

Abstract
The thiazolidinedione (TZD) family of PPARγ agonists, especially troglitazone and ciglitazone, induce cell cycle arrest, differentiation, and apoptosis in cancer cells. Mounting evidence indicates that TZDs interfere with multiple signaling mechanisms independently of PPARγactivation, which affect many aspects of cellular functions governing cell cycle progression and survival of cancer cells. Here, we review the “off-target” mechanisms that underlie the antitumor effects of TZDs with emphasis on three key pathways, namely, inhibition of Bcl-2/Bcl-xL function, proteasomal degradation of cell cycle- and apoptosis-regulatory proteins, and transcriptional repression of androgen receptor (AR) through Sp1 degradation. Relative to tumor cells, nonmalignant cells are resistant to these PPARγ-independent antitumor effects, which underscores the translational potential of these agents. Furthermore, dissociation of these antitumor effects from their PPARγ agonist activity provides a rationale for using TZDs as scaffolds for lead optimization to develop a novel class of antitumor agents with a unique mode of mechanism.
Keywords: peroxisome proliferator-activated receptor gamma, thiazolidinediones, beta-TrCP, Sp1, androgen receptor
1. Introduction
Thiazolidinediones (TZDs), including rosiglitazone, pioglitazone, troglitazone, and ciglitazone, are high-affinity ligands for the peroxisome proliferator-activated receptor γ (PPARγ) [1], a transcription factor preferentially expressed in adipose tissue [2]. These TZDs improve insulin sensitivity by regulating many aspects of adipose tissue function through the transcriptional activation of certain insulin-sensitive genes involved in glucose homeostasis, fatty acid metabolism, and triacylglycerol storage in adipocytes [3, 4]. Moreover, TZD-mediated PPARγ activation has been shown to promote the differentiation of preadipocytes by mimicking certain genomic effects of insulin on adipocytes, and to modulate the expression of adiponectin, pro-inflammatory cytokines like IL-6 and TNFα, and a host of endocrine regulators in adipocytes and macrophages [4]. Through these beneficial effects, TZDs offer a new type of oral therapy for type II diabetes by reducing insulin resistance and assisting glycemic control. Troglitazone (Rezulin), however, was withdrawn from the market due to idiosyncratic hepatotoxicity, which was thought to be attributable to the induction of mitochondrial stress in liver cells by high doses of the drug [5].
2. PPARγ-dependent and –independent antitumor effects of TZDs
Like adipocytes, many human cancer cell lines have been reported to exhibit high levels of PPARγ expression. In vitro exposure of these tumor cells to high doses (≥ 10 μM) of TZDs, especially troglitazone and ciglitazone, led to cell cycle arrest, apoptosis and/or redifferentiation [review: [6–10]], suggesting a putative link between PPARγ signaling and TZD’s antitumor activities. Furthermore, the in vivo anticancer efficacy of troglitazone was demonstrated in a few clinical cases that involved patients with liposarcomas [11] or prostate cancer [12]. To date, the identities of target genes that contribute to the antiproliferative activities of PPARγ agonists remain elusive, in part, because the genomic responses to PPARγ activation are complex and will be highly dependent upon cellular context in cancer cells [7]. Reported PPARγ-specific target genes in colorectal cancer cells included genes linked to growth regulatory pathways, colon epithelial cell maturation, immune modulation, and intercellular adhesion [13]. However, the functional role of these PPARγ target genes in mediating TZDs’ antiproliferative activities in cancer cells is unclear.
In contrast, several lines of evidence argue against the dependence of the antitumor effect of TZDs on PPARγ activation. First, TZDs’ antitumor effects seem to be structure-specific irrespective of potency in PPARγ activation, i.e., troglitazone and ciglitazone are more active in inducing apoptosis in cancer cells relative to rosiglitazone and pioglitazone. Second, there exists a 3-orders-of-magnitude discrepancy between the concentration required to mediate antitumor effects and that for PPARγ activation. Third, there lacks a correlation between the susceptibility of tumor cells to TZDs and the expression level of PPARγ. For example, LNCaP prostate cancer and MCF-7 breast cancer cells, both of which exhibit low PPARγ expression levels, were more sensitive to the effects of troglitazone and ciglitazone on suppressing cell viability than their PPARγ-overexpressing counterparts, PC-3 and MDA-MB-231 cells, respectively [14, 15]. Fourth, Δ2TG and Δ2CG, PPARγ-inactive analogues of troglitazone and ciglitazone, respectively (Fig. 1), were modestly more potent than their parent compounds in suppressing cell proliferation in cancer cells [14, 15]. Fifth, siRNA-mediated knockdown of PPARγ in PC-3 cells did not affect the ability of troglitazone or Δ2TG to induce apoptotic death (Chen, unpublished data).
Figure 1.
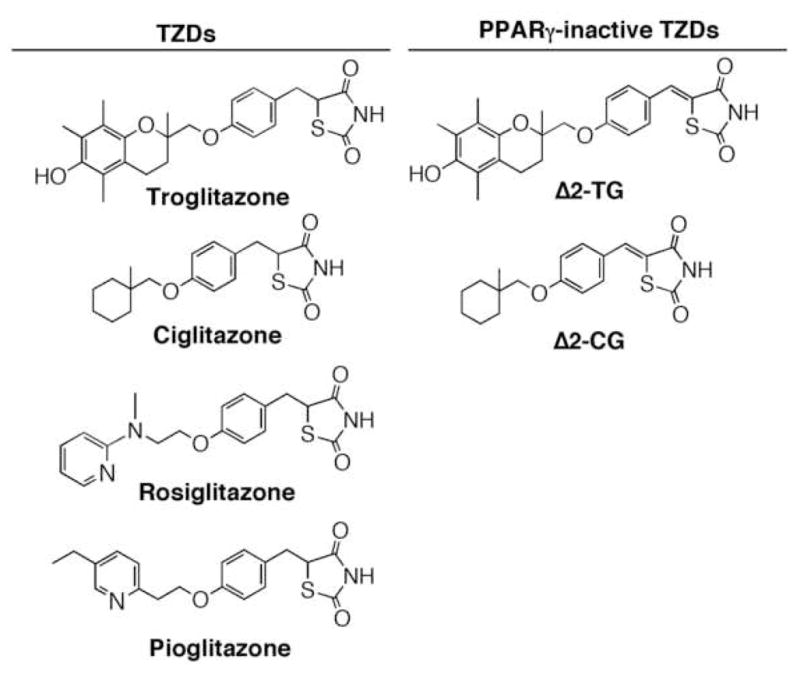
Structures of troglitazone, ciglitazone, their PPARγ-inactive analogues Δ2TG and Δ2CG, rosiglitazone, and pioglitazone. Our data indicate that introduction of a double bond adjacent to the thiazolidinedione ring abolished the ability of the resulting molecule to activate PPARγ (14).
3. Pleiotropic effects of TZDs on multiple signaling targets
Although the functional role of PPARγ in regulating cell proliferation and differentiation varies in different cellular contexts, mounting evidence indicates that the effect of TZDs on inducing apoptotic death in cancer cells is, to a large extent, attributable to “off-target” mechanisms [10]. To date, an array of signaling targets have been linked to the antitumor activities of troglitazone and ciglitazone, which affect many aspects of cellular functions governing cell cycle progression and survival of cancer cells (Table 1).
Table 1.
Signaling mechanisms that underlie TZD-mediated antitumor effects
| Signaling targets | Reported effects of TZDs |
|---|---|
| Intracellular Ca2+ store [37] | TZDs caused partial depletion of intracellular Ca2+ stores in ES cells, resulting in phosphorylating deactivation of the α-subunit of eukaryotic initiation factor (eIF2α) and subsequent inhibition of translation initiation. |
| JNK MAP kinase [38] | Troglitazone caused apoptosis in HepG2 cells by activating the JNK-dependent cell death pathway accompanied by increased Bid cleavage and elevation of Bad and Bax. |
| The CDK inhibitors p21 and p27 [39, 40] | Troglitazone mediated growth inhibition in HepG2 cells by increasing levels of p27 and p21, accompanied by reduced Rb phosphorylation. Troglitazone-induced p27 up-regulation was mediated through the downregulation of the E3 ligase Skp2. |
| P53 and GADD45, a p53-responsive stress protein [41] | Troglitazone induced apoptosis in vascular smooth muscle cells by activating the p53-GADD45 pathway. |
| Bcl-2/Bcl-xL [14] | Troglitazone and ciglitazone blocked the BH3 domain- mediated interaction of Bcl-2 and Bcl-xL with Bak and other proapoptotic proteins with IC50 of approximately 25 μM, which might underlie the apoptosis-inducing effect of these TZDs in cancer cells. |
| FLIP (FLICE inhibitory protein) [16, 32] | Troglitazone and ciglitazone facilitated ubiquitin-dependent, proteasomal degradation of FLIP, an apoptosis-suppressing protein that blocks death receptor signaling, via a PPARγ-independent mechanism. This FLIP downregulation underlies the ability of troglitazone to sensitize glioma cells to TRAIL. |
| β-Catenin [17–20] | Troglitazone facilitated the ubiquitin-dependent, proteasomal degradation of β-catenin via a PPARγ-dependent or – independent mechanism in different cell systems. |
| Cyclin D1 [15, 21–24] | Troglitazone and ciglitazone down-regulated cyclin D1 expression via proteasomal degradation as part of the mechanism for causing cell-cycle arrest and growth inhibition in breast cancer cells. |
| Prostate-specific antigen (PSA) and androgen receptor (AR) [26, 31] | Troglitazone and ciglitazone mediated the transcriptional repression of AR by facilitating the proteasomal degradation of the transcriptional factor Sp1 in LNCaP prostate cancer cells. |
| Estrogen receptor α (ERα) [15, 24] | Troglitazone and ciglitazone mediated the transcriptional repression of ERα in breast cancer cells. |
From a mechanistic perspective, some of the aforementioned targets appear to be cell type-specific due to differences in signaling pathways regulating cell proliferation and survival in different cancer cell systems. However, three aspects of the PPARγ-independent antitumor activities of TZDs are noteworthy because they are amenable to pharmacologic exploitation that could foster novel strategies for cancer treatment/prevention, namely, inhibition of Bcl-2/Bcl-xL function, proteasomal degradation of target proteins, and transcriptional repression of AR through Sp1 degradation (Fig. 2).
Figure 2.
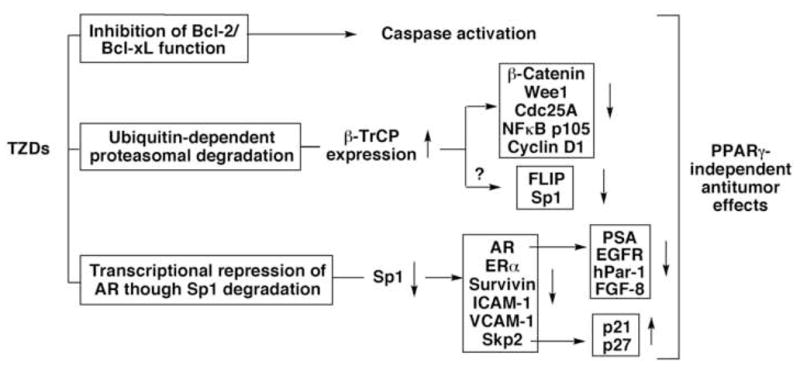
A schematic diagram depicting the major PPARγ-independent antitumor mechanisms of TZDs.
3.1. Inhibition of Bcl-2/Bcl-xL function
Evidence suggests that troglitazone and ciglitazone inhibit the antiapoptotic functions of Bcl-xL and Bcl-2 independently of PPARγ [14]. Treatment of cancer cells with these TZDs reduced intracellular association of Bcl-2 and Bcl-xL with Bak, leading to caspase-dependent apoptosis. Moreover, Bcl-xL overexpression protected cancer cells from TZD-induced apoptosis. In contrast, rosiglitazone and pioglitazone lacked the ability to disrupt mitochondrial integrity and, consequently, showed marginal effects on apoptosis even at high concentrations. Molecular docking analysis indicated that troglitazone competed with the Bak BH3 peptide for BH3 domain-mediated interactions with Bcl-xL (Fig. 3), with an IC50 of approximately 20 μM.
Figure 3.
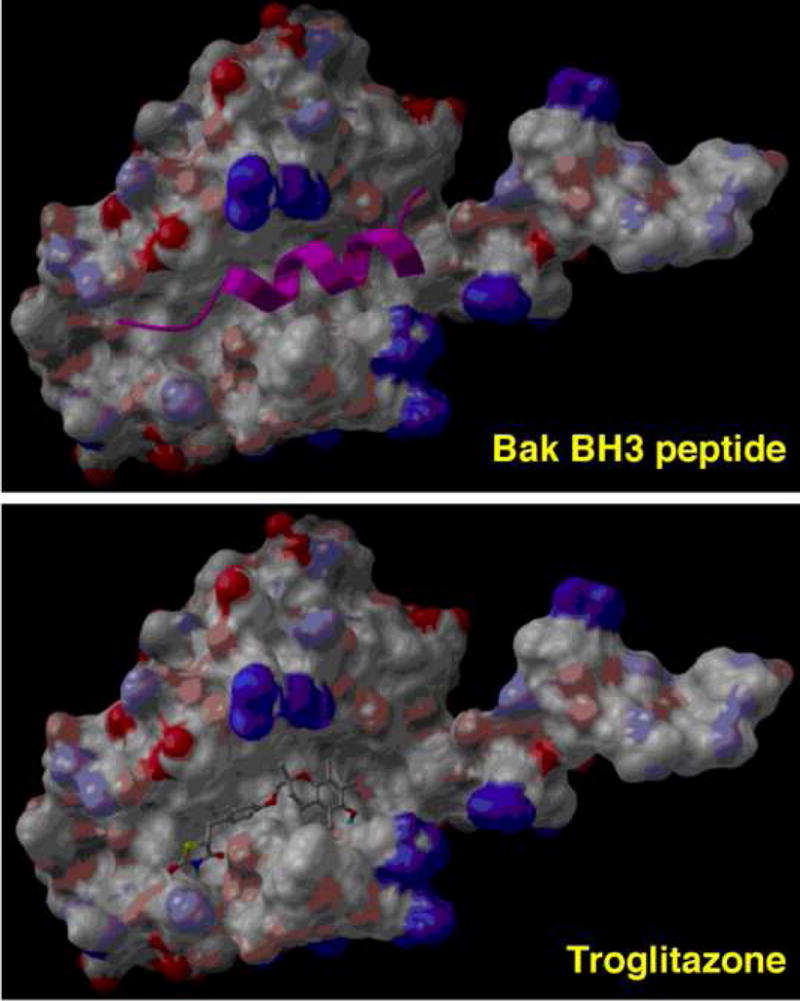
Molecular docking of Bak BH3 peptide (top; represented by a ribbon structure) and troglitazone (bottom) into the Bak BH3 peptide-binding site of Bcl-xL.
3.2. Proteasomal degradation of cell cycle- and apoptosis-regulatory proteins
Data from this and other laboratories show that troglitazone and ciglitazone exhibited a unique ability to facilitate the ubiquitin-dependent proteasomal degradation of a series of cell cycle- and apoptosis-regulatory proteins, including FLIP [16], β-catenin [17–20], cyclin D1 [15, 21–25], and Sp1 [26], independently of PPARγ activation. As each of these target proteins plays a role in regulating carcinogenesis and tumor progression, the effect of these TZDs on selectively activating the ubiquitin-proteasome system provide a novel framework to account for their antitumor activities.
To shed light onto this novel pharmacological action, we previously examined the pathway by which troglitazone and its PPARγ-inactive analogs Δ2TG and STG28 promoted β-catenin degradation in prostate cancer cells [20]. The ability of these thiazolidinediones to repress β-catenin in PC-3 cells was not affected by siRNA-mediated knockdown of PPARγ, indicating the dissociation of these two pharmacological activities (Fig. 4A).
Figure 4.
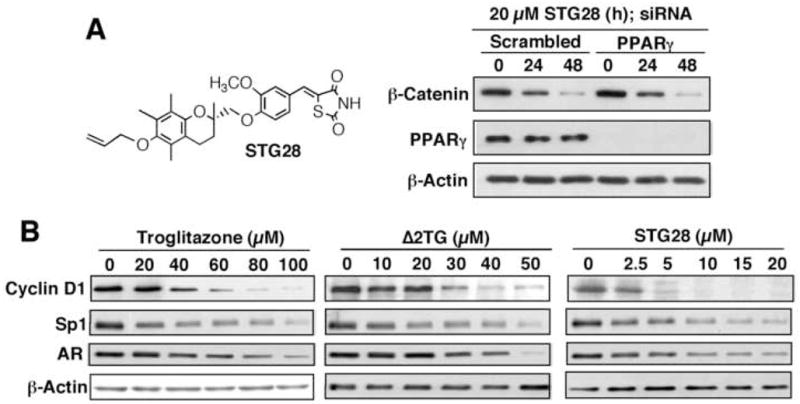
A, structure of STG28 (left panel) and siRNA-mediated knockdown of PPARγ does not interfere with the effect of STG28 on β-catenin repression. B, Western blot analysis of the dose-dependent effect of STG28 vis-à-vis troglitazone and Δ2TG on attenuating the expression levels of cyclin D1, Sp1, and AR.
Several lines of evidence indicate that TZD-facilitated β-catenin proteolysis was mediated through the upregulation of β-transducin repeat-containing protein (β-TrCP), an F-box component of the Skp1-Cul1-F-box protein (SCF) E3 ligase. This finding was validated by the concurrent down-regulation of a number of other known β-TrCP substrates, including Wee1, IκBα, cdc25A, and NFκB/p105, in TZD-treated cancer cells. In addition, while ectopic β-TrCP expression enhanced TZD-facilitated β-catenin ablation, its siRNA-mediated knockdown protected cells from this degradation. Equally important, nonmalignant cells were resistant to the effect of these TZDs on up-regulating β-TrCP expression, suggesting the translational potential of these agents in chemoprevention.
Although some TZD-targeted proteins such as cyclin D1 lack a consensus sequence of D(pS)GXn(pS) (X is any amino acid; n = 2 – 4) for β-TrCP recognition [27], we hypothesize that they are also targeted by β-TrCP through unconventional recognition motifs. For example, our recent mutational and modeling analyses indicate that cyclin D1 binds to the WD40 domain of β-TrCP through an unconventional recognition site, 279EEVDLACpT286 [25].
Substantial evidence suggests that the SCF complexes and the anaphase promoting complex/cyclosome (APC/C) represent two major classes of E3 ubiquitin ligases that reciprocally regulate cell cycle progression and proliferation by controlling the proteolysis of cell-cycle regulatory proteins [28–30]. To the best of our knowledge, TZDs represent the first class of agents with the ability to perturb the ubiquitin E3 ligase signaling network.
3.3. Transcriptional repression of AR through Sp1 degradation
Our data indicate that troglitazone and ciglitazone might interfere with AR function through two PPARγ-independent mechanisms at different dose ranges [26, 31]. At ~10 μM concentrations, these TZDs suppressed PSA expression by blocking AR recruitment to the PSA promoter, and at higher concentrations, they were capable of down-regulating AR expression by activating the proteasomal degradation of Sp1. The effect of TZDs on repressing AR expression is noteworthy considering the clinical relevance of AR in androgen-refractory prostate cancer. In addition, the ability of these TZDs to ablate Sp1 might account for the reported effect of troglitazone on down-regulating the expression of estrogen receptor [15, 24], epidermal growth factor receptor (EGFR) [26], survivin [32], the cell adhesion molecules ICAM-1 and VCAM-1 [33], and the oncogenic protein Skp2 [20], all of which are downstream targets of this transcription factor. Sp1 and other Sp family members have been reported to play a crucial role in the growth and metastasis of many tumor types by regulating expression of cell cycle genes and vascular endothelial growth factor, thus representing potential targets for cancer therapy [34]. Our preliminary data suggest that thiazolidinedione-facilitated proteasomal degradation of Sp1 was also mediated through a β-TrCP-dependent mechanism (Wei and Chen, unpublished).
4. Pharmacological exploitation of troglitazone and ciglitazone to develop novel antitumor agents
Dissociation of the aforementioned antitumor effects of troglitazone and ciglitazone from their PPARγ agonist activity provides a rationale for using these TZDs as scaffolds for lead optimization to develop novel agents for cancer therapy or prevention. The proof-of-principle of this premise was demonstrated by STG28 [26, 35] and OSU-CG12 [36], PPARγ-inactive derivatives of Δ2TG and Δ2CG, respectively, with improved antitumor potency. For example, STG28 exhibited an-order-of-magnitude higher potency in suppressing the expression of cyclin D1, Sp1, and AR through proteasomal degradation or transcriptional repression in prostate cancer cells than the parent molecules troglitazone and Δ2TG (Fig. 4B).
It is noteworthy that normal prostate epithelial cells were resistant to the ablating effect of STG28 and OSU-CG12 on these target proteins [26]. This differential effect between malignant versus nonmalignant cells underlies the translational potential of this novel class of antitumor agents.
5. Conclusion
Although thiazolidinedione PPARγ agonists exhibit multiple mechanisms for inducing PPARγ-independent antitumor effects, this finding does not conflict with the general notion regarding the pivotal role of PPARγ in carcinogenesis. From a therapeutic standpoint, pharmacological exploitation of these “off-target” mechanisms might foster novel strategies for cancer treatment and prevention. It is especially noteworthy that the TZD’s effect on promoting β-TrCP-mediated degradation of target proteins parallels that of glucose starvation [25], suggesting a resemblance among the respective signaling networks. As dysregulated expression of cyclin D1, Sp1, and other cell-cycle regulatory proteins has been linked to tumorigenesis and drug resistance in human cancers, targeting β-TrCP by small-molecule agents might represent a viable approach in cancer therapy.
Acknowledgments
The authors acknowledge support from National Cancer Institute grant (CA112250), William R. Hearst Foundation, Prostate Cancer Foundation, and the Lucius A. Wing Endowed Chair Fund at The Ohio State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med. 1999;16:179–192. doi: 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 2.Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 3.Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest. 2000;106:467–472. doi: 10.1172/JCI10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma AM, Staels B. Review: Peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. 2007;92:386–395. doi: 10.1210/jc.2006-1268. [DOI] [PubMed] [Google Scholar]
- 5.Jaeschke H. Troglitazone hepatotoxicity: are we getting closer to understanding idiosyncratic liver injury? Toxicol Sci. 2007;97:1–3. doi: 10.1093/toxsci/kfm021. [DOI] [PubMed] [Google Scholar]
- 6.Kopelovich L, Fay JR, Glazer RI, Crowell J. Peroxisome proliferator-activated receptor modulators as potential chemopreventive agents. Mol Cancer Ther. 2002;1:357–363. [PubMed] [Google Scholar]
- 7.Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 8.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 9.Jiang M, Shappell SB, Hayward SW. Approaches to understanding the importance and clinical implications of peroxisome proliferator-activated receptor gamma (PPARgamma) signaling in prostate cancer. J Cell Biochem. 2004;91:513–527. doi: 10.1002/jcb.10770. [DOI] [PubMed] [Google Scholar]
- 10.Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS. Beyond peroxisome proliferator-activated receptor gamma signaling: the multi-facets of the antitumor effect of thiazolidinediones. Endocr Relat Cancer. 2006;13:401–413. doi: 10.1677/erc.1.01182. [DOI] [PubMed] [Google Scholar]
- 11.Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman SB, Singer S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A. 1999;96:3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. Down-Regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor gamma in human prostate cancer. Cancer Res. 2000;60:5494–5498. [PubMed] [Google Scholar]
- 13.Gupta RA, Brockman JA, Sarraf P, Willson TM, DuBois RN. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J, Biol Chem. 2001;276:29681–29687. doi: 10.1074/jbc.M103779200. [DOI] [PubMed] [Google Scholar]
- 14.Shiau CW, Yang CC, Kulp SK, Chen KF, Chen CS, Huang JW, Chen CS. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Res. 2005;65:1561–1569. doi: 10.1158/0008-5472.CAN-04-1677. [DOI] [PubMed] [Google Scholar]
- 15.Huang JW, Shiau CW, Yang YT, Kulp SK, Chen KF, Brueggemeier RW, Shapiro CL, Chen CS. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol Pharmacol. 2005;67:1342–1348. doi: 10.1124/mol.104.007732. [DOI] [PubMed] [Google Scholar]
- 16.Kim Y, Suh N, Sporn M, Reed CJ. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002;277:22320–22329. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- 17.Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer JSR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607–613. doi: 10.1042/BJ20030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279:45020–45027. doi: 10.1074/jbc.M407050200. [DOI] [PubMed] [Google Scholar]
- 19.Sharma C, Pradeep A, Wong L, Rana A, Rana B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem. 2004;279:35583–35594. doi: 10.1074/jbc.M403143200. [DOI] [PubMed] [Google Scholar]
- 20.Wei S, Lin LF, Yang CC, Wang YC, Chang GD, Chen H, Chen CS. Thiazolidinediones modulate the expression of beta-catenin and other cell-cycle regulatory proteins by targeting the F-box proteins of Skp1-Cul1-F-box protein E3 ubiquitin ligase independently of peroxisome proliferator-activated receptor gamma. Mol Pharmacol. 2007;72:725–733. doi: 10.1124/mol.107.035287. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Fu M, D’Amico M, Albanese C, Zhou JN, Brownlee M, Lisanti M, Chatterjee MP, Lazar VKMA, Pestell RG. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol. 2001;21:3057–3070. doi: 10.1128/MCB.21.9.3057-3070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin F, Wakino S, Liu Z, Kim S, Hsueh WA, Collins AR, Van Herle AJ, Law RE. Troglitazone inhibits growth of MCF-7 breast carcinoma cells by targeting G1 cell cycle regulators. Biochem Biophys Res Commun. 2001;286:916–922. doi: 10.1006/bbrc.2001.5491. [DOI] [PubMed] [Google Scholar]
- 23.Lapillonne H, Konopleva M, Tsao T, Gold D, McQueen T, Sutherland R, Madden T, Andreeff M. Activation of peroxisome proliferator-activated receptor gamma by a novel synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res. 2003;63:5926–5939. [PubMed] [Google Scholar]
- 24.Qin C, Burghardt R, Smith R, Wormke M, Stewart J, Safe S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res. 2003;63:958–964. [PubMed] [Google Scholar]
- 25.Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ, Lai MD, Chen CS. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008 doi: 10.1074/jbc.M802160200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang CC, Wang YC, Wei S, Lin LF, Chen CS, Lee CC, Lin CC, Chen CS. Peroxisome proliferator-activated receptor gamma-independent suppression of androgen receptor expression by troglitazone mechanism and pharmacologic exploitation. Cancer Res. 2007;67:3229–3238. doi: 10.1158/0008-5472.CAN-06-2759. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–333. doi: 10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 29.Yamasaki L, Pagano M. Cell cycle, proteolysis and cancer. Curr Opin Cell Biol. 2004;16:623–628. doi: 10.1016/j.ceb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 31.Yang CC, Ku CY, Wei S, Shiau CW, Chen CS, Pinzone JJ, Ringel MD, Chen CS. Peroxisome proliferator-activated receptor gamma-independent repression of prostate-specific antigen expression by thiazolidinediones in prostate cancer cells. Mol Pharmacol. 2006;69:1564–1570. doi: 10.1124/mol.105.018333. [DOI] [PubMed] [Google Scholar]
- 32.Schultze K, Bock B, Eckert A, Oevermann L, Ramacher D, Wiestler O, Roth W. Troglitazone sensitizes tumor cells to TRAIL-induced apoptosis via down-regulation of FLIP and Survivin. Apoptosis. 2006;11:1503–1512. doi: 10.1007/s10495-006-8896-3. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki M, Jordan P, Welbourne T, Minagar A, Joh T, Itoh M, Elrod JW, Alexander JS. Troglitazone, a PPAR-gamma activator prevents endothelial cell adhesion molecule expression and lymphocyte adhesion mediated by TNF-alpha. BMC Physiol. 2005;5:3. doi: 10.1186/1472-6793-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–2448. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Huang JW, Shiau CW, Yang J, Wang DS, Chiu HC, Chen CY, Chen CS. Development of small-molecule cyclin D1-ablative agents. J Med Chem. 2006;49:4684–4689. doi: 10.1021/jm060057h. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Wei S, Wang DS, Wang YC, Kulp SK, Chen CS. Pharmacological exploitation of the peroxisome proliferator-activated receptor gamma agonist ciglitazone to develop a novel class of androgen receptor-ablative agents. J Med Chem. 2008;51:2100–2107. doi: 10.1021/jm701212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–6218. [PubMed] [Google Scholar]
- 38.Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- 39.Bae MA, Rhee H, Song BJ. Troglitazone but not rosiglitazone induces G1 cell cycle arrest and apoptosis in human and rat hepatoma cell lines. Toxicol Lett. 2003;139:67–75. doi: 10.1016/s0378-4274(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 40.Koga H, Harada M, Ohtsubo M, Shishido S, Kumemura H, Hanada S, Taniguchi E, Yamashita K, Kumashiro R, Ueno T, Sata M. Troglitazone induces p27Kip1-associated cell-cycle arrest through down-regulating Skp2 in human hepatoma cells. Hepatology. 2003;37:1086–1096. doi: 10.1053/jhep.2003.50186. [DOI] [PubMed] [Google Scholar]
- 41.Okura T, Nakamura M, Takata Y, Watanabe S, Kitami Y, Hiwada K. Troglitazone induces apoptosis via the p53 and Gadd45 pathway in vascular smooth muscle cells. Eur J Pharmacol. 2000;407:227–235. doi: 10.1016/s0014-2999(00)00758-5. [DOI] [PubMed] [Google Scholar]