

Abstract
Eukaryotes possess numerous quality control systems that monitor both the synthesis of RNA and the integrity of the finished products. We previously demonstrated that Saccharomyces cerevisiae possesses a quality control mechanism, nonfunctional rRNA decay (NRD), capable of detecting and eliminating translationally defective rRNAs. Here we show that NRD can be divided into two mechanistically distinct pathways: one that eliminates rRNAs with deleterious mutations in the decoding site (18S NRD) and one that eliminates rRNAs containing deleterious mutations in the peptidyl transferase center (25S NRD). 18S NRD is dependent on translation elongation and utilizes the same proteins as those participating in no-go mRNA decay (NGD). In cells that accumulate 18S NRD and NGD decay intermediates, both RNA types can be seen in P-bodies. We propose that 18S NRD and NGD are different observable outcomes of the same initiating event: a ribosome stalled inappropriately at a sense codon during translation elongation.
Introduction
High fidelity information transmission from DNA to RNA to protein is critical for proper gene expression. Maintenance of this fidelity requires tight quality control over each individual step as well as the machinery mediating that step. An emerging realization with regard to eukaryotes is the remarkable extent to which they monitor the synthesis, processing and final integrity of the RNA species involved in translation: mRNAs, rRNAs and tRNAs (Doma and Parker, 2007). All of these species are transcribed as precursor RNAs (pre-RNAs) that must undergo extensive trimming, splicing, editing, addition of extra sequences (e.g., polyA and CCA tails) and/or chemical modifications in order to generate the mature RNAs that participate in translation.
Quality control of eukaryotic transcripts begins in the nucleus. Nuclear RNA surveillance has been most extensively studied in S. cerevisiae and relies heavily on the exosome, the cell’s major 3′->5′ exonuclease (Houseley et al., 2006). The nuclear and cytoplasmic exosomes share ten core subunits, one of which, Rrp44p, is responsible for its exonucleolytic activity (Dziembowski et al., 2007). However, nuclear and cytoplasmic exosomes differ by the presence of the exoribonuclease Rrp6p and nucleic acid binding protein Rrp47p in the nucleus versus the GTPase Ski7p in the cytoplasm. Within the nucleus, the exosome has been shown to degrade aberrant pre-mRNAs, pre-tRNAs, pre-rRNAs and pre-snoRNAs (Houseley et al., 2006). Prior to their decay, many nuclear exosome substrates undergo polyadenylation by the Trf4/5p/Air/Mtr4p polyadenylation (TRAMP) complex, which stimulates decay via exosome recruitment (Kadaba et al., 2004; LaCava et al., 2005; Vanacova et al., 2005). Decay of such polyadenylated RNAs is thought to occur within a region of the nucleolus known as the No-body (Dez et al., 2006). Some polyadenylated pre-RNAs are also thought to be decayed by the nuclear 5′->3′ exonuclease Rat1p, although where this occurs within the nucleus is unknown (Fang et al., 2005).
In the cytoplasm, several pathways have been described for mRNA quality control (Isken and Maquat, 2007). All of these pathways are translation-dependent, initiating when a ribosome stalls during translation in a context that impedes efficient elongation or termination. Nonstop mRNA decay (NSD) eliminates mRNAs lacking any in frame stop codon, such as truncated or prematurely polyadenylated transcripts. This pathway is dependent on Ski7p, which recruits the cytoplasmic exosome (Frischmeyer et al., 2002; van Hoof et al., 2002). A second quality control system, nonsense-mediated mRNA decay (NMD), eliminates mRNAs containing a stop codon in a poor context for translation termination, often a nonsense or premature termination codon (Amrani et al., 2006). Following recruitment of the Upf proteins (Upf1p, Upf2p and Upf3p) to the stalled translation complex, the mRNA is decapped and then degraded by the major cytoplasmic 5′->3′ exoribonuclease Xrn1p (Muhlrad and Parker, 1994). Nonsense transcripts are also subject to 3′->5′ degradation by the cytoplasmic exosome via interaction between Upf1p and Ski7p (Mitchell and Tollervey, 2003; Takahashi et al., 2003). In D. melanogaster and human cells, NMD can also be initiated by endonucleolytic cleavage of the mRNA (Gatfield and Izaurralde, 2004; Huntzinger et al., 2008). Finally, no-go mRNA decay (NGD) eliminates mRNAs containing a structural barrier within the open reading frame that induces ribosome stalling. Such stalling stimulates endonucleolytic cleavage of the mRNA immediately upstream of the structural barrier, followed by Xrn1p- and Ski7p-mediated decay of the 3′ and 5′ halves, respectively. NGD also involves the eukaryotic release factor (eRF) homologs Dom34p and Hbs1p, although the precise functions of these proteins are not yet understood (Doma and Parker, 2006). Within the cytoplasm, both general mRNA turnover and NMD are thought to occur in discrete structures known as processing or P-bodies. P-bodies are regions in eukaryotic cells that contain translationally repressed mRNPs proteins involved in mRNA decay. These proteins include the decapping complex Dcp1p/Dcp2p, decapping activator Dhh1p and exoribonuclease Xrn1p (Parker and Sheth, 2007). Where NSD and NGD occur, however, has not been previously examined.
In addition to the above mRNA quality control pathways, recent studies showed that S. cerevisiae also has the capacity to eliminate mature but functionally defective tRNAs and rRNAs. Hypomodified, but otherwise mature tRNAs undergo rapid tRNA decay (RTD) by a process involving Rat1p and Xrn1p (Chernyakov et al., 2008). Work from our laboratory has shown that rRNAs containing deleterious mutations in either the peptidyl transferase center of 25S rRNA or the decoding site of 18S rRNA are subject to a late-acting quality control system dubbed nonfunctional rRNA decay (NRD) (LaRiviere et al., 2006). In bacteria, the highly conserved bases A2451 and U2585 (E. coli numbering) in the peptidyl transferase center and G530 and A1492 in the decoding site are all required for proper ribosome function, and mutations at any of these positions have dominant negative phenotypes (Green et al., 1997; Powers and Noller, 1990, 1993; Thompson et al., 2001; Yoshizawa et al., 1999; Youngman et al., 2004). In contrast, mutations introduced at the analogous positions in yeast are recessive due to degradation of the rRNAs containing them. Prior to their decay, such NRD substrates are transcribed and processed with normal kinetics into mature 18S and 25S rRNAs and are contained within ribosomal subunit-sized complexes (LaRiviere et al., 2006). Thus NRD is clearly different from the early-acting, nuclear pre-rRNA quality control pathways described above.
Here we show that NRD constitutes at least two mechanistically distinct pathways, one that eliminates mutant 25S rRNAs (25S NRD) and one that eliminates mutant 18S rRNAs (18S NRD). 25S NRD substrates, which are largely contained in 60S complexes, accumulate just outside the nuclear envelope, and degradation of these rRNAs is not blocked by small molecule inhibitors of translation elongation. Conversely, mutant 18S rRNAs localize throughout the cell, and their decay is completely abolished when translation elongation is inhibited. Further, elimination of mutant 18S rRNAs involves the same set of proteins as does NGD, and both 18S NRD and NGD substrates accumulate in P-bodies.
Results
Subcellular localization of NRD substrates
We previously described a plasmid-based rDNA reporter system for expressing 35S pre-rRNAs from a Pol II promoter (Cole and LaRiviere, 2008). In this system, the resultant rRNA products are distinguishable from endogenous rRNAs by virtue of short neutral sequence tags inserted near the 5′ ends of the plasmid-based 18S and 25S genes (Figure 1A). Use of these rDNA reporter plasmids allows for expression and ready detection of tagged rRNAs in a large background of untagged endogenous rRNAs in otherwise normally growing wild-type yeast cells (Cole and LaRiviere, 2008; LaRiviere et al., 2006).
Figure 1.

In situ localization of wild type and mutant rRNAs. (A) Schematic of rDNA reporter plasmids containing neutral sequence tags at indicated sites. (B) FISH analysis of wild type cells (BY4741) carrying no plasmid, pJV12-WT (18S:wild type), pJV12-G530U (18S:G530U) or pJV12-A1492C (18S:A1492C) using Alexa 594-labeled probe FL125. (C) FISH analysis of wild type cells (BY4741) carrying no plasmid, pJV12-WT (25S:wild type), pJV12-A2451G (25S:A2451G) or pJV12-U2585A (25S:U2585A) using Alexa 594-labeled probe FL126. In both B and C, the nucleus is marked by DAPI staining.
To determine whether rRNAs subject to NRD exhibit any altered subcellular localization compared to wild type rRNAs, we employed fluorescence in situ hybridization (FISH) with oligonucleotide probes complementary to the sequence tags in the plasmid-derived rRNAs (Figure 1B, C). No FISH signal above background was detected in cells lacking an rDNA reporter plasmid, indicating no significant cross reaction of the oligonucleotide probes with endogenous nucleic acids.
As expected for rRNAs in translating ribosomes, wild type 18S and 25S rRNAs were found throughout the cell, with strong but diffuse localization in the cytoplasm. This same diffuse cytoplasmic distribution was observed for NRD substrates 18S:G530U and 18S:A1492C rRNAs (Figure 1B). In contrast, NRD substrates 25S:A2451G and 25S:U2585A rRNAs were largely confined to one or more foci adjacent to the DNA (Figure 1C). To address the possibility that these foci were coincident with the nucleolus, we combined FISH with immunofluorescence for the nucleolar protein Nop1p (Figure 2A). Whereas the α-Nop1p antibody stained a region within the nucleus, consistent with the expected location of the nucleolus, the larger accumulations of 25S:A2451G and 25S:U2585A rRNAs were consistently found to be spatially distinct from Nop1p-positive regions. The mutant 25S rRNA foci are therefore not coincident with the nucleolus. To examine the relationship between the mutant 25S rRNA foci and the nuclear envelope, we combined FISH with immunofluorescence for O-linked glycoproteins in the nuclear pore complex. The specificity of the antibody was confirmed by immunofluorescence and DAPI staining (Figure 2B). Whereas a portion of the mutant 25S rRNA FISH signals did colocalize with the nuclear pore complex, a much larger fraction was clearly external to the nucleus (Figure 2C). Thus, unlike the mutant 18S rRNAs, which exhibit the same dispersed distribution as wild type rRNAs, mutant 25S rRNAs accumulate in perinuclear foci in the cytoplasm.
Figure 2.

Comparison of 25S rRNA localization with nucleolar and nuclear pore complex markers. (A) Wild type (BY4741) cells carrying pJV12-WT (25S:wild type), pJV12-A2451G (25S:A2451G) or pJV12-U2585A (25S:U2585A) analyzed by FISH using Alexa 594-labeled probe FL126 and by indirect immunofluorescence using an antibody recognizing the nucleolar protein Nop1p plus an Alexa 488-conjugated secondary antibody. (B) Detection of the nuclear envelope by indirect immunofluorescence using an antibody against nuclear pore complex O-linked glycoproteins and an Alexa 488-conjugated secondary antibody. In both A and B, DAPI staining shows the location of the nucleus. (C) Wild type (BY4741) cells carrying pJV12-WT (25S:wild type), pJV12-A2451G (25S:A2451G) or pJV12-U2585A (25S:U2585A) were analyzed by immunofluorescence to visualize the nuclear envelope as in B and FISH to visualize 25S rRNAs as in A. Arrows indicate foci of mutant 25S rRNA outside the nucleus.
The role of translation in NRD
To assess whether NRD is dependent on ongoing translation, we measured the stability of the NRD substrates by transcriptional pulse-chase analysis in the absence or presence of cycloheximide. Cycloheximide is an antibiotic that inhibits the peptidyl transferase activity of ribosomes engaged in translation elongation, effectively locking them onto mRNA via an interaction with the large subunit (Stocklein and Piepersberg, 1980). For this and subsequent analyses, we focused on the 18S:A1492C and 25S:U2585A rRNA mutants. Wild type cells were transformed with plasmids expressing either 18S:A1492C or 25S:U2585A rRNA under control of a galactose-inducible promoter. Cultures grown in raffinose-containing media were induced with galactose and then shifted either to media containing glucose or glucose plus cycloheximide. At the indicated time points after glucose addition, samples were taken and total RNA analyzed by northern blotting with tag-specific oligonucleotide probes (Figure 3). As expected, wild type rRNAs were stable over the time course and this stability was unaffected by the addition of cycloheximide. Also, as previously observed (LaRiviere et al., 2006), both the 18S:A1492C and 25S:U2585A rRNAs were subject to elimination when no cycloheximide was present. However, in the presence of cycloheximide, 18S:A1492C rRNA was stable throughout the time course (Figure 3A). Complete stabilization of 18S:A1492C rRNA was also observed in the presence of hygromycin B, an antibiotic that interferes with translation elongation by a mechanism different from cycloheximide (Supplemental Results and Figure S1).
Figure 3.

Differential effects of cycloheximide on 18S and 25S NRD. (A) Transcriptional pulse-chase and northern analysis of wild type cells (BY4741) carrying either pSC40 (18S:wild type) or pSC40-A1492C (18S:A1492C) in the absence or presence of cycloheximide. (B) Transcriptional pulse-chase and northern analysis of wild type cells (BY4741) carrying either pSC40 (25S:wild type) or pSC40-U2585A (25S:U2585A) in the absence or presence of cycloheximide. Cycloheximide was added at transcriptional shut off. (C) Same as in B except cycloheximide was added 30 minutes after transcriptional shut off. In both A and B, times indicated are relative to transcriptional shut off (i.e., glucose addition; t=0). In C, times indicated are relative to cycloheximide addition. In B and C cultures were split at the time of cycloheximide addition and thus share the t=0 time point. Plasmid-derived rRNAs were detected with 32P-labeled probe FL125 or FL126. Endogenous scR1 RNA, which served as a loading control, was also monitored by northern blotting. Indicated half-lives are the mean of at least three independent trials (error: standard deviation).
In contrast to its stabilizing effect on 18S:A1492C rRNA, cycloheximide did not prevent decay of 25S:U2585A rRNA. Instead, cycloheximide addition coincident with transcriptional shutoff appeared to shorten 25S:U2585A rRNA half-life (Figure 3B). Because 25S maturation is significantly slower than 18S maturation (Trapman and Planta, 1976; Udem and Warner, 1973), wild type 25S rRNA reaches its maximal level 30 minutes later than does 18S rRNA during transcriptional pulse-chase analysis (Cole and LaRiviere, 2008; LaRiviere et al., 2006). This is apparent in Figure 3A and B, where 25S rRNA levels, but not 18S rRNA levels, continue to increase 30 minutes after transcription shut off. To ensure that this apparent acceleration of mutant 25S rRNA decay by cycloheximide was not due to disruption of 25S rRNA synthesis, we delayed cycloheximide addition by 30 minutes relative to the transcriptional shutoff. Nonetheless, accelerated mutant 25S rRNA decay was still observed (Figure 3C). Additionally, at a concentration of hygromycin B that completely inhibited decay of 18S:A1492C rRNA, decay of 25S:U2585A rRNA was still observed (Figure S1). Therefore decay of the mutant 18S rRNA (18S NRD) is more sensitive to translation elongation inhibitors than decay of the mutant 25S rRNA (25S NRD).
The role of exonucleases in NRD
We next tested a set of known exonucleases for their involvement in 18S and 25S NRD. Using transcriptional pulse-chase analysis as above, we measured the stability of plasmid-derived 18S:A1492C and 25S:U2585A rRNAs in the absence of the nonessential exonucleases Rrp6p and Xrn1p and the cytoplasmic exosome recruitment factor Ski7p (Figure 4A and B). To account for any growth rate differences between strains, all half-life calculations were corrected for cell division. Deletion of RRP6 had no significant effect on the decay rate of either 18S:A1492C or 25S:U2585A rRNA. Similarly, no significant change in 25S:U2585A rRNA half-life was observed in the ski7Δ and xrnlΔ strains. In contrast, deletion of SKI7 led to a twofold stabilization of mature 18S:A1492C rRNA, while deletion of XRN1 led to the appearance of an approximately 1kb 18S:A1492C rRNA fragment († in Figure 4A). Despite this apparent decay intermediate, the half-life of full-length 18S:A1492C rRNA was unchanged in this S288C-based xrn1Δ strain. However, in a different strain background (W303), deletion of XRN1 did significantly stabilize full-length 18S:A1492C rRNA compared with its isogenic wild type strain (Figure S2).
Figure 4.

Analysis of 18S and 25S NRD in exonuclease-deficient strains. (A) Transcriptional pulse-chase and northern analysis of 18S:A1492C rRNA expressed from pSC40-A1492C in wild type strain BY4741 or the indicated isogenic exonuclease-deficient strains. † indicates an 18S rRNA decay intermediate. (B) Same as A, except strains contained pSC40-U2585A expressing 25S:U2585A rRNA. (C) Transcriptional shut off and northern analysis of 18S:A1492C rRNA expressed from pSC40-A1492C in the temperature-sensitive rrp44-1 strain grown in galactose at the permissive temperature (23°C) or shifted to the non-permissive temperature (37°C) 3 hours prior to glucose addition. (D) Same as C, except strains contained pSC40-U2585A expressing 25S:U2585A rRNA. † indicates a 25S rRNA decay intermediate. In all panels, times indicated are relative to transcriptional shut off (i.e., glucose addition; t=0). Plasmid-derived rRNAs and endogenous scR1 RNA, which served as a loading control, were detected with 32P-labeled probes (FL125, FL126 and FL217). Histograms report average rRNA half-lives from at least three independent trials (error: standard deviation). Mutant strains exhibiting statistically significant differences (unpaired student t-test; p<0.01) in 18S:A1492C rRNA half-lives are indicated (*).
Because a portion of the 25S:U2585A FISH signal was coincident with the nuclear envelope, we also examined 25S:U2585A rRNA stability in a strain carrying a temperature sensitive allele of the essential nuclear 5′->3′ exonuclease Rat1p (Figure S3). At the non-permissive temperature (37°C), we observed no significant change in 25S:U2585A rRNA stability by transcriptional shutoff when compared to cells grown at the permissive temperature.
To directly test whether the core exosome participates in 18S and/or 25S NRD, we measured by transcriptional shut off the stability of 18S:A1492C rRNA and 25S:U2585A rRNA in a strain carrying a temperature sensitive allele of RRP44 (rrp44-1). At the restrictive temperature (37°C) there was no significant change in decay of either full-length 18S:A1492C or 25S:U2585A rRNA when compared to decay at the permissive temperature (23°C) (Figure 4C and D). However, at 37°C we did observe a decrease in smaller RNA products seen as a smear below both full-length 18S:A1492C and 25S:U2585A rRNAs (Figure S4), as well as stabilization of an approximately 2 kb fragment of 25S:U2585A rRNA (Figure 4D). This fragment is unique to 25S rRNA containing the U2585A mutation, as it was not observed upon analysis of wild type 25S rRNA in the rrp44-1 strain at the restrictive temperature (Figure S4). Taken together, these results indicate that while not rate limiting for the decay of full-length NRD substrates, the core exosome does contribute to both 18S and 25S NRD. Further, Ski7p and Xrn1p contribute to 18S NRD but not to 25S NRD.
The roles of mRNA decay factors in NRD
Since efficient 18S NRD requires the cytoplasmic mRNA decay proteins Xrn1p and Ski7p as well as ongoing mRNA translation, we hypothesized that it might be mechanistically related to one of the known mRNA quality control pathways. We therefore assessed the effects of deleting mRNA decay factors UPF1, DOM34 and HBS1 on 18S:A1492C rRNA half-life by transcriptional pulse-chase (Figure 5). Compared to isogenic wild type and upf1Δ strains, both of which degraded the 18S:A1492C rRNA with a half-life of approximately 100 minutes, stability of this rRNA was increased approximately two-fold in both the dom34Δ and hbs1Δ strains. Deletion of both DOM34 and HBS1 together had no additional effect on 18S:A1492C rRNA stability, suggesting that Dom34p and Hbs1p act together in the same pathway. However, deletion of both SKI7 and HBS1 resulted in a dramatic stabilization of 18S:A1492C rRNA, with no half-life detected during the time course. Deletion of both DOM34 and XRN1 also greatly increased the half-life of the 18S NRD substrate, although unlike in the ski7Δhbs1Δ strain, 18S:A1492C rRNA decay still occurred. Further, in the absence of both Dom34p and Xrn1p, the 18S:A1492C rRNA intermediate seen in the absence of Xrn1p alone was still observed (Figure 5C). Lastly, none of the single or double deletions tested significantly affected 25S:U2585A rRNA half-life (Figure 5B and S5).
Figure 5.

Analysis of 18S and 25S NRD in strains lacking mRNA decay proteins. (A) Transcriptional pulse-chase analysis of 18S:A1492C rRNA expressed from pSC40-A1492C in wild type strain BY4741 or indicated isogenic mutant strains. (B) Same as A, except strains contained pSC40-U2585A expressing 25S:U2585A rRNA. (C) Same as A. Double mutant strains are isogenic to BY4741. In A and B, the isogenic wild type time courses shown are the same as those in Figure 4A and B; experiments in these panels were preformed concurrently. In all panels, times indicated are relative to transcriptional shut off (i.e., glucose addition; t=0). Plasmid-derived rRNAs and endogenous scR1 RNA, which served as a loading control, were detected with 32P-labeled probes (FL125, FL126 and FL217). Histograms report average rRNA half-lives from at least three independent trials (error: standard deviation). Mutant strains exhibiting statistically significant differences (unpaired student t-test; p<0.01) in 18S:A1492C rRNA half-lives are indicated (*).
Taken together with the exonuclease data, the above results confirm that 18S NRD is mechanistically distinct from 25S NRD. Further, the dependence of 18S NRD on Ski7p, Xrn1p, Dom34p and Hbs1p, which have all been implicated in NGD, indicates that 18S NRD is mechanistically related to NGD.
Localization of 18S NRD and NGD substrates
In eukaryotic cells, mRNA decay has been linked to P-bodies (Parker and Sheth, 2007). P-body size and abundance increases upon XRN1 deletion, which stabilizes mRNA decay intermediates (Sheth and Parker, 2003, 2006). Since an apparent 18S rRNA decay intermediate was observed in xrn1Δ strains, we used FISH to determine whether 18S NRD substrate localization changes in the absence of Xrn1p. In contrast to their diffuse cytoplasmic localization in wild type cells, 18S:G530U and 18S:A1492C rRNAs accumulated in distinct cytoplasmic foci xrn1Δ cells (Figure 6A). In contrast, no significant change in wild type 18S rRNA, wild type 25S rRNA or 25S NRD substrate localization was observed in xrn1Δ cells (Figure 6A and S6).
Figure 6.

FISH analysis of 18S NRD and NGD substrates. (A) xrn1Δ strain (YGL173C) carrying pJV12-WT (18S:wild type), pJV12-G530U (18S:G530U) or pJV12-A1492C (18S:A1492C) analyzed by FISH using Alexa 594-labeled probe FL125. (B) FISH analysis of xrn1Δ strain (HFY1081) carrying reporter plasmid PGK1 (pRP469) or PGK1-SL (pRP1251) using Alexa 647-labeled probe RP141. (C) xrn1Δ strain (HFY1081) carrying pWL160-A1492C and PGK1-SL (pRP1251) analyzed by FISH using Alexa 647-labeled probe RP141 and Cy3-labeled probe FL125. (D) FISH analysis of xrn1Δ DCP2-GFP strain (yRP1923) carrying pWL160-A1492C using Cy3-labeled probe FL125. (E) FISH analysis of dcp1Δ DHH1-GFP strain (yRP1736) carrying pSC39 or pWL160-A1492C using Alexa 594-labeled probe FL125.
Deletion of XRN1 is also known to stabilize the 3′ half of mRNAs cleaved during NGD (Doma and Parker, 2006). Since efficient 18S NRD requires the same proteins as NGD, we next examined the spatial relationship between 18S:A1492C rRNA and PGK1-SL mRNA in the absence of Xrn1p. PGK1-SL mRNA is identical to PGK1 reporter mRNA except for a stem loop within the open reading frame that induces NGD. Both PGK1-SL and PGK1 reporters are distinguishable from endogenous PGK1 mRNA via a unique 3′-UTR sequence tag (Doma and Parker, 2006). In cells lacking Xrn1p, the PGK1 reporter had a low but detectable signal that was dispersed throughout the cytoplasm, consistent with the localization of endogenous PGK1 mRNA by FISH (Hurt et al., 2000). In contrast, the PGK1-SL mRNA accumulated in distinct cytoplasmic foci (Figure 6B), which colocalized with foci containing 18S:A1492C rRNA (Figure 6C).
To determine whether the cytoplasmic foci containing 18S NRD substrates are P-bodies, we performed FISH for 18S:A1492C rRNA in an xrn1Δ strain expressing GFP-tagged Dcp2p. Dcp2p is a component of the Dcp1p/Dcp2p decapping complex that removes 5′ cap structures from mRNAs prior to their decay, and its localization is highly concentrated in P-bodies (Dunckley and Parker, 1999; Sheth and Parker, 2003, 2006). It was previously reported that P-bodies, as monitored by GFP-tagged Dcp2p localization, are sensitive to in situ hybridization conditions, resulting in decreased P-body abundance (Brengues and Parker, 2007). Under our standard FISH conditions, we also observed this same phenomenon (data not shown). However, by modifying the fixation and hybridization protocols (see Methods), we were able to establish conditions in which Dcp2p-positive P-bodies could still be observed in most cells, albeit at reduced numbers per cell (data not shown). Using these conditions, we observed that the surviving P-bodies marked by GFP-tagged Dcp2p were coincident with the foci containing 18S:A1492C rRNA (Figure 6D). We therefore conclude that both 18S NRD and NGD substrates colocalize in the same cytoplasmic foci, and these foci are P-bodies.
It is possible that the accumulation of 18S NRD substrates in P-bodies in the absence of Xrn1p is merely a consequence of the increased P-body size and abundance in this strain. To address this possibility, we performed FISH for 18S:A1492C rRNA in cells lacking the mRNA decapping factor Dcp1p. These cells have large and abundant P-bodies (Figure S6) presumably due to a general inhibition of mRNA decay (Sheth and Parker, 2003). In this strain 18S:A1492C rRNA exhibited diffuse cytoplasmic staining indistinguishable from wild type 18S rRNA (Figure 6E). This indicates that accumulation of 18S NRD substrates in P-bodies in the xrn1Δ strain is a specific effect of deleting XRN1 and not an indirect consequence of increasing P-body abundance.
Discussion
Although much attention has been paid to cytoplasmic mRNA surveillance pathways, it is now becoming apparent that stable RNAs such as rRNAs and tRNAs are also subject to quality control in the cytoplasm. For example, a recent study in S. cerevisiae examining tRNA stability revealed that otherwise mature but hypomodified tRNAs are subject to elimination via the RTD pathway. Involvement of the cytoplasmic 5′->3′ exonuclease Xrn1p in this process suggests that least a portion of RTD occurs in the cytoplasm (Chernyakov et al., 2008). Our findings here indicate that mutant rRNAs capable of assembling into ribosomal subunits but incapable of supporting efficient translation are also eliminated in the cytoplasm.
Previous data supporting the idea that 18S NRD is a cytoplasmic process included the observation that NRD acts on mature 18S rRNA (LaRiviere et al., 2006), which is only generated upon 20S pre-rRNA cleavage in the cytoplasm (Fromont-Racine et al., 2003). Further, we had found that 18S NRD substrates co-sediment with both monosomes and polysomes (LaRiviere et al., 2006). Consistent with the latter, we show here that in wild type cells 18S NRD substrates exhibit an intracellular localization pattern indistinguishable from wild type rRNAs. Additionally, inhibition of 18S NRD by both cycloheximide and hygromycin B indicates that the process requires ongoing translation elongation. We also identified the major cytoplasmic 5′->3′ exonuclease Xrn1p, the core exosome, the cytoplasmic exosome recruitment factor Ski7p and the cytoplasmic eRF3- and eRF1-like proteins Hbs1p and Dom34p as key 18S NRD participants. Finally, in xrn1Δ strains, we find that 18S NRD substrates accumulate in P-bodies. Therefore, we conclude that 18S NRD is a cytoplasmic process.
18S NRD factors Xrn1p, Ski7p, Dom34p and Hbs1p were previously shown to participate in the NGD mRNA quality control pathway (Doma and Parker, 2006). However, where in the cytoplasm NGD occurs had not been previously examined. We find here that an NGD substrate colocalizes with our 18S NRD substrate in P-bodies. In addition to implicating these structures as the site of NGD substrate decay, our data indicate that rRNA can also localize to P-bodies. Prior to this work, the presence of other RNAs in P-bodies had not been examined. It was thought that ribosomes are excluded from these structures, since ribosomal proteins fail to colocalize with known P-body components, and P-bodies contain non-translating mRNAs (Teixeira et al., 2005). Consistent with the idea that P-bodies do not contain significant quantities of ribosomes, we find that the diffuse cytoplasmic localization pattern of wild type rRNAs is unchanged even when P-body size and abundance are increased by the absence of either Xrn1p or Dcp1p. We propose that the presence of 18S NRD substrates or decay intermediates in P-bodies is a consequence of these rRNAs being specifically targeted there, and P-bodies also function in aberrant rRNA elimination.
Given all the similarities between 18S NRD and NGD, it is likely that what is being recognized in both cases is a ribosome inappropriately stalled on an mRNA due to an inability to elongate. In the case of 18S NRD, this stalling is due to a defect in the decoding center of the small ribosomal subunit, whereas in the case of NGD, it is due to some structural barrier in the mRNA that prevents ribosome translocation. In both cases, the stalled ribosome contains a sense codon within the A-site. Hbs1p and Ski7p are both members of the eEF-1A-like GTPase family, which includes elongation factor eEF-1A (EF-Tu in bacteria) and termination factor eRF3 (RF3 in bacteria) (Benard et al., 1999; Nelson et al., 1992; van Hoof et al., 2002). Our finding that deletion of both HBS1 and SKI7 enhanced the stabilization of 18S:A1492C rRNA compared to deletion of either gene alone suggests that these two proteins facilitate 18S NRD via separate pathways. One possibility is that Hbs1p and Ski7p compete for interaction with the empty A-site of a ribosome stalled at a sense codon and that interaction with either protein is sufficient to initiate 18S NRD/NGD. Although our current data do not directly address either the fate of the mRNA bound to a NRD substrate or the fate of the ribosome bound to an NGD substrate, it seems reasonable to propose that both the bound mRNA and at least the 18S rRNA of the stalled ribosome are targeted together to P-bodies where they are degraded (Figure 7).
Figure 7.
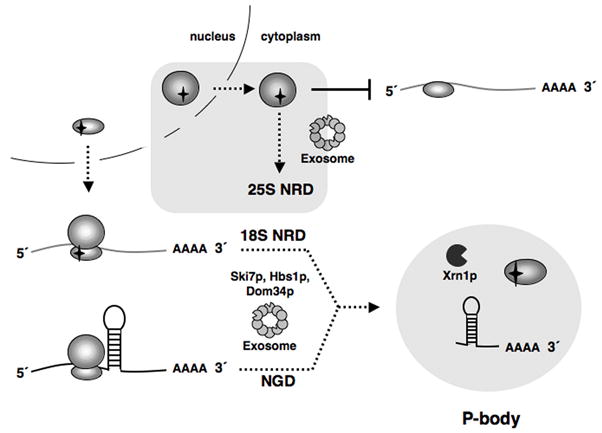
Models for 18S NRD and 25S NRD. Upon export from the nucleus, 18S NRD substrates are able to engage in translation. However, deleterious decoding site mutations cause these defective 40S subunits to stall on mRNAs, a scenario similar to ribosome stalling on an NGD substrate due to a structural hindrance in the mRNA. In both cases, the stalled translation complexes are eliminated through the combined action of Dom34p, Hbs1p, Ski7p, Xrn1p and the cytoplasmic exosome. 5′->3′ decay of 18S NRD and NGD substrates occurs in P-bodies. Conversely, 25S NRD substrates, which accumulate around the nuclear envelope, are eliminated after export to the cytoplasm in a process not requiring ongoing translation elongation but involving the core exosome.
In contrast to the combination of HBS1 and SKI7 deletions, double deletion of HBS1 and DOM34 had no additive effect on 18S NRD over deletion of either gene alone. This is consistent with Dom34p being an accessory factor for Hbs1p. Dom34p is homologous to eRF1 (Davis and Engebrecht, 1998) and has been shown to interact with Hbs1p in vivo, as well as to enhance Hbs1p’s affinity for GTP in vitro (Carr-Schmid et al., 2002; Graille et al., 2008). Interestingly, Dom34p has been shown to have endonuclease activity in vitro, and this endonuclease activity was proposed to be responsible for NGD substrate cleavage (Lee et al., 2007). The presence of an 18S rRNA decay intermediate in the absence of Xrn1p could suggest that the 18S:A1492C rRNA is cleaved endonucleolytically during 18S NRD. However, because this intermediate is still observed in xrn1Δdom34Δ cells, its generation does not require Dom34p.
Enhanced stabilization of 18S:A1492C rRNA in the xrn1Δdom34Δ strain compared to the single mutant strains suggests that Xrn1p’s function in 18S NRD is independent of Dom34p (and by inference Hbs1p). Unfortunately, we were unable to determine the effect of simultaneously deleting XRN1 and SKI7 as these genes proved synthetically lethal in our strain background (Figure S7).
In contrast to 18S NRD, 25S NRD appears unrelated to any of the known translation-dependent mRNA decay pathways. Unlike NMD, NSD, NGD and 18S NRD, 25S NRD still occurs in the presence of translation elongation inhibitors. Further, 25S NRD substrates accumulate around the nuclear envelope as opposed to the dispersed cytoplasmic localization observed for 18S NRD substrates in wild type cells. Finally, 25S NRD is independent of all mRNA decay factors tested, with the exception of the core exosome exonuclease Rrp44p. Thus 25S NRD occurs by a different pathway from 18S NRD.
That the nuclear exosome exonuclease Rrp6p is dispensable for 25S NRD suggests that 25S NRD is not a nuclear process related to TRAMP-mediated pre-rRNA decay. Supporting this idea, 25S:A2451G and 25S:U2585A rRNAs do not accumulate in either the nucleolus or nucleoplasm. The lack of any discernible effect of inactivating Rat1p on 25S NRD lends further support to the idea that 25S NRD occurs subsequent to nuclear export. Since we saw neither stabilization of 25S:U2585A rRNA nor appearance of the Rrp44p-dependent degradation intermediate upon deletion of Ski7p, 25S NRD must recruit the exosome independent of Ski7p.
Previously we had shown that, at steady state, 25S NRD substrates co-sedimented primarily with 60S ribosomal subunits and were largely absent from monosome and polysome fractions (LaRiviere et al., 2006). Here we showed that 25S NRD substrates accumulate close to the nuclear envelope, as opposed to the dispersed cytoplasmic distribution observed for translating wild type 25S rRNAs. Both of these observations are consistent with our finding that 25S NRD occurs in the presence of cycloheximide and hygromycin B. Our data suggest that ribosomal subunits containing 25S NRD substrates fail to enter the translationally active pool. Once exported from the nucleus, 60S subunits undergo a number of cytoplasmic maturation steps involving the release of some nuclear acquired proteins and acquisition of cytoplasmic factors prior to becoming translationally competent (Zemp and Kutay, 2007). It is possible that 25S NRD is a consequence of the failure of one or more of these late maturation proteins to either efficiently bind or release newly synthesized 60S subunits containing a NRD substrate. Such late quality control steps could function to prevent 60S subunits with a functional defect in the peptidyl transferase center from interfering with normal protein synthesis.
A common feature of the NMD, NGD and NSD mRNA quality control pathways is that they all rely on the same basic decay machinery involved in bulk mRNA turnover, but with the addition of specific targeting factors. Yet in comparison to our in depth understanding of general and targeted mRNA turnover, remarkably little is known about cytoplasmic rRNA decay. This is due in part to the extreme stability of wild type rRNAs in normally growing cells. Although bulk ribosome turnover in eukaryotic cells is known to occur during apoptosis, starvation, UV and chemical stress (Casati and Walbot, 2004; Kraft et al., 2008; Mroczek and Kufel, 2008), the pathways involved are just beginning to be understood. It will be of interest to determine if and how 18S and 25S NRD relate to these pathways, and whether there exist even more surveillance mechanisms that ensure the integrity of the translational machinery.
Materials and Methods
Yeast strains and plasmids
Strains and plasmids are described in the Supplemental Data and listed in Table S1.
Fluorescent In Situ Hybridization and Immunofluorescence
FISH analysis was adapted from Singer and colleagues (Long et al., 1995). Oligonucleotides probes are listed in Table S2. For FISH analysis of plasmid-derived rRNA, cells were grown to early log phase (OD600 of 0.3–0.5) in the appropriate dropout medium and fixed with 4% formaldehyde and 10% glacial acetic acid for 15 minutes at room temperature. Cells were pelleted three times and resuspended in 1.2 M sorbitol, 0.1M potassium phosphate, pH 6.5 (Buffer A) and adhered to poly-L-lysine coated slides for 60 minutes at 4°C. Cells were washed for 5 minutes at room temperature in Buffer A plus 0.2% β-mercaptoenthanol. Spheroblasts were generated by incubating cells in Buffer A plus 20 mM vanadyl-ribonucleoside, 5% zymolyase 20T, 1% DTT and 1000 U/mL of lyticase for 20 minutes at 30°C. Cells were washed twice for 5 minutes at room temperature in Buffer A, twice in 0.1 M potassium phosphate, pH 6.5 and incubated overnight at −20°C in 70% ethanol. After rehydration in two washes of 2 × SSC, cells were incubated for a minimum of 15 minutes at room temperature in 0.4 M urea, 2 × SSC. Cells were hybridized overnight at 30°C in a solution of 20% dextran sulfate, 2 mM vanadyl-ribonucleoside, 0.02% RNAse-free BSA, 25 μg yeast tRNA, 25 μg salmon sperm DNA, 0.5 M urea, 2 × SSC and 30 ng of Alexa 594 5′-end labeled oligonucleotide FL125 or FL126 (IDT DNA). Cells were washed twice for 15 minutes at 30°C in 0.4 M urea, 2 × SSC and then once for 15 minutes in 2 × SSC, 0.1% Triton X-100, twice for 15 minutes in 1 × SSC, and once for 5 minutes in 1 × PBS, all at room temperature. Coverslips were mounted with Prolong Gold Antifade reagent with DAPI (Invitrogen).
FISH analysis of PGK1 and PGK1-SL mRNA was done as above using Alexa 647 5′-end labeled oligonucleotide RP141 (IDT DNA). In cells analyzed for both plasmid-derived 18S rRNAs and PGK1 mRNAs, Cy3 5′-end labeled FL125 and Alexa 647 5′-end labeled RP141 oligonucleotides were used. FISH for plasmid-derived rRNAs in xrn1Δ DCP2-GFP strain (yRP1923) was preformed as above except acetic acid was omitted from fixation and cells were hybridized in buffer containing 25% formamide instead of urea with Cy3 5′-end labeled oligonucleotide FL125 (IDT DNA).
Immunofluorescence was preformed prior to FISH. After overnight incubation in 70% ethanol, cells were rehydrated in 1X PBS and blocked in 1% RNAse-free BSA, 1X PBS (Buffer B) for one hour. To detect Nop1p, cells were incubated with Mouse IgG1 anti-Fibrillarin antibody (Novus Biologicals) diluted in Buffer B for one hour and washed three times in Buffer B. Cells were then incubated with Alexa 488 Goat anti-Mouse IgG (Invitrogen) diluted in Buffer B for one hour and washed twice in Buffer B and 1X PBS. The protocol for nuclear pore O-linked glycoprotein immunofluorescence was the same except washes in Buffer B contained 0.1% NP-40, primary antibody (Abcam) was diluted in 1% RNase-free BSA, 1.2 × PBS, and the secondary antibody was an Alexa 488 Goat anti-Mouse IgM (Invitrogen). All washes were for 5 minutes at room temperature; all incubations were at room temperature.
Cells lacking an rDNA or mRNA reporter plasmid were used as negative controls for FISH. Primary antibody was omitted for immunofluorescence negative controls. Images were acquired on an inverted Olympus IX70 microscope in multiple focal planes (z = 0.2 μm, 25–30 planes), deconvolved and projected in two dimensions using a Delta Vision microscopy system and accompanying software.
Northern analysis
For northern blots, 2.5–5 μg total RNA was separated on 1% agarose-formaldehyde gels as described (Brown, 2004). Membranes were hybridized with 32P-end-labeled probes (Table S2) for 4–12 hours in ExpressHyb (BD Biosciences). Signals were visualized and quantified using a Storm 840 Phosphorimager (Molecular Dynamics) and normalized for loading to endogenous scR1 RNA (Galani et al., 2004).
Half-life analysis
Transcriptional pulse-chases analyses of plasmid-derived rRNAs were done as previously described (Cole and LaRiviere, 2008). For transcriptional pulse-chase analyses in the presence of cycloheximide, cycloheximide was added at the indicated time points to a final concentration of 0.1 mg/mL. For transcriptional shut off analysis in the rrp44-1 and rat1-1 strains (Amberg et al., 1992; Bousquet-Antonelli et al., 2000), cells were grown in SC-ura containing 2% galactose to early log phase (OD600 = 0.30-0.40) at 23°C. Prior to transcription inhibition, half of the cultures were resuspended in SC-ura plus 2% galactose pre-warmed to 42°C and placed at 37°C for 3 hours as described previously. Transcription was inhibited by the addition of glucose to a final concentration of 2%. RNA from aliquots taken at indicated time points was analyzed by northern blot as above. During transcriptional pulse-chase and shut off experiments, cell division was monitored by OD600 to account for dilution of plasmid-derived rRNA during the time course. Corrected data were fit to single or to double rate equations using KaleidaGraph® (Synergy Software); half-lives were calculated using these equations. Reported half-lives are the averages of at least three independent trials.
Supplementary Material
Acknowledgments
We thank A. Jacobson (University of Massachusetts, Worcester), R. Parker (University of Arizona, Tucson), D. Tollervey (University of Edinburgh) and J. Haber and M. Rosbash (Brandeis University) for yeast strains and plasmids. We thank J. Haber for hosting S.E.C. in his laboratory and J. Lydeard and N. Ramirez for technical assistance. We also thank A. Jacobson for critically reading this manuscript. F.J.L. was a postdoctoral fellow of the Damon Runyon Cancer Research Foundation. S.E.C. was supported by NIH training grant # 2 T32 GM007122-31A1. M.J.M. is an HHMI Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amberg DC, Goldstein AL, Cole CN. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- Benard L, Carroll K, Valle RC, Masison DC, Wickner RB. The ski7 antiviral protein is an EF1-alpha homolog that blocks expression of non-Poly(A) mRNA in Saccharomyces cerevisiae. J Virol. 1999;73:2893–2900. doi: 10.1128/jvi.73.4.2893-2900.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Presutti C, Tollervey D. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000;102:765–775. doi: 10.1016/s0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Brengues M, Parker R. Accumulation of polyadenylated mRNA, Pab1p, eIF4E, and eIF4G with P-bodies in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2592–2602. doi: 10.1091/mbc.E06-12-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T, Mackey K, Du T. Analysis of RNA by Northern and Slot Blot Hybridization. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2004. [Google Scholar]
- Carr-Schmid A, Pfund C, Craig EA, Kinzy TG. Novel G-protein complex whose requirement is linked to the translational status of the cell. Mol Cell Biol. 2002;22:2564–2574. doi: 10.1128/MCB.22.8.2564-2574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati P, Walbot V. Crosslinking of ribosomal proteins to RNA in maize ribosomes by UV-B and its effects on translation. Plant Physiol. 2004;136:3319–3332. doi: 10.1104/pp.104.047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008;22:1369–1380. doi: 10.1101/gad.1654308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SE, LaRiviere FJ. Analysis of non-functional rRNA decay in Saccharomyces cerevisiae. Methods Enzymol. 2008;449:239–259. doi: 10.1016/S0076-6879(08)02412-9. [DOI] [PubMed] [Google Scholar]
- Davis L, Engebrecht J. Yeast dom34 mutants are defective in multiple developmental pathways and exhibit decreased levels of polyribosomes. Genetics. 1998;149:45–56. doi: 10.1093/genetics/149.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dez C, Houseley J, Tollervey D. Surveillance of nuclear-restricted pre-ribosomes within a subnucleolar region of Saccharomyces cerevisiae. Embo J. 2006;25:1534–1546. doi: 10.1038/sj.emboj.7601035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. RNA quality control in eukaryotes. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. Embo J. 1999;18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziembowski A, Lorentzen E, Conti E, Seraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nature structural & molecular biology. 2007;14:15–22. doi: 10.1038/nsmb1184. [DOI] [PubMed] [Google Scholar]
- Fang F, Phillips S, Butler JS. Rat1p and Rai1p function with the nuclear exosome in the processing and degradation of rRNA precursors. Rna. 2005;11:1571–1578. doi: 10.1261/rna.2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M, Senger B, Saveanu C, Fasiolo F. Ribosome assembly in eukaryotes. Gene. 2003;313:17–42. doi: 10.1016/s0378-1119(03)00629-2. [DOI] [PubMed] [Google Scholar]
- Galani K, Nissan TA, Petfalski E, Tollervey D, Hurt E. Rea1, a dynein-related nuclear AAA-ATPase, is involved in late rRNA processing and nuclear export of 60 S subunits. J Biol Chem. 2004;279:55411–55418. doi: 10.1074/jbc.M406876200. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature. 2004;429:575–578. doi: 10.1038/nature02559. [DOI] [PubMed] [Google Scholar]
- Graille M, Chaillet M, van Tilbeurgh H. Structure of yeast Dom34: a protein related to translation termination factor eRF1 and involved in No-Go decay. J Biol Chem. 2008 doi: 10.1074/jbc.M708224200. [DOI] [PubMed] [Google Scholar]
- Green R, Samaha RR, Noller HF. Mutations at nucleotides G2251 and U2585 of 23 S rRNA perturb the peptidyl transferase center of the ribosome. J Mol Biol. 1997;266:40–50. doi: 10.1006/jmbi.1996.0780. [DOI] [PubMed] [Google Scholar]
- Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nat Rev Mol Cell Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. Rna. 2008;14:2609–2617. doi: 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt E, Strasser K, Segref A, Bailer S, Schlaich N, Presutti C, Tollervey D, Jansen R. Mex67p mediates nuclear export of a variety of RNA polymerase II transcripts. J Biol Chem. 2000;275:8361–8368. doi: 10.1074/jbc.275.12.8361. [DOI] [PubMed] [Google Scholar]
- Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell. 2006;24:619–626. doi: 10.1016/j.molcel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Lee HH, Kim YS, Kim KH, Heo I, Kim SK, Kim O, Kim HK, Yoon JY, Kim HS, Kim do J, et al. Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol Cell. 2007;27:938–950. doi: 10.1016/j.molcel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Long RM, Elliott DJ, Stutz F, Rosbash M, Singer RH. Spatial consequences of defective processing of specific yeast mRNAs revealed by fluorescent in situ hybridization. Rna. 1995;1:1071–1078. [PMC free article] [PubMed] [Google Scholar]
- Mitchell P, Tollervey D. An NMD pathway in yeast involving accelerated deadenylation and exosome-mediated 3′-->5′ degradation. Mol Cell. 2003;11:1405–1413. doi: 10.1016/s1097-2765(03)00190-4. [DOI] [PubMed] [Google Scholar]
- Mroczek S, Kufel J. Apoptotic signals induce specific degradation of ribosomal RNA in yeast. Nucleic Acids Res. 2008;36:2874–2888. doi: 10.1093/nar/gkm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. Premature translational termination triggers mRNA decapping. Nature. 1994;370:578–581. doi: 10.1038/370578a0. [DOI] [PubMed] [Google Scholar]
- Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell. 1992;71:97–105. doi: 10.1016/0092-8674(92)90269-i. [DOI] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Powers T, Noller HF. Dominant lethal mutations in a conserved loop in 16S rRNA. Proc Natl Acad Sci U S A. 1990;87:1042–1046. doi: 10.1073/pnas.87.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Noller HF. Evidence for functional interaction between elongation factor Tu and 16S ribosomal RNA. Proc Natl Acad Sci U S A. 1993;90:1364–1368. doi: 10.1073/pnas.90.4.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 2006;125:1095–1109. doi: 10.1016/j.cell.2006.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocklein W, Piepersberg W. Binding of cycloheximide to ribosomes from wild-type and mutant strains of Saccharomyces cerevisiae. Antimicrobial agents and chemotherapy. 1980;18:863–867. doi: 10.1128/aac.18.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Araki Y, Sakuno T, Katada T. Interaction between Ski7p and Upf1p is required for nonsense-mediated 3′-to-5′ mRNA decay in yeast. Embo J. 2003;22:3951–3959. doi: 10.1093/emboj/cdg374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, Parker R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. Rna. 2005;11:371–382. doi: 10.1261/rna.7258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Kim DF, O’Connor M, Lieberman KR, Bayfield MA, Gregory ST, Green R, Noller HF, Dahlberg AE. Analysis of mutations at residues A2451 and G2447 of 23S rRNA in the peptidyltransferase active site of the 50S ribosomal subunit. Proc Natl Acad Sci U S A. 2001;98:9002–9007. doi: 10.1073/pnas.151257098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapman J, Planta RJ. Maturation of ribosomes in yeast. I Kinetic analysis by labelling of high molecular weight rRNA species. Biochim Biophys Acta. 1976;442:265–274. doi: 10.1016/0005-2787(76)90301-4. [DOI] [PubMed] [Google Scholar]
- Udem SA, Warner JR. The cytoplasmic maturation of a ribosomal precursor ribonucleic acid in yeast. J Biol Chem. 1973;248:1412–1416. [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon-anticodon helix by ribosomal RNA. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- Youngman EM, Brunelle JL, Kochaniak AB, Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- Zemp I, Kutay U. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 2007;581:2783–2793. doi: 10.1016/j.febslet.2007.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.