

Abstract
Arylphosphonium salts (APS) are compounds that have both lipophilic and cationic character, allowing them facile transport through plasma membranes or cell walls to accumulate in the cytoplasm or mitochondria of cells. APS molecules preferentially accumulate in tumor cells and are therefore under investingation as tumor imaging agents and mitochondrial targeting molecules. We have generated a systematic set of APS to study their ability to associate with DNA. The chemical structure of the APS determines the extent of its interaction with DNA and therefore its ability to aggregate the DNA. Also, APS compounds blocked DNA amplification in vitro at concentrations below the aggregation threshhold, corraborating the structure/interaction relationship. Furthermore, the extent of APS:DNA interaction strongly correlates with bacterial toxicity, implying that APS molecules may deter cellular metabolic DNA pathways. Finally, DNA repair deficient and DNA bypass polymerase deficient bacterial strains were screened for sensitivity to APS. Interestingly, no single pathway for the repair or tolerance of these compounds was solely responsible for APS mediated toxicity. Taken together, these findings suggest that APS compounds may be capable of targeting and regulating unchecked cell growth and therefore show potential applications as a chemotherapeutic agent.
Keywords: Lipophilic cation, phosphonium salt, DNA, toxicity
1. Introduction
Small molecules that interact with DNA have been and continue to be intensely investigated as potential therapeutics for ailments as diverse as cancer, psychosis and malaria [1]. However, these molecules can also lead to undesired genetic changes by inducing cell-cycle delays, mutations, genomic instability, and/or cytotoxicity, usually via covalent modification of DNA, RNA and protein. Molecules that associate non-covalently with DNA may also disrupt DNA structure and/or chromatin function. These molecules fall into three main categories: those that undergo electrostatic attraction to the anionic DNA backbone, those that interact with functional groups within the helical grooves and those that intercalate between two adjacent base pairs.
Lipophilic cations (salt compounds that are both hydrophobic and hydrophilic) act as phase transition mediators. Their cationic nature attracts these molecules to cell walls or lipid bilayers with negative energy potentials. The lipophilic nature then allows easy transport through the membrane. The net result is rapid accumulation of this family of compounds in the interior of cells or cellular compartments. Mitochondrial accumulation is of particular interest as a target for the disruption of cellular power supplies [2].
Substituted arylphosphonium salts (APS) are a class of positively-charged lipophilic cation compounds that accumulate in the mitochondria by this mechanism. The high proliferation rate of carcinoma cells produces more mitochondrial byproducts than normal eukaryotic cells and thus exacerbates the mitochondrial membrane potential difference. The accumulation of APS compounds into the mitochondria of cancerous cells is further increased [3], making them possible tumor imaging agents [4-6] and/or targets for chemotherapeutics. APS compounds selectively penetrate cells as diverse as rat glioma, human breast cancer, melanoma and cervical cancer over non-cancerous (normal) epithelial cells to accumulate in the higher negative environment within the mitochondria [6]. Indeed, the APS compound Tetraphenylphosphonium Chloride (TPP-Cl) accumulation in carcinoma cells is approximately fifty times greater than in their non-cancerous counterparts. This corresponds to an overall IC50 value below 50uM, within the pharmaceutical parameters for active compounds [3]. Moreover, novel phosphonium salts in which the lipophilic cation is conjugated to a known antioxidant species have recently been studied for their potential to decrease mitochondrial byproducts that cause cellular damage [7]. In this way, it may be possible to diminish cellular levels of reactive oxygen species, precluding oxidative damage to mitochondria and potentially mitigating neurodegenerative diseases.
In the studies described herein, we have synthesized a set of APS compounds similar to TPP-Cl [3] and evaluated each for the ability to associate with DNA and to induce bacterial toxicity. Using two independent assays, we found that APS compounds associate with DNA and that the association is directly dependent on APS chemical structure. The chemical structure of the APS molecules also determines their toxicity in bacteria. Furthermore, cellular tolerance of APS compounds was independent of mutation in BER, HR, NER and MMR DNA repair pathways and was not altered by defects in any single DNA bypass polymerase. These studies demonstrate a negative cellular outcome as a result of arylphosphonium salt association with DNA. Further studies with this class of compound are clearly warranted to assess their affect on tumor progression.
2. Materials and methods
2.1 Chemicals and solvents
Triphenylphosphine (CAS# 603-35-0), Benzylbromide (CAS# 100-39-0), (2-bromoethyl) benzene (CAS# 103-63-9), (3-bromopropyl) benzene (CAS# 637-59-2), Tetraphenyl phosphonium Bromide (TPP-Br; CAS# 2751-90-8), Cinnamyltriphenyl phosphonium Chloride (CTP-Cl; CAS# 1530-35-4), Methyl methanesulfonate (MMS, CAS# 66-27-3), Toluene (CAS# 108-88-3) and Dimethyl sulfoxide (DMSO, CAS# 67-68-5) were obtained from Sigma Aldrich (St. Louis, MO). DNA plasmid, p3X-FLAG was obtained from Sigma Aldrich (St. Louis, MO) and linearized with restriction endonuclease XbaI (NE Biolabs, Ipswich, MA).
2.2 Synthesis
For the synthesis of TPP bromine salts, molar equivalents of bromine compounds (benzylbromide, (2-bromoethyl)benzene, (3-bromopropyl)benzene) were added to triphenylphosphine in a minimum amount of hot toluene. The mixture was refluxed for 1-18 hours until a white precipitate appeared. The solid APS salt product was collected via vacuum filter, washed with cold toluene and dried overnight. All structures were confirmed by MP, IR spectroscopy (Perkin-Elmer, Sprectum 100 FT-IR Spectrometer), NMR spectroscopy (Bruker, 400MHz NMR Spectrometer) and MS spectroscopy (Applied Biosystems Inc, Mariner System 5311 Mass Spectrometer).
2.3 Molecular Mechanics Optimization
The MM+ molecular mechanics program in HyperChem Professional 7.5 software (Hypercube, Inc. Gainesville, FL) was used to generate geometrically optimized structures (gradient ≤ 0.09). The distances were measured from the phosphonium ion to the para carbon on the phenyl ring in group “Y”. Bond angles were measured to all four groups attached to the phosphonium ion. Settings for MM+ were as follows: MM+ options: electrostatic; bond dipoles, cutoffs; none, geometry optimization: algorithm; Polak-Ribiere options; termination code; RMS gradient 0.1 kcal/(Å mol) or 500 maximum cycles.
2.4 APS characterization
Benzyltriphenylphosphonium Bromide
White solid; mp 258°C; 1H NMR (d6-DMSO): δ5.17(d, 2H, J=16.0Hz), 6.96(d, 2H, J=2.0Hz), 7.21-7.32(m, 3H), 7.60-7.77 (m, 12H), 7.91 (m, 3H); IR: 3042.21 cm-1 (w), 2917.54 cm-1 (w), 2841087 cm-1 (w), 1591.34 cm-1 (w), 1491.52 cm-1 (w), 1436.0 cm-1 (m), 1400.9 cm-1 (w), 1109.27 cm-1 (s), 745.47 cm-1 (s), 687.52 cm-1 (s); ESI TOF MS: Parent Ion C25H22PBr: 353.0759 m/z.
2-phenylethyltriphenylphosphonium Bromide [8]
Glassy Solid; 1H NMR (d6-DMSO): δ2.88(m, 2H), 3.90(m, 2H), 7.10-7.30(m, 5H), 7.50-7.90 (m, 15H); IR: 3055.67 cm-1 (w), 2913.14 cm-1 (w), 2850.77 cm-1 (w), 1580.30 cm-1 (w), 1474.79 cm-1 (m), 1430.01 cm-1 (m), 1089.39 cm-1 (m), 1024.73 cm-1 (m), 740.80 cm-1 (s), 685.52 cm-1 (s); ESI TOF MS: Parent Ion C26H24PBr 367.4643 m/z.
3-phenylpropyltriphenylphosphonium Bromide
White solid; mp 180-210°C; 1H NMR (d6-DMSO): δ1.82(m, 2H), 2.80(m, 2H), 3.70(m, 2H), 7.10-7.61 (m, 10H), 7.69-7.89(m, 16H); IR: 3060.13 cm-1 (w), 3012.22 cm-1 (w), 1580.34 cm-1 (w), 1474.96 cm-1 (m), 1433.98 cm-1 (m), 1089.46 cm-1 (m), 1024.89 cm-1 (m), 741.05 cm-1 (s), 685.52 cm-1 (s); ESI TOF MS: Parent Ion C27H26PBr 381.4832 m/z.
2.5 Bacterial strains and media
All Escherichia coli strains were grown in Luria-Bertani (LB) broth or on LB agar plates supplemented with standard concentrations of antibiotic if appropriate. Repair deficient strains, AB2500 (urvA-, thyA-, deoB-), GM5560 (recA-), BW528 (xth-, nfo-) were generated from the parental strain AB1157 (Genotype: F- thr-1 leu-6 thi-1 sup E44 lac Y1 mal A1 ls gal K2 ara-14 xyl-5 mtl-1 pro A2 his-4 arg E3 str-31 tsx-33 sup-37) and were a gift from Dr. B.P. Engelward [9] (MIT, Cambridge, MA). All other strains were obtained from The Coli Genetic Stock Center (CGSC), Yale University (New Haven, CT)
2.6 DNA Gel Shift Assay
DNA plasmid p3X-FLAG was digested with XbaI restriction endonuclease and agarose gel purified. Stock concentrations of APS compounds were diluted in either de-ionized H2O or DMSO depending on compound solubility to 2X working solutions in order to maintain a standard volume throughout each experiment. The concentration range for each compound was experimentally determined. APS working dilutions were added to 76.9 ng of DNA and incubated at RT for 10 minutes. Aggregated DNA was separated from free DNA by electrophoresis through a 1% agarose gel (TAE, 115V, 75 min) and quantified with BioRad Quantity One software. Results represent mean values of a minimum of three independent trials.
2.7 qPCR Assay
Quantitative PCR was performed following manufacturer's instructions (BioRad, Inc., Hercules, CA) using purified plasmid DNA as template. Individual PCR wells were run on iCycler machinery and analyzed by iQ5 software. Each trial of APS dose was completed in triplicate. Three independent trials were completed. An rfu reading at approximately 100 was used as a reference point to determine mean threshold cycles.
2.8 Cytotoxicity Assay
Bacterial overnight cultures (2.0 ml) were inoculated into 50 ml fresh LB broth and cultured for 2-2.5 hours to log phase (OD600 of 0.4-0.6). Cells were used for experiments without washing. Aliquots of log-phase culture (100-500 ul) were exposed to APS salt dilutions for 1 hour at 37°C with gentle shaking. DMSO was used as the normalizing control culture. Final concentrations of APS salts and MMS controls are expressed in the corresponding figures. After exposure, each cell culture was diluted into fresh LB broth (1:10) and then serially diluted out to 1:1,000,000. Bacterial dilutions were then plated in 10ul dots onto LB agar plates and incubated overnight at 37°C. Viable cell colonies were imaged and counted using Quantity One software (BioRad, Inc.; Hercules, CA). All results were normalized to DMSO controls and presented as mean values of a minimum of three independent trials.
3. Results
3.1 APS structure suggests an alleviation of the sterically crowded cationic environment
To study the effect of APS chemical structure on their ability to interact with double stranded DNA, we synthesized a set of APS by systematically extending the distance to a fourth phenyl ring. The structures of tetraphenylphosphonium bromide (TPP-Br) and cinnamyltriphenylphosphonium chloride (CTP-Cl) (Sigma-Aldrich, St. Louis, MO) are shown in Fig 1. Bz-TPP, Et-TPP and Pr-TPP were synthesized by refluxing equimolar quantities of the target bromide compound with triphenylphosphine in a minimal quantity of toluene, Fig 1A. After 1-18 hours, the white phosphonium salt precipitated, was washed briefly in cold toluene and collected by vacuum filtration. Melting point, IR, 1H NMR, and LC-MS spectroscopy verified each target (not shown). APS compounds, as shown in Fig 1B, contain increasing numbers of methylene groups to elongate the distance between the positively charged phosphorus ion and the para carbon of the fourth phenyl group. Bond angles and atom distances were measured using Hyperchem software on structures that were geometrically optimized with molecular mechanics, Fig 1B. Measurements from the phosphonium ion to the para carbon of the three phenyl rings remained stable at ∼4.5 angstrom compared to distances to the fourth phenyl ring that increased from 4.5 in TPP-Br to a maximum of 8.0 angstrom in Pr-TPP. The average increase for each additional C-C bond was 1.15 angstrom, less than the expected 1.54, suggesting a sterically crowded chemical environment.
Figure 1.

Synthesis and structure of triarylphosphonium salts (APS). (A) Synthesis scheme: Reflux triphenylphosphine and the corresponding bromide (Benzyl bromide shown) in hot toluene until arylphosphonium salt precipitates. Wash with cold toluene, dry and characterize by IR, NMR and LC-MS. (B) Table of compounds, Y represents the starting bromide compound. Structures were geometrically optimized using molecular mechanics with Hyperchem software. Angstrom measurements from the phosphonium cation to the para carbon of the benzene ring are presented on the right. Abbreviations: TPP-Br, Tetraphenylphosphonium Bromide; Bz-TPP, Benzyl triphenylphosphonium Bromide; Et-TPP, 2-phenylethyl triphenylphosphonium Bromide; Pr-TPP, 3-phenylpropyl triphenylphosphonium Bromide; CTP-Cl, Cinnamyl triphenylphosphonium Chloride.
Bond angles surrounding the phosphonium ion also imply a sterically crowded environment. The TPP-Br compound is the most constricted, producing a bowtie-like geometry with 2 angles at 103.4° and 2 angles at 122.2°. This suggests that the cationic character of the TPP-Br may be partially obscured by the close proximity of four phenyl substituents thereby decreasing any association with the negatively charged DNA. Exchanging one phenyl group for the benzyl moiety alleviated some of this stress, decreasing the angle differential from 18.8° to 5.6° and producing the tetrahedral geometry expected for a quaternary phosphorus cation. Further extension of the arm did not dramatically alter this tetrahedral geometry, with angle differentials ranging from 0.97° to 4.1° (data not shown).
3.2 APS structure modulates the ability to associate with double stranded DNA
We tested the strength of APS association with DNA by measuring the ability of each compound to alter dsDNA mobility through an agarose gel matrix. Gel electrophoresis was used to separate the freely mobile DNA from the aggregated DNA remaining in the well. High concentrations of APS are required to coat the strands with hydrophobic APS compounds thereby facilitating the aggregation of multiple strands. The intermediate stages of aggregation were identifiable as a laddering of the DNA between the aggregated and free DNA extremes (Fig 2A). Free DNA was quantified via densitometry and plotted against APS concentration to obtain predictive equations (Fig. 2B). It is important to note that the association of APS with the DNA did not alter the ability of ethidium bromide to intercalate into the DNA, as overall DNA content per well remained constant. The decreased cationic nature of TPP-Br and Bz-TPP reduced their solubility in DMSO and hindered complete aggregation of the DNA. In contrast, Et-TPP, Pr-TPP, and CTP-Cl all associated with the DNA to sufficient levels for full aggregation to occur.
Figure 2.

Evaluation of DNA aggregation capabilities of APS compounds. (A) Representative data of CTP-Cl mediated aggregation of DNA. (B) Plots of the ratio of free DNA versus APS compound concentration. Mean values with error bars indicated were calculated from triplicate experiments. Lanes representing the absence of APS were set at 1.0 ratio free DNA. A background intensity of 0.4 persists. (C) Plot showing APS concentration needed to maintain 0.8 ratio of free DNA. (D) Correlation between APS chemical structures as shown by the angstrom distance of the P+-Y arm and the DNA aggregating ability. Linear trend reported r2 value of 0.9064.
To simplify comparisons, a plot of the concentration required to maintain a 0.8 ratio of free DNA was generated in Fig 2C. TPP-Br clearly demonstrated the weakest electrostatic association with DNA, needing 78.7 mM to reach the 0.8 ratio mark, while CTP-Cl exhibited the strongest association, needing only 2.8 mM. The DNA concentrations decreased in response to the extended arm length (Fig 2C), indicating a stronger association with the DNA as the arm is elongated. To assess the significance of the chemical structure on DNA association, we plotted the distance between the phosphonium cation and the extended phenyl para carbon (arm length) against the concentration needed to maintain a 0.8 ratio of free DNA (Fig 2D). There is a clear linear relationship with a strong correlation (r2 =0.9064).
3.3 APS compounds inhibit DNA amplification
We further probed the nature of the DNA:APS interaction by monitoring PCR product formation from plasmid DNA template as a function of APS concentration. Initial experiments using a standard thermocycler showed a complete block of DNA amplification at high concentrations of both CTP-Cl and TPP-Br (Fig 3A). This could be due to inhibition of the polymerase by APS directly or decreased availability of template resulting from APS induced aggregation of DNA. Alternatively, the inhibition of amplification may be due to APS compounds interfering with primer annealing. Therefore, we evaluated DNA amplification by qPCR to monitor DNA product formation in real time. DNA amplification via qPCR at high APS concentration confirmed the abrogation of amplification seen previously, in other words no amplification of DNA was detected (data not shown). We next evaluated equimolar APS concentrations below the aggregation threshold. These dilutions produced 100% freely mobile DNA by electrophoresis. Figure 3B is a plot of the differential amplification reported as total fluorescence per PCR cycle, while Fig 3C quantifies the end product for the same experiments. The greater the cycle number required to reach a fluorescence threshold indicates that there is less DNA amplification. CTP-Cl (circles) hindered amplification to a greater extent than did equimolar TPP-Br (squares), corroborating the greater strength of the CTP-Cl:DNA association. This result suggests either a reduced level of DNA template or a disruption of DNA-primer annealing as the polymerase responsible for amplification functioned well at TPP-Br concentrations well above polymerase concentration (Fig 3B and 3C).
Figure 3.
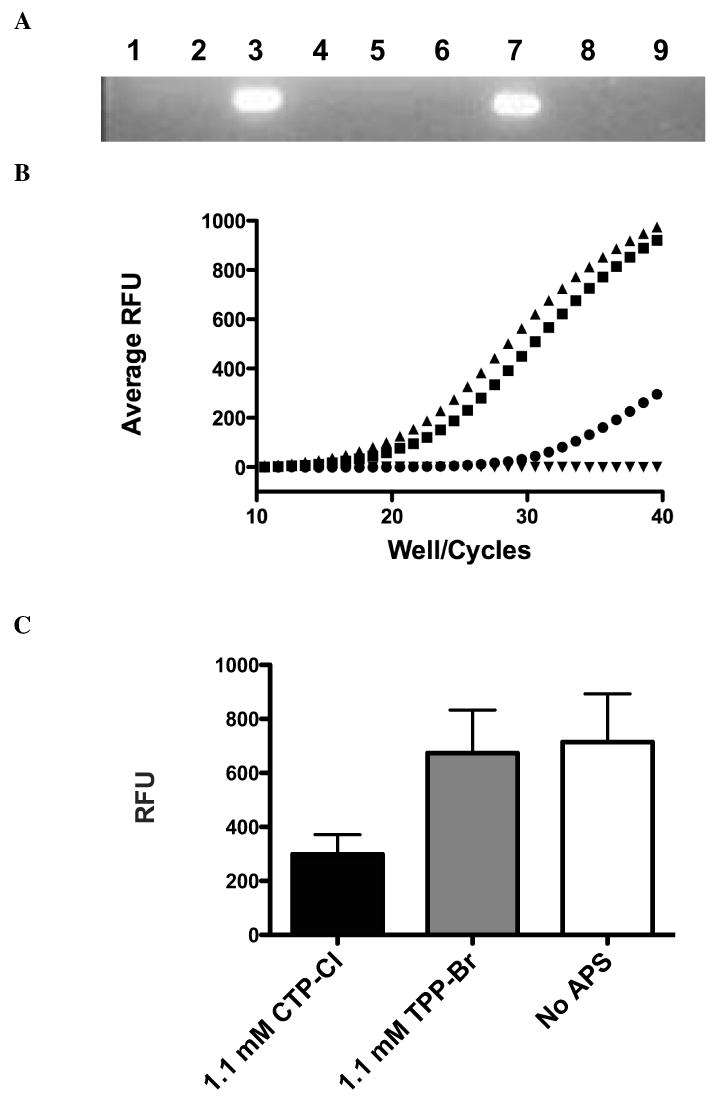
DNA Amplification Block by APS compounds: A) PCR amplification of plasmid DNA in the presence of 176mM CTP-Cl (lane 1), 17.6mM CTP-Cl (lane 2), No APS compounds (lane 3), 17.6mM CTP-Cl pre-incubated with primers (lane 4), no template (lane 5), 107mM TPP-Br (lane 6), No APS (lane 7), 10.7mM TPP-Br pre-incubated with primer (lane 8), 107mM TPP-Br (lane 9). Data presented is representative of three independent trials. B) DNA amplification via qPCR in the presence of 1.1 mM CTP-Cl (circles), 1.1 mM TPP-Br (squares), No APS compound (triangles) and no template (inverted triangles). Independent trials were run in triplicate. Representative sample of three trials is shown. C) Analysis of qPCR amplification endpoints for equimolar doses presented in 3B. The mean values of three independent trials with standard deviations indicated is shown.
3.4 Bacterial cytotoxicity increases in response to APS:DNA association
To study the effects of APS:DNA association on cellular growth, we conducted cytotoxicity assays. AB1157 bacterial cultures were exposed to 12 mM (final concentration) of each APS compound for one hour. Cells were then serially diluted and plated to assess the percentage of viable cells. Individual colonies were counted, normalized to DMSO exposed cells and the data is presented in Figure 4A. An equimolar concentration of the well-characterized DNA alkylating agent methylmethane sulfonate (MMS) was used to establish a baseline of toxicity. All APS compounds studied were significantly more toxic than MMS, possibly due to the phase-transitioning capabilities of APS compounds that allows greater access into the cellular compartment. CTP-Cl was the most toxic, 1000 times more toxic than TPP-Br and 10,000 times more toxic that MMS. Similar to the DNA aggregation data, cytotoxicity decreases with shorter arm lengths as Pr-TPP, Et-TPP and Bz-TPP produced viable cells between the extremes (Fig 4A). The APS concentration at 0.8 ratio of DNA binding (Fig 2C) was plotted against the percent growth of AB1157 at 12 mM for each target compound (Fig 4B), demonstrating a high correlation between APS compound's ability to associate with DNA and their cytotoxic characteristics. The r2 value calculated for this relationship is 0.8416.
Figure 4.
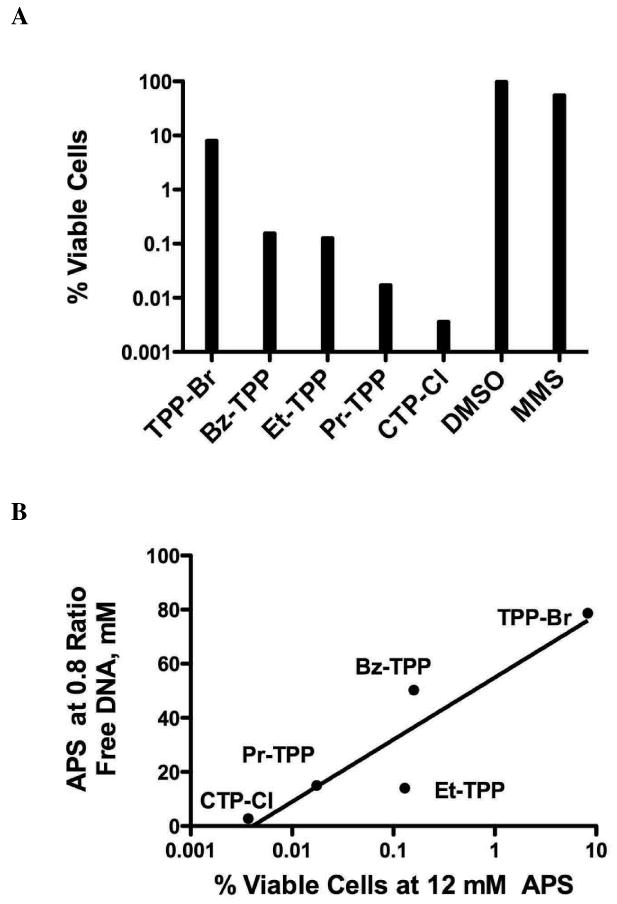
Cytotoxicity study. (A) Cytotoxicity at 12 mM APS. All compounds were normalized to DMSO. Methylmethanesulfonate (12mM), a known DNA alkylating agent, served as a toxicity baseline. Discrete colonies were counted from three independent trials. Results represent mean values of all trials. (B) Correlation between DNA interaction, shown in Figure 2C, and APS cytotoxicity. Logarithmic r2 value equals 0.8416.
3.5 DNA repair activity does not protect from APS-induced cytotoxicity
In general, molecules that interact through non-covalent means have been reported as mutagenic [10]. Mutation is generated through one of two processes: exposure to agents that induce DNA damage or nucleotide mis-incorporation during DNA replication. Therefore, to further evaluate the affect of DNA binding on cytotoxicity, isogenic E. coli strains deficient in known DNA damage repair pathways were tested with TPP-Br and CTP-Cl representing the range of DNA association strengths. AB1157 was used as the parental strain for GM5560, a homologous recombination deficient (recA-) strain; AB2500, a nucleotide excision repair (NER) deficient (uvrA-) strain and BW528, a base excision repair (BER) deficient (xth-/nfo-) strain (Fig 5A and Table 1). A DNA helicase deficient (recQ-) strain was screened against its isogenic parent W3110 (Fig 5B). Finally mismatch repair deficient strain JW4128-1 was compared to its parental strain of BW25113 (Fig 5C). Each strain was evaluated against a DMSO control to normalize for variations in cell growth due to genetic alterations. It should be noted that the bacterial growth assay, as performed herein, can not differentiate between growth inhibition and cell death. In other words, APS exposure may delay population doubling during the treatment period, allowing a normal growth pattern to resume upon removal of the APS compounds. For this assay, the percentage of viable cells resulting from a total growth inhibition would be 12.5%. Strains subjected to either 2.5 mM TPP-Br or 0.5 mM CTP-Cl grew to approximately 30-50% of their DMSO exposed counterparts, within the growth inhibition parameter. However, no genetic repair defect exhibited a significant increase in sensitivity as a function of APS concentration, including the GM5560 strain that initially appeared to foster cell growth post CTP-Cl exposure (Fig 5A). The two-tailed p value between GM5560 and its AB1157 parental cells is 0.2649 and therefore not statistically significant. Mean values for three independent trials are shown. The data is summarized in Table 1.
Figure 5.
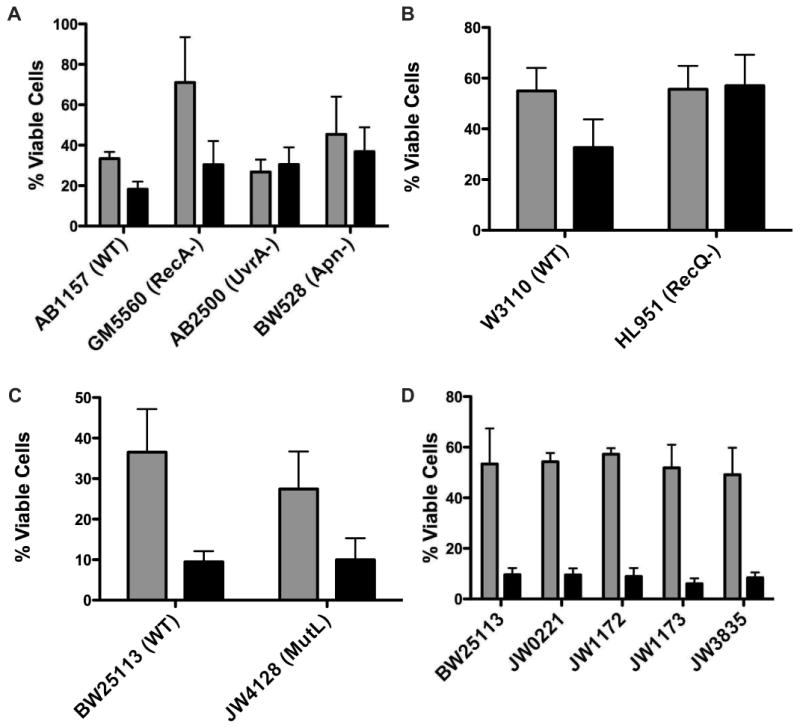
Bacterial Cytotoxicity of DNA Repair and Bypass Polymerase Deficient Strains. Gray bars report bacterial growth after exposure to 0.5 mM CTP-Cl while black bars report growth after exposure to 2.5 mM TPP-Br. Results are mean values of three independent experiments with standard error indicated. (A) AB1157 is parental strain for GM5560, AB2500 and BW528. GM5560 is deficient in recA, a critical Homologous Recombination repair pathway protein. AB2500 contains three mutations (uvrA, thyA and deoB) rendering these cells deficient in Nucleotide Excision Repair. BW528 has the two AP endonucleases, nth and xfo, deleted making this strain deficient in Base Excision Repair. (B) W3110 serves as parent strain for deletion of the replication fork stall stabilizing protein recQ (HL951). (C) BW25113 is parent strain for JW4128 that is deficient in mismatch repair (mutL). (D) Strains were obtained from Yale E. Coli database (Keio Collection) with single deletions as follows: BW25113, Parental strain; JW0221, dinB deficient; JW1172, umuC deficient; JW1173, umuD deficient and JW3835, polA deficient. Results are the mean values of three independent experiments with standard deviations indicated.
Table 1.
Summary of bacterial strains sensitivity to APS agents plotted in Figure 5.
| Strain Name | Genotype | Pathway Deficiency | Percent Viability to 0.5 mM CTP-Cl | Percent Viability to 2.5 mM TPP-Br |
|---|---|---|---|---|
| AB1157 | WT | 33.4 | 18.2 | |
| GM5560 | recA- | HRR | 71.0 | 30.4 |
| AB2500 | uvrA-/thyA-/deoB- | NER | 26.8 | 30.5 |
| BW528 | xth-/nfo- | BER | 45.3 | 36.8 |
| W3110 | WT | 55.0 | 32.7 | |
| HL951 | recQ- | HRR/DNA Stability | 55.6 | 57.0 |
| BW25113 | WT | 36.5 | 9.5 | |
| JW4128 | mutL- | MMR | 27.4 | 10.0 |
| BW25113 | WT | 53.4 | 9.6 | |
| JW0221 | dinB- | Bypass | 54.3 | 9.5 |
| JW1172 | umuC- | Bypass | 57.3 | 8.9 |
| JW1173 | umuD- | Bypass | 51.8 | 6.0 |
| JW3835 | polA- | Exonuclease involved in Bypass | 49.1 | 8.4 |
3.6 Bypass polymerases do not influence the mechanism of APS toxicity
We next explored the possibility that DNA bypass polymerases could influence the growth of bacteria exposed to APS compounds. Isogenic strains of the parental BW25113, containing single mutations in DinB (JW0221), umuC (JW1172), umuD (JW1173) or PolA (JW3835), were exposed to 2.5 mM TPP-Br or 0.5 mM CTP-Cl and evaluated for growth compared to a DMSO control (Fig 5D and Table 1). Deletion of any single bypass polymerase did not significantly effect the growth of bacteria when challenged with APS. It is possible that more than one polymerase may influence bacterial tolerance to APS compounds and therefore the complementary nature of bypass polymerase specificity may be mitigating the overall effect.
4. Discussion
Molecules that non-covalently interact with DNA are a large and diverse chemical group. As a general rule, this diverse family of compounds contain an electrostatic component and have been reported to block replication and induce genetic mutations [11,12]. Consequently, these compounds have been intensely investigated for mechanisms that will disrupt disease progression. Arylphosphonium salts are lipophilic cations that easily cross the lipid bilayer to accumulate inside cells. APS compounds are now being developed as potential tumor imaging agents [5,6] as well as derivatives of APS being developed to target mitochondrial DNA [2]. We investigated the potential of a systematic set of APS to associate with DNA as well as the repercussions of this interaction.
The cationic nature of APS molecules, i.e. the chemical structure surrounding the phosphonium ion directly relates to their ability to associate with DNA. The bond angles of the phosphonium ion in TPP-Br, and to a lesser extent Bz-TPP, suggest an environment that is sterically hindered by four large phenyl rings, thus shielding the cationic character of the phosphonium ion. This is confirmed by TPP-Br insolubility in water; requiring DMSO to obtain a concentration comparable to CTP-Cl. The shielding of the phosphonium cation would also account for the diminutive electrostatic association with DNA. Addition of one methylene group to the benzyl derivative reduced the stress surrounding the phosphonium ion, allowing Bz-TPP to adopt a more natural tetrahedral-like geometry. Consequently, the electrostatic association with DNA increases as the cationic nature of the compound is exposed. Indeed, the solubility in water and the strength of APS-induced DNA aggregation increased as a function of increasing arm length from TPP-Br to Bz-TPP (Fig 2C and 2D). Furthermore, it is possible that the high degree of association seen in the CTP-Cl compound could, in part, be augmentation of binding through intercalation of the fully conjugated (i.e., planar) cinnamyl moiety.
PCR experiments on plasmid DNA confirm the APS:DNA association trends. Both TPP-Br and CTP-Cl induced a complete replication block at very high concentrations. At lower, equimolar concentrations however, the strongest association (CTP-Cl) corresponded to decreased amplification while TPP-Br had no effect (Fig 3B and 3C). The differential amplification may result from a decrease in the availability of template, either through an aggregation of DNA strands or an increase of melting temperature for DNA:APS complexes. Alternatively, APS compounds may be masking the recognition sites for primer annealing thereby decreasing the total product formed. Finally, APS molecules may be blocking the elongation of the polymerase as it progresses across the template. It is unlikely that APS inhibit the polymerase directly as DNA amplification was unaffected by TPP-Br addition (Fig 3B and 3C), even at levels significantly above the polymerase concentration.
To understand the in vivo consequences of APS exposure and the possible pathways by which a cell can tolerate them, we next investigated the bacterial toxicity of our APS compounds. All APS compounds studied were significantly more toxic at equimolar concentrations than MMS, a well-characterized and highly toxic DNA alkylating agent. The increased toxicity of the APS compounds may be partially due to the phase-transitioning nature of these compounds. The combination of both lipophilic and cationic character of the APS family enables them to permeate through the plasma membrane of mammalian cells to accumulate in the cytoplasm [2]. A similar mechanism likely enables passage through the bacterium cell wall. Moreover, the general structure of a tetra-substituted phosphonium cation is relatively stable to further chemical modification in vivo. This implies that the degradation of these compounds is comparatively slow. A strong correlation was calculated between APS structure and bacterial toxicity as a result of APS exposure (Fig 4B), clearly suggesting that the chemical structure of these compounds (and their ability to interact with DNA) affect the toxicity profile in vivo. Taken together, a picture emerges of a family of compounds that have easy access to cellular compartments, are electrostatically attracted to DNA, and remain relatively stable in vivo. Under these circumstances, APS could accumulate to high enough concentrations for an association with the DNA to occur.
It should be noted that toxicities in bacteria generally occur at higher concentrations than those reported for mammalian cells; bacterial cell walls are more resistant to uptake of environmental components than plasma membranes found in multi-cellular organisms. MMS exemplifies this trend, 12mM MMS treated for just 15 minutes corresponds to ∼65% bacterial survival [13] while mouse embryonic fibroblasts required only 1mM MMS for similar survival [14,15] and CHO-K1 hamster cells respond to MMS at an IC50 value of .75 uM [16]. In fact, an IC50 value of 46 uM was reported for TPP-Cl [3]. Based on the studies herein, the IC50 value for CTP-Cl should be significantly lower.
Given the degree of toxicity resulting from APS exposure coupled with the decrease of DNA amplification in vitro, we probed for the mechanism of that toxicity by evaluating whether the DNA:APS interaction could be recognized, and hence corrected, by one of the well characterized DNA damage repair or damage tolerance pathways. Therefore, we screened both TPP-Br and CTP-Cl for growth sensitivity in E. coli strains deficient in DNA repair pathways (Fig 5A-C and Table 1). Homologous recombination is the primary pathway responsible for repair of double strand breaks and collapsed replication fork restart. RecA is the central player in the HR pathway and thus a good candidate for screening HR activity when a cell is exposed to APS compounds. Additionally, RecQ, the homolog of BLM that is responsible for Bloom Syndrome and is classified as a HR protein, was also screened for a possible role. Neither RecA null nor RecQ null strains showed any increased sensitivity to either TPP-Br or CTP-Cl. Furthermore, NER, the pathway for repair of bulky DNA lesions, and BER, the repair pathway for small metabolic and exogenous agents that covalently modify DNA, were both screened as potential pathways for the tolerance of APS compounds. The mutagenic potential of APS was evaluated by screening mutL deficient bacteria, a protein necessary for the DNA mismatch repair pathway. Again, this pathway was not necessary for the robust growth of cells after being subjected to APS molecules. Finally, we assessed bypass-polymerase deficient bacteria for increased sensitivity to APS exposure. Single deletions of known bypass polymerases did not show any dramatic or significant effect in response to APS challenge (Fig 5D), suggesting that PCR amplification attenuation is not due to APS blocking lesions on the DNA template. It is possible that any single bypass polymerase deletion would not result in dramatic effects due to a complementarity of known bypass polymerase functions. In this case, depletion of more than one bypass polymerase may be necessary to detect sensitivity to APS molecules.
It is interesting that neither single mutation of DNA bypass polymerases nor DNA repair pathway depletion appear to protect cells from APS compounds, despite the high correlation between DNA association and cytotoxicity. If APS molecules can accumulate in vivo to threshold levels for DNA aggregation, there may be no mechanism to reverse this process and proceed through replication. This could explain the high levels of toxicity within this class of compounds. The lipophilic character of the chemical structure provides a mechanism to cross the cell wall, where the cationic character of the APS compounds facilitates association with DNA. Without an apparent pathway to repair or tolerate this association, replication slows or stalls to induce cell death. Further studies are clearly necessary to elucidate the precise mechanism by which APS compounds produce their potent cytotoxic effect.
Acknowledgments
The authors thank Dr. Robert W. Sobol, Jr. for his helpful discussions and critical reading of this manuscript. We also thank Dr. Bevin P. Engelward for kind gifts of E. Coli strains and Dr. Bongsup Cho for providing the proton NMR spectra of the arylphosphonium salts reported. This work was supported by the RI-INBRE Grant #P20 RR16457 from National Center for Research Resources/National Institutes of Health to KHA and JCW and by the Rhode Island College faculty research fund to KHA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hendry LB, Mahesh VB, Bransome ED, Jr, Ewing DE. Small molecule intercalation with double stranded DNA: implications for normal gene regulation and for predicting the biological efficacy and genotoxicity of drugs and other chemicals. Mutat Res. 2007;623:53–71. doi: 10.1016/j.mrfmmm.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rideout DC, Calogeropoulou T, Jaworski JS, Dagnino R, Jr, McCarthy MR. Phosphonium salts exhibiting selective anti-carcinoma activity in vitro. Anticancer Drug Des. 1989;4:265–280. [PubMed] [Google Scholar]
- 4.Madar I, Anderson JH, Szabo Z, Scheffel U, Kao PF, Ravert HT, Dannals RF. Enhanced uptake of [11C]TPMP in canine brain tumor: a PET study. J Nucl Med. 1999;40:1180–1185. [PubMed] [Google Scholar]
- 5.Madar I, Ravert H, Nelkin B, Abro M, Pomper M, Dannals R, Frost JJ. Characterization of membrane potential-dependent uptake of the novel PET tracer 18F-fluorobenzyl triphenylphosphonium cation. Eur J Nucl Med Mol Imaging. 2007;34:2057–2065. doi: 10.1007/s00259-007-0500-8. [DOI] [PubMed] [Google Scholar]
- 6.Min JJ, Biswal S, Deroose C, Gambhir SS. Tetraphenylphosphonium as a novel molecular probe for imaging tumors. J Nucl Med. 2004;45:636–643. [PubMed] [Google Scholar]
- 7.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 8.Okuma K, Yoshitake K, Izaki T, Yoshida K, Shioji K. Unusual reaction course of styrenes to 2-arylethyltriphenylphosphonium salts. Bull Chem Soc Jpn. 2007;80:1785–1790. [Google Scholar]
- 9.Spek EJ, Vuong LN, Matsuguchi T, Marinus MG, Engelward BP. Nitric oxide-induced homologous recombination in Escherichia coli is promoted by DNA glycosylases. Journal of Bacteriology. 2002;184:3501–3507. doi: 10.1128/JB.184.13.3501-3507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner PR, Denny WA. The mutagenic properties of DNA minor-groove binding ligands. Mutat Res. 1996;355:141–169. doi: 10.1016/0027-5107(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 11.Strekowski L, Wilson B. Noncovalent interactions with DNA: an overview. Mutat Res. 2007;623:3–13. doi: 10.1016/j.mrfmmm.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Snyder RD, McNulty J, Zairov G, Ewing DE, Hendry LB. The influence of N-dialkyl and other cationic substituents on DNA intercalation and genotoxicity. Mutat Res. 2005;578:88–99. doi: 10.1016/j.mrfmmm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Nowosielska A, Smith SA, Engelward BP, Marinus MG. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 2006;34:2258–2268. doi: 10.1093/nar/gkl222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, Prasad R, Samson LD, Wilson SH. Base Excision Repair Intermediates Induce p53-independent Cytotoxic and Genotoxic Responses. Journal of Biological Chemistry. 2003;278:39951–39959. doi: 10.1074/jbc.M306592200. [DOI] [PubMed] [Google Scholar]
- 16.Muller C, Calsou P, Frit P, Cayrol C, Carter T, Salles B. UV sensitivity and impaired nucleotide excision repair in DNA-dependent protein kinase mutant cells. Nucleic Acids Res. 1998;26:1382–1389. doi: 10.1093/nar/26.6.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]