 |
| Fat mass (kg) | 31.1 ± 6.1 | 22.0 ± 3.0 | 0.20 |
| Fat-free mass (kg) | 43.7 ± 3.6 | 49.2 ± 3.7 | 0.30 |
| Truncal fat (kg) | 13.4 ± 1.8 | 10.0 ± 1.7 | 0.20 |
| Fasting glucose (mmol/l) | 5.1 ± 0.2 | 5.2 ± 0.1 | 0.73 |
| 2-h glucose (mmol/l) | 6.9 ± 0.3 | 6.8 ± 0.3 | 0.62 |
| A1C (%) | 5.6 ± 0.1 | 5.5 ± 0.2 | 0.72 |
Data are means ± SE.
FIG. 1.

Plasma glucose insulin and C-peptide concentrations during the oral glucose tolerance test (A) and intravenous GGI (B) in the K/K (○) and E/E (●) groups. P values indicate the significance of the differences between AUC values between groups. Values are means ± SE.
FIG. 2.

A: Insulin secretory indexes during the OGTT and intravenous GGI in the K/K (□) and E/E groups (■). *P < 0.05 for the differences between groups. B: Insulin secretory rate glucose dose-response curve as constructed from the intravenous GGI in the K/K (○) and E/E groups (●). P value indicates the significance of the differences between AUC values between groups. Values are means ± SE.
Alterations in ATP sensitivity of reconstituted E23K channels in vitro.
To examine the molecular basis for the differences in insulin secretion, we transiently expressed Kir6.2 with residue 23, being either glutamate (E23) or lysine (K23), together with the SUR1 subunit and measured ATP sensitivity in excised membrane patches (Fig. 3A). Homomeric K23 channels (K/K) exhibit a modest, yet significant, decrease in ATP inhibition compared with homomeric E23 (E/E) channels (K1/2,ATP = 16 μmol/l [n = 28 patches] and 7.5 μmol/l [n = 24 patches], respectively). A similar relative shift in ATP sensitivities was observed in the presence of physiological (2 mmol/l) Mg2+ for E/E and K/K channels (K1/2,ATP = 19 μmol/l [n = 19 patches] and 26 μmol/l [n = 17 patches], respectively). In contrast, the averaged KATP channel density was not different between cells expressing E/E or K/K channels (201 ± 59 and 128 ± 50 channels/patch, respectively; n = 11–19 patches). To recapitulate the heterozygous E23K genotype (E/K), cells were transfected with a 1:1 mixture of E/E and K/K cDNAs. Since four subunits generate the channels, five different ratios of subunits will be present in the resultant channels (1 of 16 channels will be homozygous E/E and homozygous K/K). The ensemble of expressed channels display intermediate ATP sensitivity (K1/2,ATP = 10.0 μmol/l [n = 10 patches] compared with homomeric K/K and E/E channels) (Fig. 3B). Our data are similar to those reported by Schwanstecher et al. (5) for recombinant K/K channels. For comparison, the dose-response curves are shown in Fig. 3B for two Kir6.2 mutations that impair insulin release and underlie NDM. The I182V mutation underlies a transient NDM, whereas the I296L mutation underlies a syndromic form of NDM (32,33). Importantly, the ATP sensitivities of mutant channels correlate with the severity of the disease (K1/2,ATP = 39 μmol/l for homomeric I182V [n = 17 patches] and 771 μmol/l for homomeric I296L channels [n = 5 patches]).
FIG. 3.

Reduced ATP and sulfonyurea sensitivity of mutant E23K channels. A: Representative currents (at −50 mV) from inside-out membrane patches from COS cells expressing KATP channel (Kir6.2 + SUR1): homomeric E23 channels (E/E), K23 channels (K/K), or heteromeric E23 and K23 channels (E/K). Patches were exposed to differing [ATP], and baseline current was determined by exposure to ATP (5 mmol/l). B: Steady-state dependence of membrane current on [ATP] (relative to current in zero ATP [Irel]) for E23- and K23-containing channels. K1/2 ATP = 7.5 μmol/l (E/E) and 16 μmol/l (K/K). Data points represent means ± SE (n = 24–28 patches). The fitted lines correspond to least-squares fits of a Hill equation (see research design and methods). **P < 0.01 vs. E/E channels by unpaired Student's t test (two tailed assuming equal variance). C: Representative currents recorded from inside-out membrane patches containing homomeric E/E or mutant K/K channels at −50 mV and in response to varying [tolbutamide]. Zero-channel current was determined by application of ATP (5 mmol/l). D: Steady-state dependence of current on [tolbutamide] (relative to current in zero tolbutamide [Irel]) for E/E (○) and K/K (●) variant channels (from records such as those shown in C). Data points represent the means ± SE (n = 6–19 patches). For all channels, the lines are fits of the product of two Hill components, each of the form (Irel = 1/([1 + {[Tolb]/K1/2}H]), with H fixed at 1.3 in each case (see research design and methods). The relative fraction and K1/2 values of each component were varied. The high-affinity component was 53 and 44% for wild-type and K/K channels, respectively. *P < 0.05 vs. wild-type KATP channel by unpaired Student's t test. The shaded box shows the reported range of serum tolbutamide concentrations from a cohort of 37 type 2 diabetic subjects receiving sulfonylurea therapy (49).
Increased cellular activity of reconstituted E23K channels.
KATP channel activity in metabolically intact cells was screened by 86Rb+-efflux from transfected COSm6 cells. Efflux from cells transfected with homomeric E/E channels was low under basal conditions and was activated by metabolic inhibition to lower cellular [ATP]/[ADP] (Fig. 4A). Rb+-efflux from cells transfected with recombinant K/K channels was also low under basal conditions and increased with metabolic inhibition. Quantitative estimation of KATP channel conductance (see research design and methods) indicates that for K/K channels, basal conductance was a higher fraction of fully activated conductance than E/E channels (Fig. 4B). In the β-cell, the increased basal flux is expected to impair glucose sensing and account for the association of K/K genotype with reduced insulin secretion.
FIG. 4.

Increased basal activity in intact cells expressing K/K variant channels. A: Representative efflux of 86Rb+ as a function of time in basal conditions or in the presence of metabolic inhibition for reconstituted E/E and variant K/K channels and untransfected controls. C: Ratio of KATP channel–dependent efflux rate constant (k2) in basal conditions relative to metabolic inhibition (MI) for E/E or homomeric K/K channels (see research design and methods). Graphs show compiled data (means ± SE) from six experiments in which each transfection was done in triplicates. *P < 0.05 vs. E/E channels by paired one-tailed Student's t test.
E23K variant decreases ATP sensitivity by stabilizing the open state of the channel.
Kir6.2 mutations can reduce ATP sensitivity by directly reducing ATP binding to the Kir6.2 subunit or indirectly by affecting the intrinsic opening ability (28,29,34). In the latter case, an increase in the open probability (Po,zero) decreases the frequency with which the channel enters the ATP-accessible closed state, resulting in a decrease in ATP sensitivity. We have modeled this nonlinear relationship between Po,zero and K1/2,ATP (29), and this kinetic model describes the diabetes-causing effects of Kir6.2 mutations that underlie NDM (e.g., Q52R, I296L) (Fig. 5C). To estimate the open probability of KATP channels, both nonstationary noise analysis (NA) and phosphatidylinositol biphosphate (PIP2) application were used to independently examine KATP channel gating in isolated membrane patches. As shown in Fig. 5A, the estimated open probability (Po,zero) of K/K channels in the absence of ATP (0.68 ± 0.04 [NA method]; 0.67 ± 0.08 [PIP2 method]) is higher than that of E/E channels (0.49 ± 0.05 [NA method]; 0.46 ± 0.06 [PIP2 method], P < 0.05), and this increase can fully account for the shifted ATP sensitivity (K1/2,ATP = 9.4 ± 1.6 μmol/l [E/E] and 21.6 ± 3.3 μmol/l [K/K] from curve fit of individual membrane patches, P < 0.01) (Fig. 5B). A similar increase in open probability was reported for K/K channels by Schwanstecher et al. (5), using analysis of single-channel records. That multiple methodologies reiterate the same findings strengthens the conclusion that the changes associated with the E23K variant are significant and real. As with more severe NDM, the E23K variant indirectly affects ATP sensitivity by increasing the Po,zero (Fig. 5C). The predicted consequence will be reduced excitability of the β-cell, with increasingly severe consequences for insulin secretion (E23K < Q52R < I296L).
FIG. 5.

An increase in maximum open probability underlies the reduced ATP-sensitivity of K/K variant channels. A: Open probability in zero ATP (Po zero) calculated using the PIP2 method and noise analysis (NA) (see research design and methods) for membrane patches expressing either homomeric E/E or K/K channels. Data points represent means ± SE (n = 21–44 patches for NA method; n = 6–8 patches for PIP2 method). *P < 0.05 and **P < 0.01 by two-tailed Student's t test assuming equal variance. B: Relationship between Po zero (calculated using the NA method) and ATP sensitivity (K1/2 ATP) for individual membrane patches expressing E/E or K/K channels together with averaged data (triangles). For E/E: K1/2 ATP = 9.4 ± 1.6 μmol/l, Po zero = 0.49 ± 0.05 (n = 18 patches). For K/K: K1/2 ATP = 21.6 ± 3.3 μmol/l, Po zero = 0.0.68 ± 0.04 (n = 18 patches). **P < 0.01 for both K1/2 ATP and Po zero values of E/E compared with K/K channels (unpaired Student's t test). Data points represent means ± SE. C: Relationship between Po zero and K1/2 ATP (mmol/l). Solid line represents prediction of kinetic model II (inset) of Enkvetchakul et al. (29). Symbols represent data points as in B together with mean values for Q52R and I296L channels. The key feature of the model is that ATP acts by binding to a closed state and in consequence ATP sensitivity is reduced by shifting the equilibrium between the Cin and O states toward the open state (increasing KCO).
Sulfonylurea sensitivity is reduced in E23K channels.
Mutations in Kir6.2 that allosterically decrease ATP sensitivity by stabilizing the open state of the channel also reduce high-affinity block by sulfonylureas, a feature of NDM-associated mutations (28). We next examined the effect of the first-generation sulfonyurea, tolbutamide, on channel activity (Fig. 3C). As shown in Fig. 3D, homomeric E/E channels exhibit a typical biphasic block by tolbutamide, with both a high-affinity (IC50 = 1.1 μmol/l) and a low-affinity (IC50 = 2 mmol/l) site. The fractional block by the high-affinity therapeutically relevant component is ∼53%. For the K/K channels, the value for the high-affinity block (IC50 = 1.0 μmol/l) is unaltered; however, the fractional block is significantly decreased from 53 to 44%.
Insulin sensitivity in subjects with the E23K variant.
The clinical findings described above (of reduced insulin secretion with normal glucose concentrations) suggest a simultaneous change in insulin sensitivity. Hyperinsulinemic-euglycemic clamp experiments revealed that hepatic insulin sensitivity was significantly greater (2.4 ± 0.4 vs. 1.5 ± 0.2 [1,000/μmol · kg fat-free mass−1 · min−1 · pmol−1 · l−1]; P < 0.05) in subjects with the K/K genotype (Fig. 6A). There was also a strong trend (0.19 ± 0.03 vs. 0.14 ± 0.2 μmol · kg fat-free mass−1 · min−1 · pmol−1 · l−1; P < 0.10) for an increase in peripheral insulin sensitivity, although the differences were not statistically significant. Similar trends were observed for whole-body insulin sensitivity as assessed from the OGTT (Fig. 6B). Glucose infusion rate, plasma glucose, insulin, glucagon, and tracer-to-tracee ratio are presented in the online appendix (available at http://diabetes.diabetesjournals.org/cgi/content/full/db09-0025/DC1).
FIG. 6.

Hepatic insulin sensitivity and peripheral insulin sensitivity from the hyperinsulinenimic-euglycemic clamp with stable-isotope–tracer infusion (A) and whole-body insulin sensitivity from the OGTT (B) in the K/K (□) and E/E (■) groups. Values are means ± SE. *P < 0.05 for the differences between groups.
Confirmation of changes of insulin secretion and action.
The human studies described above were conducted in a small group of E/E or K/K subjects. To confirm the findings on insulin secretion and action in a larger cohort, we carried out 5-h OGTTs in 461 additional subjects (Table 2). Subjects were divided into three groups based on E23K genotype. The three groups had similar glucose concentrations during fasting and 2-h post-glucose challenge. The relative frequencies of E/K (40%) and K/K (13%) in our sample are comparable with those reported (7,8,35), conferring a relative diabetes risk of 1.15–1.65 (3). Consistent with the above findings, the K/K subjects had reduced insulin and C-peptide concentrations, reduced insulin secretory responses, reduced β-cell responsiveness to glucose, and enhanced insulin sensitivity and clearance rates. Interestingly, β-cell responsiveness was also lower in the E/K variant than the E/E.
TABLE 2.
Characteristics of study participants (study 2)
| Kir6.2 genotype |
||||
|---|---|---|---|---|
| E/E | E/K | K/K | P value | |
| n | 216 | 184 | 61 | |
| Women (%) | 158 (73) | 120 (65) | 45 (74) | 0.40 |
| Caucasian (%) | 127 (59) | 155 (84) | 55 (90) | 0.001 |
| Age (years) | 38.0 ± 0.9 | 38.2 ± 1.1 | 36.6 ± 12.4 | 0.74 |
| BMI (kg/m2) | 29.5 ± 0.5 | 27.5 ± 0.5* | 27.1 ± 0.9* | 0.001 |
| Fat mass (kg) | 33.5 ± 0.7 | 31.9 ± 0.7 | 31.8 ± 1.3 | 0.23 |
| Truncal fat (kg) | 13.2 ± 0.5 | 12.2 ± 0.4 | 11.4 ± 0.9 | 0.16 |
| A1C (%) | 5.6 ± 0.0 | 5.5 ± 0.0* | 5.5 ± 0.1* | 0.002 |
| OGTT | ||||
| Fasting glucose (mmol/l) | 5.3 ± 0.1 | 5.1 ± 0.0 | 5.2 ± 0.1 | 0.28 |
| 2-h glucose (mmol/l) | 7.8 ± 0.1 | 7.6 ± 0.1 | 7.4 ± 0.2 | 0.43 |
| Glucose AUC × 104 (min · mmol−1 · l−1) | 19.6 ± 0.2 | 19.1 ± 0.3 | 18.8 ± 0.4 | 0.34 |
| Insulin AUC × 103 (min · pmol−1 · l−1) | 90.2 ± 4.2 | 73.6 ± 3.5 | 62.5 ± 4.9* | <0.001 |
| C-peptide AUC × 104 (min · pmol−1 · l−1) | 51.6 ± 1.4 | 48.9 ± 1.3 | 43.4 ± 2.2* | 0.010 |
| Insulin sensitivity (SI) | ||||
| SIMM × 10−4 (dl · kg−1 · min−1 · pmol−1 · l−1)† | 1.7 ± 0.1 | 1.8 ± 0.7 | 2.6 ± 0.2† | <0.001 |
| SIComposite‡ | 5.1 ± 0.2 | 5.8 ± 0.3 | 6.7 ± 0.5* | 0.007 |
| Insulin secretion | ||||
| ISR AUC × 103 | 30.0 ± 0.9 | 28.1 ± 0.8 | 25.4 ± 1.2* | 0.01 |
| β-Cell responsivity (Φo) (109/min) | 13.7 ± 0.84 | 13.3 ± 0.4* | 11.5 ± 0.7* | 0.02 |
| Insulin clearance (IC) | ||||
| IC1 (ISR AUC/insulin AUC) | 2.8 ± 0.1 | 3.1 ± 0.1* | 3.2 ± 0.1* | 0.001 |
| IC2 (C-peptide AUC/insulin AUC) | 47.8 ± 1.1 | 53.3 ± 1.4* | 55.9 ± 2.3* | 0.001 |
DISCUSSION
Association of Kir6.2 E23K variant with impaired insulin secretion.
Type 2 diabetes is a multifactorial disease in which genetic and environmental factors interact to determine the level of predisposition, and, recently, genome-wide association studies have identified common polymorphisms in various genes that are associated with increased diabetes susceptibility (2,36–39). These studies often involve the comparison of genetic and phenotypic characteristics of diabetic and control subjects. They have made important contributions to identifying novel genetic loci that determine diabetes risk. However, studies of subjects with diabetes do not provide information on the effects of the respective polymorphisms prior to the onset of glucose intolerance and diabetes. Since hyperglycemia, per se, adversely affects insulin secretion, it is difficult to differentiate between the effects of hyperglycemia and the effects of the polymorphism in studies of established diabetes.
The present study was focused on subjects without a prior history of diabetes who had normal glucose tolerance. Our results demonstrate that the Kir6.2 E23K variant is associated with multiple insulin secretory defects, including a reduction in overall β-cell responsiveness to glucose and a rightward shift in the dose-response curve between glucose and insulin secretion. Overall insulin secretory responses to glucose were reduced by ∼40%, and the extent of the reduction was similar to both oral and intravenous glucose.
Association of E23K variant with enhanced insulin sensitivity.
We did not anticipate that severe reductions in insulin secretion would be compatible with normal glucose tolerance. The significant increase in insulin sensitivity is presumably the mechanism that makes this possible, and the results of the hyperinsulinemic-euglycemic clamp suggest that the major effects are in the liver. This is the first study to report the coexistence of decreased insulin secretion with increased insulin action in subjects with the E23K polymorphism. Previous studies that evaluated insulin action yielded inconsistent results. One study (11) suggested an increase in insulin sensitivity, while others (8,12,13) found no effect. Moreover, findings from the hyperinsulinemic clamp suggested an increase in peripheral (skeletal muscle) insulin sensitivity, although this did not reach statistical significance (P < 0.10) because of the small sample size. This interpretation is supported by the results obtained in the larger cohort study in which statistically significant increases in insulin sensitivity were observed after oral glucose ingestion. Although BMI was different between groups, measurements of total and regional fat (truncal fat) showed no differences. Nevertheless, because of the differences in BMI, we controlled for BMI using ANCOVA. The differences in insulin sensitivity remained significant (P < 0.001), despite similar and normal glucose tolerance.
The mechanistic basis of this change in insulin sensitivity is not clear. Kir6.2 is expressed in multiple tissues, including skeletal muscle, brain, and heart. The increase in insulin sensitivity could be due to direct effects of the altered KATP channel activity in one or more of these tissues, and a link between the KATP channel in the hypothalamus and the liver has recently been established (40). An increase in activation of KATP channels in the hypothalamus decreases hepatic gluconeogenesis, and this central effect could serve to counterbalance peripheral actions to maintain glucose homeostasis (40,41). It is interesting to note that basal and insulin-stimulated muscle glucose transport is increased in Kir6.2-null mice, which suggests a role for this channel in the regulation of muscle glucose metabolism (10).
Whether the enhanced insulin sensitivity results from direct effects of KATP channel activity in muscle or is secondary to effects on insulin secretion is unclear. There is precedent for an increase in insulin sensitivity secondary to reduction in insulin secretion. In the evolution of type 1 diabetes, insulin secretion is reduced before glucose levels rise (42), and increased insulin sensitivity has been demonstrated in normoglycemic carriers of HNF1α mutations with reduced insulin secretion prior to diabetes onset (43). These observations, coupled with those of the present study, suggest that in the evolution of type 2 diabetes, increased insulin sensitivity may compensate for a reduction in insulin secretion, resulting in normal glucose tolerance. Over time, glucose intolerance develops due to progression in the severity of the secretion defect and/or exposure to factors that reduce insulin sensitivity. However, we also cannot completely exclude the likelihood that these E23K variants have not developed diabetes because of their increased insulin sensitivity. In such cases, it might reflect a feature of nondiabetic E23K variants rather than of subjects with E23K variants, per se. Although Kir6.2 levels are high in pancreatic α-cells, we did not find differences in glucagon secretion. An additional new finding is the association of E23K variant with an increase in insulin clearance, as demonstrated in study 2. An increase in insulin clearance may contribute to the reduction in peripheral insulin concentrations that is due largely to the decrease in insulin secretion. The liver is the major site of insulin clearance under physiologic circumstances, and this change in insulin clearance is likely due to a change in hepatic insulin metabolism (44). A link between hepatic insulin receptor binding and action and degradation by the liver has been reported (45).
Molecular basis of the E23K phenotype.
In heterologous expression studies, reconstituted E23K channels exhibit a mild, yet significant, decrease in ATP sensitivity and a relative increase in basal activity in the intact cell. In the β-cell, decreased ATP inhibition and consequent channel overactivity is predicted to suppress glucose sensing. In contrast to activating mutations in Kir6.2 that underlie NDM (28), the E23K variant has a less radical effect on channel activity and is associated with type 2 diabetes.
The molecular consequence of the E23K variant has been controversial. Given the high KATP channel density in β-cells, it is predicted that a change in KATP channel activity (<1%) could significantly affect insulin secretion, and, therefore, subtle effects of E23K on channel activity could be physiologically relevant. In support of the conclusion that E23K does not alter channel activity, whole-cell K+-currents were similar in Xenpous oocytes expressing E/E or K/K channels (4). A subsequent study (6) reported an increase in ATP sensitivity of K/K channels, relative to E/E, but a subsequent decrease in ATP inhibition upon application of long-chain acyl-CoAs. The most detailed study was carried out by Schwanstecher and colleagues (5,46). Our data parallel their initial findings of an approximately twofold reduction in ATP sensitivity and increase in open probability and reduced sulfonylurea sensitivity (46) associated with the E23K variant. The similarity of our findings, in a completely independent study, utilizing different methodologies to assess open probability, strongly supports the conclusion that the E23K variant decreases ATP inhibition of the KATP channel.
Mechanistically, the observed increase in open probability can account for both the reduced insulin secretion and decrease in sulfonylurea inhibition of E23K channels. Whether the E23K variant affects sulfonylurea dosing is unknown, but it is notable that the E23K variant is associated with risk for secondary failure to sulfonylureas in type 2 diabetic patients (47,48), and human islets isolated from E23K donors (E/K and K/K) exhibit a decrease in sulfonylurea-induced insulin secretion (47).
Conclusions.
Subjects with the Kir6.2 E23K variant have multiple insulin secretory defects to glucose and decreased responsiveness of the β-cell over a physiologic range of glucose concentrations, and these can be explained by the molecular properties of the E23K channels. These defects in insulin secretion are accompanied by an increase in insulin sensitivity, possibly a compensatory response to reduced insulin secretion. The β-cell secretory defects are present prior to diabetes onset and are likely responsible for the increased risk of type 2 diabetes in subjects with the at-risk E23K genotypes, if superimposed on lifestyle factors causing insulin resistance.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health (NIH) Grants DK031842 (to K.S.P.) and DK069445 (to C.G.N.), the Washington University Clinical Nutrition Research Unit and Clinical and Translational Science Award (NIH Grants DK56341 and UL1RR0224992, respectively), the Washington University and University of Chicago Diabetes Research and Training Centers (NIH Grants DK20579 and DK20595, respectively), and a gift from the Kovler Family Foundation. No potential conflicts of interest relevant to this article were reported.
We thank the participants for their cooperation and the staff of the intensive research unit of the Institute of Clinical and Translational Sciences for their skilled assistance in the performance of this study. We thank Veronica Paz for her assistance with the genotyping.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Flanagan SE, Clauin S, Bellanne-Chantelot C, de LP, Harries LW, Gloyn AL, Ellard S: Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat 2008 [DOI] [PubMed] [Google Scholar]
- 2.Perry JR, Frayling TM: New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care 2008; 11: 371– 377 [DOI] [PubMed] [Google Scholar]
- 3.Riedel MJ, Steckley DC, Light PE: Current status of the E23K Kir6.2 polymorphism: implications for type-2 diabetes. Hum Genet 2005; 116: 133– 145 [DOI] [PubMed] [Google Scholar]
- 4.Sakura H, Wat N, Horton V, Millns H, Turner RC, Ashcroft FM: Sequence variations in the human Kir6.2 gene, a subunit of the beta-cell ATP-sensitive K-channel: no association with NIDDM in while Caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia 1996; 39: 1233– 1236 [DOI] [PubMed] [Google Scholar]
- 5.Schwanstecher C, Meyer U, Schwanstecher M: K(IR)6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta-cell ATP-sensitive K(+) channels. Diabetes 2002; 51: 875– 879 [DOI] [PubMed] [Google Scholar]
- 6.Riedel MJ, Boora P, Steckley D, de VG, Light PE: Kir6.2 polymorphisms sensitize beta-cell ATP-sensitive potassium channels to activation by acyl CoAs: a possible cellular mechanism for increased susceptibility to type 2 diabetes? Diabetes 2003; 52: 2630– 2635 [DOI] [PubMed] [Google Scholar]
- 7.Nielsen EM, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glumer C, Thorsteinsson B, Borch-Johnsen K, Hansen T, Pedersen O: The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes 2003; 52: 573– 577 [DOI] [PubMed] [Google Scholar]
- 8.Florez JC, Burtt N, de Bakker PI, Almgren P, Tuomi T, Holmkvist J, Gaudet D, Hudson TJ, Schaffner SF, Daly MJ, Hirschhorn JN, Groop L, Altshuler D: Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes 2004; 53: 1360– 1368 [DOI] [PubMed] [Google Scholar]
- 9.Chistiakov DA, Potapov VA, Khodirev DC, Shamkhalova MS, Shestakova MV, Nosikov VV: Genetic variations in the pancreatic ATP-sensitive potassium channel, beta-cell dysfunction, and susceptibility to type 2 diabetes. Acta Diabetol 2008 [DOI] [PubMed] [Google Scholar]
- 10.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S: Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A 1998; 95: 10402– 10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen L, Echwald SM, Hansen T, Urhammer SA, Clausen JO, Pedersen O: Amino acid polymorphisms in the ATP-regulatable inward rectifier Kir6.2 and their relationships to glucose- and tolbutamide-induced insulin secretion, the insulin sensitivity index, and NIDDM. Diabetes 1997; 46: 508– 512 [DOI] [PubMed] [Google Scholar]
- 12.Doi Y, Kubo M, Ninomiya T, Yonemoto K, Iwase M, Arima H, Hata J, Tanizaki Y, Iida M, Kiyohara Y: Impact of Kir6.2 E23K polymorphism on the development of type 2 diabetes in a general Japanese population: the Hisayama Study. Diabetes 2007; 56: 2829– 2833 [DOI] [PubMed] [Google Scholar]
- 13.Florez JC, Jablonski KA, Kahn SE, Franks PW, Dabelea D, Hamman RF, Knowler WC, Nathan DM, Altshuler D: Type 2 diabetes-associated missense polymorphisms KCNJ11 E23K and ABCC8 A1369S influence progression to diabetes and response to interventions in the Diabetes Prevention Program. Diabetes 2007; 56: 531– 536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer ND, Langefeld CD, Bryer-Ash M, Rotter JI, Taylor KD, Bowden DW: Association of the Kir6.2 E23K variant with reduced acute insulin response in African-Americans. J Clin Endocrinol Metab 2008; 93: 4979– 4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrne MM, Sturis J, Polonsky KS: Insulin secretion and clearance during low-dose graded glucose infusion. Am J Physiol 1995; 268: E21– E27 [DOI] [PubMed] [Google Scholar]
- 16.Byrne MM, Sturis J, Clement K, Vionnet N, Pueyo ME, Stoffel M, Takeda J, Passa P, Cohen D, Bell GI: Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 1994; 93: 1120– 1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, Mohammed BS: Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med 2004; 350: 2549– 2557 [DOI] [PubMed] [Google Scholar]
- 18.Reeds DN, Yarasheski KE, Fontana L, Cade WT, Laciny E, DeMoss A, Patterson BW, Powderly WG, Klein S: Alterations in liver muscle and adipose tissue insulin sensitivity in men with HIV infection and dyslipidemia. Am J Physiol Endocrinol Metab 2006; 290: E47– E53 [DOI] [PubMed] [Google Scholar]
- 19.Allison DB, Paultre F, Maggio C, Mezzitis N, Pi-Sunyer FX: The use of areas under curves in diabetes research. Diabetes Care 1995; 18: 245– 250 [DOI] [PubMed] [Google Scholar]
- 20.Van Cauter E, Mestrez F, Sturis J, Polonsky KS: Estimation of insulin secretion rates from C-peptide levels: comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992; 41: 368– 377 [DOI] [PubMed] [Google Scholar]
- 21.Breda E, Cavaghan MK, Toffolo G, Polonsky KS, Cobelli C: Oral glucose tolerance test minimal model indexes of β-cell function and insulin sensitivity. Diabetes 2001; 50: 150– 158 [DOI] [PubMed] [Google Scholar]
- 22.Deivanayagam S, Mohammed BS, Vitola BE, Naguib GH, Keshen TH, Kirk EP, Klein S: Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am J Clin Nutr 2008; 88: 257– 262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuda M, DeFronzo RA: Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999; 22: 1462– 1470 [DOI] [PubMed] [Google Scholar]
- 24.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K: A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006; 355: 2297– 2307 [DOI] [PubMed] [Google Scholar]
- 25.Dalla MC, Caumo A, Basu R, Rizza R, Toffolo G, Cobelli C: Minimal model estimation of glucose absorption and insulin sensitivity from oral test: validation with a tracer method. Am J Physiol Endocrinol Metab 2004; 287: E637– E643 [DOI] [PubMed] [Google Scholar]
- 26.Dalla MC, Caumo A, Cobelli C: The oral glucose minimal model: estimation of insulin sensitivity from a meal test. IEEE Trans Biomed Eng 2002; 49: 419– 429 [DOI] [PubMed] [Google Scholar]
- 27.Tillil H, Shapiro ET, Miller MA, Karrison T, Frank BH, Galloway JA, Rubenstein AH, Polonsky KS: Dose-dependent effects of oral and intravenous glucose on insulin secretion and clearance in normal humans. Am J Physiol 1988; 254: E349– E357 [DOI] [PubMed] [Google Scholar]
- 28.Koster JC, Remedi MS, Dao C, Nichols CG: ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: implications for pharmacogenomic therapy. Diabetes 2005; 54: 2645– 2654 [DOI] [PubMed] [Google Scholar]
- 29.Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG: The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys J 2000; 78: 2334– 2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neher E, Stevens CF: Conductance fluctuations and ionic pores in membranes. Annu Rev Biophys Bioeng 1977; 6: 345– 381 [DOI] [PubMed] [Google Scholar]
- 31.Sigworth FJ: The variance of sodium current fluctuations at the node of Ranvier. J Physiol 1980; 307: 97– 129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van RA, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT: Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350: 1838– 1849 [DOI] [PubMed] [Google Scholar]
- 33.Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, Temple IK, Mackay DJ, Shield JP, Freedenberg D, Noyes K, Ellard S, Ashcroft FM, Gribble FM, Hattersley AT: Relapsing diabetes can result from moderately activating mutations in KCNJ11. Hum Mol Genet 2005; 14: 925– 934 [DOI] [PubMed] [Google Scholar]
- 34.Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM: Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci U S A 2004; 101: 17539– 17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM: Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003; 52: 568– 572 [DOI] [PubMed] [Google Scholar]
- 36.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS:; Wellcome Trust Case Control Consortium (WTCCC) McCarthy MI, Hattersley AT: Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316: 1336– 1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, Jorgensen T, van Vliet-Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K: A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 2007; 39: 770– 775 [DOI] [PubMed] [Google Scholar]
- 38.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Boström K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Råstam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjögren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S: Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007; 316: 1331– 1336 [DOI] [PubMed] [Google Scholar]
- 39.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P: A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445: 881– 885 [DOI] [PubMed] [Google Scholar]
- 40.Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L: Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005; 434: 1026– 1031 [DOI] [PubMed] [Google Scholar]
- 41.Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB: Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 2007; 449: 228– 232 [DOI] [PubMed] [Google Scholar]
- 42.Fernandez-Castaner M, Molina A, Lopez-Jimenez L, Gomez JM, Soler J: Clinical presentation and early course of type 1 diabetes in patients with and without thyroid autoimmunity. Diabetes Care 1999; 22: 377– 381 [DOI] [PubMed] [Google Scholar]
- 43.Stride A, Ellard S, Clark P, Shakespeare L, Salzmann M, Shepherd M, Hattersley AT: β-Cell dysfunction insulin sensitivity and glycosuria precede diabetes in hepatocyte nuclear factor-1α mutation carriers. Diabetes Care 2005; 28: 1751– 1756 [DOI] [PubMed] [Google Scholar]
- 44.Duckworth WC, Hamel FG, Peavy DE: Hepatic metabolism of insulin. Am J Med 1998; 85: 71– 76 [DOI] [PubMed] [Google Scholar]
- 45.Terris S, Steiner DF: Binding and degradation of 125I-insulin by rat hepatocytes. J Biol Chem 1975; 250: 8389– 8398 [PubMed] [Google Scholar]
- 46.Schwanstecher C, Schwanstecher M: Nucleotide sensitivity of pancreatic ATP-sensitive potassium channels and type 2 diabetes. Diabetes 2002; 51( Suppl. 3): S358– S362 [DOI] [PubMed] [Google Scholar]
- 47.Sesti G, Laratta E, Cardellini M, Andreozzi F, Del GS, Irace C, Gnasso A, Grupillo M, Lauro R, Hribal ML, Perticone F, Marchetti P: The E23K variant of KCNJ11 encoding the pancreatic beta-cell adenosine 5′-triphosphate-sensitive potassium channel subunit Kir6.2 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91: 2334– 2339 [DOI] [PubMed] [Google Scholar]
- 48.Holstein A, Hahn M, Stumvoll M, Kovacs P: The E23K variant of KCNJ11 and the risk for severe sulfonylurea-induced hypoglycemia in patients with type 2 diabetes. Horm Metab Res 2009; 41( 5): 387– 390 [DOI] [PubMed] [Google Scholar]
- 49.Melander A, Sartor G, Wahlin E, Schersten B, Bitzen PO: Serum tolbutamide and chlorpropamide concentrations in patients with diabetes mellitus. Br Med J 1978; 1: 142– 144 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.