

Abstract
Hair cells of the mammalian inner ear are the mechanoreceptors that convert sound-induced vibrations into electrical signals. The molecular mechanisms that regulate the development and function of the mechanically sensitive organelle of hair cells, the hair bundle, are poorly defined. We link here two gene products that have been associated with deafness and hair bundle defects, protocadherin 15 (PCDH15) and myosin VIIa (MYO7A), into a common pathway. We show that PCDH15 binds to MYO7A and that both proteins are expressed in an overlapping pattern in hair bundles. PCDH15 localization is perturbed in MYO7A-deficient mice, whereas MYO7A localization is perturbed in PCDH15-deficient mice. Like MYO7A, PCDH15 is critical for the development of hair bundles in cochlear and vestibular hair cells, controlling hair bundle morphogenesis and polarity. Cochlear and vestibular hair cells from PCDH15-deficient mice also show defects in mechanotransduction. Together, our findings suggest that PCDH15 and MYO7A cooperate to regulate the development and function of the mechanically sensitive hair bundle.
Keywords: hair cell, deafness, mechanotransduction, PCDH15, MYO7A, cochlea
Introduction
Hair cells of the vertebrate inner ear are mechanosensors that transduce mechanical forces arising from sound waves and head movement to provide our senses of hearing and balance. The mechanically sensitive organelle of a hair cell is the hair bundle, which consists of actin-rich stereocilia that are connected by extracellular linkages into a bundle (Hudspeth, 1997; Gillespie and Walker, 2001; Müller and Evans, 2001; Steel and Kros, 2001). The molecular mechanisms that control the development and maintenance of the hair bundle are not well understood, but the study of genes that are linked to deafness has provided first insights. These studies have shown that espin crosslinks filamentous actin (F-actin) in stereocilia and that whirlin and myosin XV cooperate to regulate stereociliary growth (Zheng et al., 2000; Mburu et al., 2003; Belyantseva et al., 2005; Delprat et al., 2005; Kikkawa et al., 2005). The transmembrane receptors cadherin 23 (CDH23) and protocadherin 15 (PCDH15), the adaptor proteins harmonin and sans, and the molecular motor myosin VIIa (MYO7A) are also implicated in hair bundle development, maintenance, and/or function. Mutations in the genes encoding these proteins have been linked to inherited forms of deafness and deaf blindness in humans, and similar mutations in the orthologous murine genes lead to structural defects in hair bundles (Ahmed et al., 2003a; Whitlon, 2004). As members of the cadherin superfamily, CDH23 and PCDH15 are candidates to form extracellular linkages in hair bundles. Consistent with this hypothesis, CDH23 is localized at transient lateral links, kinociliary links, and tip links (Boeda et al., 2002; Siemens et al., 2002, 2004; Sollner et al., 2004; Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). CDH23 binds to harmonin, whereas harmonin binds to MYO7A, F-actin, and sans, suggesting that harmonin assembles a protein complex mediating CDH23 function (Boeda et al., 2002; Siemens et al., 2002; Adato et al., 2005). The function of PCDH15 is less clear. Like CDH23, PCDH15 can bind to harmonin (Adato et al., 2005; Reiners et al., 2005), suggesting that the two cadherins use similar downstream effectors. However, although mutations in CDH23 and MYO7A lead to hair bundle defects in cochlear and vestibular hair cells shortly after birth, only the development of cochlear hair cells has been reported to be affected in PCDH15-deficient mice. Structural defects in vestibular hair cells of PCDH15-deficient mice have been observed only in the adult (Alagramam et al., 2000, 2001a, 2005; Raphael et al., 2001; Hampton et al., 2003). It therefore has remained unclear whether PCDH15 regulates hair bundle development or maintenance or whether it has different functions in cochlear and vestibular hair cells.
To gain insights into PCDH15 function in hair cells, we have now identified proteins that bind to the PCDH15 cytoplasmic domain, studied the PCDH15 distribution in hair cells, and analyzed hair cell development and function in PCDH15-deficient mice. We show here that PCDH15 binds not only to harmonin but also to MYO7A. Like MYO7A, PCDH15 is expressed toward the base of stereocilia, and the distribution of PCDH15 is perturbed in MYO7A-deficient mice, whereas the distribution of MYO7A is affected in PCDH15-deficient mice. Similar to MYO7A-deficient mice (Self et al., 1998), PCDH15-deficient mice show defects in the development and function of both cochlear and vestibular hair cells. Together, these findings suggest that PCDH15 and MYO7A cooperate to regulate hair bundle development and function.
Materials and Methods
Animals and antibodies.
Ames waltzer C57BL/6J-Pcdh15av3J/J (Alagramam et al., 2001a) and waltzer C57BL/6J-Cdh23v-2J/J mice (Di Palma et al., 2001) were obtained from The Jackson Laboratory (Bar Harbor, ME). shaker-1 Myo7a4626SB mice have been obtained as described previously (Liu et al., 1999) and were backcrossed onto the C57BL/6J genetic background. Rabbit polyclonal PCDH15 antisera were raised against a synthetic peptide corresponding to residues 1927–1943 (RVV EGI DVQ PHS QST SL) and against a glutathione S-transferase (GST)-fusion protein consisting of amino acids 1823–1943 of the mouse protein. Antibodies were affinity purified before use. Additional antibodies were as follows: mouse anti-hemagglutinin (HA).11 (Covance Research Products, Berkeley, CA), rabbit anti-MYO7A (Hasson et al., 1997), mouse anti-tubulin (Sigma, St. Louis, MO), rabbit anti-green fluorescent protein (GFP) and anti-CDH23*cyto (Siemens et al., 2002, 2004), Alexa Fluor 594 anti-rabbit and anti-mouse (Invitrogen, Carlsbad, CA), and Alexa Fluor 488–phalloidin (Invitrogen), and horseradish peroxidase-conjugated anti-rabbit and anti-mouse (Amersham Biosciences, Arlington Heights, IL).
Whole-mount immunocytochemistry.
Inner ears of embryonic day 18 (E18), postnatal day 0 (P0), P5, and P21 mice were dissected from temporal bones. The cochlear shell was removed, and the organ of Corti or vestibular patches were fixed for 45 min with 4% paraformaldehyde in PBS, followed by permeabilization and blocking for 1 h in 10% goat serum containing 0.2% Triton X-100 in PBS. Primary antibodies were incubated overnight at 4°C, and secondary antibodies were incubated for 3 h at room temperature. Where indicated, whole-mount explants were treated before fixation for 20 min with 5 mm EGTA or for 10 min with 50 μg/ml subtilisin, 10 μg/ml elastase, or 5 mm LaCl3. Whole-mount preparations were imaged on a Deltavision Deconvolution Microscope and processed with SoftWoRx software (Applied Precision, Issaquah, WA).
Electron microscopy.
For transmission electron microscopy (TEM), cochleas were removed by dissection from P5 mice and fixed by immersion with 2.5% glutaraldehyde and 1% tannic acid in 0.1 m cacodylate buffer for 2 h at room temperature. After several washes with buffer alone, the cochleas were osmicated, dehydrated, and embedded in Epon resin. Ultrathin sections were placed on copper grids and analyzed in a Philips (Aachen, Germany) 208 electron microscope. For scanning electron microscopy (SEM), inner ear sensory epithelia were fixed with 2.5% glutaraldehyde, 0.05 m sucrose, and 1% tannic acid in 0.05 m N,N-bis[2-hydroxyethyl]2-aminoethanesulfonic acid buffer, pH 7.4, for 3 weeks at ∼4°C as described previously (Osborne et al., 1984). The samples were dehydrated with an ethanol series (50, 70, 80, 90, and 100%), critical-point dried from liquid CO2, sputter coated with gold–palladium, and examined using a scanning electron microscope (FEI XL-30 ESEM FEG; FEI/Phillips, Hillsboro, OR; Scripps Institution of Oceanography San Diego, Unified Laboratory Facility, La Jolla, CA).
DNA constructs.
The full-length cDNA encoding mouse PCDH15 was amplified from adult brain RNA by reverse transcription (RT)-PCR and inserted into KpnI and EcoRI sites of pcDNA3 (Invitrogen). GST-fusion constructs GST-PCDH15-CT, containing the last 120 amino acids of PCDH15 (1823–1943), and GST-PCDH15-CTDPBI, a truncated form of GST-PCDH15-CT lacking the C-terminal seven amino acids, were obtained by PCR and inserted in-frame into BamHI and NotI sites of pGEX-4T-1 (Amersham Biosciences). The PCDH15 cytodomain construct (encoding amino acids 1403–1943) was amplified by RT-PCR from mouse brain RNA and inserted in-frame into NdeI and SalI sites of pGBKT7 (Clontech, Cambridge, UK). GFP–harmonin-a has been described previously (Siemens et al., 2002). GFP-fusion constructs containing the individual postsynaptic density-95/Discs large/zona occludens-1 (PDZ) domains of harmonin were generated by PCR using the aforementioned harmonin full-length construct as a template and inserting the PDZ1 (amino acids 1–191), the PDZ2 (amino acids 168–446), and the PDZ3 (amino acids 367–541) domains into pEGFP-C1 (Clontech). MYO7A constructs were as follows: GFP-MYO7A full length (amino acid 1–2175, human sequence in pEGFP-C3), GFP-MYO7A tail (1022–2175 in pEGFP-C3), GFP- MYO7A-SM (amino acids 1573–1820 in pEGFP-C3), GST-MYO7A-SM (amino acids 1573–1820 in pGEX4T-1), and GST-MYO7A-Src homology 3 (SH3) (amino acids 1573–1708 in pGEX4T-1). The cytodomain of E-cadherin (Ecad) was deleted after the transmembrane domain at amino acid 738 and replaced in-frame by either a 3xHA peptide (TYPYDVPDYAYPYDVPDYAYPYDVPDYA) to obtain pcDNA3 Ecad-HA or the PCDH15 cytodomain (amino acids 1403–1943) to obtain pcDNA3 Ecad-PCDH15cyto. All constructs were verified by DNA sequencing.
In vitro binding assays and GST pull-downs.
In vitro binding assays were performed using GST-tagged fusion proteins and radiolabeled proteins generated by in vitro translation as described previously (Siemens et al., 2002). To test for interactions of PCDH15 with MYO7A and harmonin, bacterial lysates containing the GST-fusion constructs or GST alone were incubated with preequilibrated glutathione-Sepharose beads (Amersham Biosciences) for 2 h at 4°C. Expression levels of GST-fusion proteins were determined by SDS-PAGE and Coomassie blue staining. Equal amounts of fusion proteins or GST alone were used for pull-down assay. Incubation of 35S-labeled proteins with GST proteins was performed for 2.5 h at 4°C. The beads were washed 3× with 20 mm Tris-HCl, pH 8.0, 1 mm EDTA, 240 mm NaCl, 0.5% Triton X-100, 1 mm DTT, and 1 mm PMSF containing protease inhibitor cocktail (Roche Products, Welwyn Garden City, UK). Sepharose beads were resuspended in 1× Laemmeli’s sample buffer containing 5% β-mercaptoethanol and analyzed by SDS-PAGE.
Immunoprecipitation and Western blot analysis.
HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen). After 24 h, extracts were prepared in 50 mm HEPES, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, 10% glycerol, 1% Triton X-100, 1 mm EDTA containing 10 mm sodium fluoride, 10 μg/ml aprotinin, 1 mm phenylmethylsulfonylfluoride, and protease inhibitor cocktail (Roche Products). Whole-cell lysates were incubated for 4 h with primary antibody, followed by 2 h incubation with 30 μl of protein G Sepharose (Amersham Biosciences). Immunocomplexes were washed with lysis buffer and 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, and 1% Triton X-100 before addition of Laemmeli’s sample buffer and SDS-PAGE. For expression controls, transfected HEK293T cells were directly lysed in Laemmeli’s sample buffer.
Immunohistochemistry of transfected cells.
HeLa cells were split the day before transfection and cultured in DMEM (4.5 g/L glucose; Invitrogen) containing 10% fetal bovine serum (FBS) and penicillin, streptomycin, and glutamine (Invitrogen) at 37°C/5% CO2. Cells were transfected using Lipofectamine Plus reagent (Invitrogen) and analyzed 18 h later. Solutions for subsequent steps were prepared in PBS. Before the addition of antibodies, cells were fixed for 20 min in 3.7% formaldehyde, permeabilized for 2 min in 0.5% Triton X-100, and then blocked for 1 h in 5% FBS. Primary antibodies were incubated for 2 h at 37°C in blocking solution, and secondary antibodies were incubated for 1 h at 37°C in PBS.
FM1-43 labeling and electrophysiology.
For FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridiniumdibromide] labeling, cochlear and utricular sensory epithelia were excised from P5–P10 animals, mounted on glass coverslips, and bathed in MEM (Invitrogen) supplemented with 10 mm HEPES. FM1-43FX at 5 μm (Invitrogen) was bath applied for 10 s, followed by three rinses in MEM. The tissue was fixed in 4% paraformaldehyde overnight, rinsed three times with PBS, counterstained with a 1:100 dilution of Alexa Fluor 633–phalloidin, and imaged on a Zeiss (Oberkochen, Germany) 510 confocal microscope. Whole-cell recordings and mechanical stimulation of mouse utricular and cochlear hair cells were performed as described previously (Holt et al., 2002; Stauffer et al., 2005). Briefly, we used the whole-cell, tight-seal technique in voltage-clamp mode to record mechanotransduction currents while deflecting hair bundles with stiff glass probes mounted on a piezoelectric bimorph. The bimorph stimulator had a 10–90% rise time of 0.6 ms. Utricle hair cells were stimulated by drawing the kinocilium into a pipette filled with extracellular solution (Holt et al., 2002), holding the kinocilium in place using constant suction. Cochlear cells were stimulated using a blunt-ended glass pipette sized to fit snugly into the V-shaped outer hair cell bundles. Bundle stimulation was monitored by video microscopy using a CCD camera (Hamamatsu, Shizouka, Japan). Our standard extracellular solution contained the following (in mm): 137 NaCl, 5.8 KCl, 10 HEPES, 0.7 NaH2PO4, 1.3 CaCl2, 0.9 MgCl2, and 5.6 d-glucose, vitamins, and amino acids as in MEM, pH 7.4 (311 mOsm/kg). Recording electrodes were pulled with resistances of 3–5 MΩ from R-6 glass (Garner Glass, Claremont, CA) and were filled with an intracellular solution that contained the following (in mm): 135 KCl, 5 EGTA-KOH, 5 HEPES, 2.5 Na2ATP, 2.5 MgCl2, and 0.1 CaCl2, pH 7.4 (284 mOsm/kg). We used an Axopatch 200B amplifier (Molecular Devices, Palo Alto, CA) to hold the cells at −64 mV. Transduction currents were filtered at 1 kHz with a low-pass Bessel filter, digitized at ≥5 kHz with a 12-bit acquisition board (Digidata 1322A), and collected using pClamp 8.2 software (Molecular Devices).
Results
PCDH15 expression in haircells
To define the expression pattern of PCDH15 in the inner ear, we raised in rabbits antisera against a peptide encompassing the C-terminal 17 amino acids of the PCDH15 protein. A second set of antisera was raised against a fusion protein between GST and the C-terminal 120 amino acids (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material). Two rabbits were immunized with each immunogen, and the antibodies were affinity purified for subsequent experiments. All antibodies gave similar results. To control for the specificity of the antibodies, we performed Western blots with extracts from HEK293T cells that were transfected with a PCDH15 expression vector. All antibodies recognized a protein with an apparent molecular mass >250 kDa, in good agreement with the predicted molecular mass of full-length PCDH15 (∼216 kDa) (supplemental Fig. S1b, available at www.jneurosci.org as supplemental material). The difference in the predicted from the observed molecular mass likely can be explained by glycosylation of the PCDH15 extracellular domain and the high proline content of the cytoplasmic domain (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material). The antisera also stained transfected HeLa and Madin-Darby canine kidney cells but not untransfected control cells (supplemental Fig. S1c, available at www.jneurosci.org as supplemental material). These findings suggest that the antibodies specifically recognize PCDH15 and are suitable for immunolocalization studies.
Previous studies have shown that PCDH15 is expressed in hair cells, including stereocilia (Ahmed et al., 2003b). To analyze the expression pattern of PCDH15 in hair cells at different developmental time points at higher resolution, we stained cochlear and vestibular sensory epithelia from C57BL/6J mice as whole mounts (Fig. 1). At P5, when the stereocilia of cochlear hair cells are elongating, we observed PCDH15 staining in outer hair cells and inner hair cells (Fig. 1a) (supplemental Fig. S1d, available at www.jneurosci.org as supplemental material). Weak PCDH15 expression was observed within the cell bodies (data not shown). More prominent expression was detectable in the hair bundles. PCDH15 was localized in a restricted area toward the base of the longest stereocilia, in which they were in close apposition to the next row of shorter stereocilia (Fig. 1a,c). This distribution contrasts with that of CDH23, which was localized toward the tips of all stereocilia as reported previously (Fig. 1c) (Siemens et al., 2004; Rzadzinska et al., 2005). Staining was not observed with secondary antibody alone and was not detectable in PCDH15-deficient Ames waltzerav3J mice, confirming the specificity of the signal (Fig. 1b and data not shown). In vestibular hair cells of P5 mice (Fig. 1d) and in hair cells in the basilar papilla of newborn chickens (Fig. 1d), PCDH15 localization was restricted even more toward the base of stereocilia close to their insertion point into the cuticular plate. Finally, at P21, PCDH15 was no longer confined to the base of stereocilia but distributed along the stereocilia in a pattern of dots (Fig. 1e). We conclude that PCDH15 shows a dynamic expression in hair cell stereocilia, with localization toward the base during early stages of hair bundle development and a more broad distribution as hair cells mature.
Figure 1.
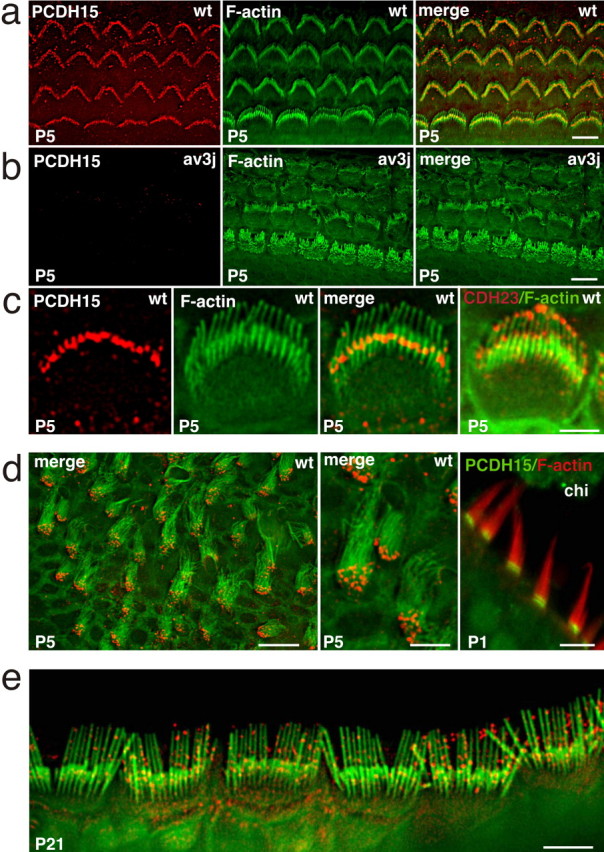
PCDH15 localization in hair cells. Cochlear whole mounts of wild-type (wt) (a, c) and Ames waltzerav3J (av3J) (b) mice at P5 were stained with antibodies to PCDH15 (red) and phalloidin (green). PCDH15 expression was detected in outer and inner hair cells and localized toward the base of the longest stereocilia. The rightmost panel in c shows a staining for CDH23 (red) and phalloidin (green), revealing CDH23 at stereociliary tips. d, Hair cells from the vestibule of P5 mice were stained with antibodies to PCDH15 (red) and phalloidin (green); (leftmost panel in d). Similar stainings were performed in P1 chickens (chi) (PCDH15, green; phalloidin, red). e, At P21, PCDH15 (red) was more widely distributed in the stereocilia of cochlear hair cells. Size bars: a, b, e, 5 μm; c, 3 μm; d, left and right, 10 μm; d, middle, 5 μm.
PCDH15 and ankle links: common and distinguishing features
Recent studies have provided evidence that ankle links in murine cochlear hair cells are transient structures that disappear at approximately P14 (Goodyear et al., 2005 and references therein). The transient localization of PCDH15 toward the base of developing stereocilia suggests that it may be a component of ankle links. Previous studies have also shown that ankle links can be distinguished from other linkages in hair cells by biochemical means. Ankle links are disrupted by treatment of hair cells with EGTA and subtilisin but not by La3+ and elastase; tip links are disrupted by treatment with EGTA, La3+, and elastase but not by subtilisin; and finally, transient lateral links and top connectors are essentially unaffected by treatment with subtilisin and EGTA (Osborne and Comis, 1990; Assad et al., 1991; Goodyear and Richardson, 1999; Kachar et al., 2000; Goodyear et al., 2005). We therefore tested the effect of these agents on PCDH15 immunolocalization in hair cells (Fig. 2). Similar to ankle link, PCDH15 localization in hair cells was sensitive to treatment with EGTA and subtilisin but resistant to treatment with La3+ (Fig. 2a). Unlike the ankle link, PCDH15 was also sensitive to elastase treatment (Fig. 2a). During treatment with EGTA, subtilisin, and elastase, PCDH15 immunoreactivity could no longer be observed in hair cells. Similar observations have been made previously for CDH23 and TrpA1 (transient receptor potential ankyrin 1) (Corey et al., 2004; Siemens et al., 2004), suggesting that hair cells may rapidly shed unfolded and degraded proteins or transport them to the cell body. We next expressed full-length PCDH15 in HEK293T cells and tested its sensitivity to protease treatment. In agreement with the studies in hair cells, PCDH15 was sensitive to proteolytic digestion by the two proteases (Fig. 2b). These findings demonstrate that PCDH15 shares many, but not all, properties with the ankle links; its biochemical properties are distinct from other linkages, such as transient lateral links and horizontal top connectors.
Figure 2.
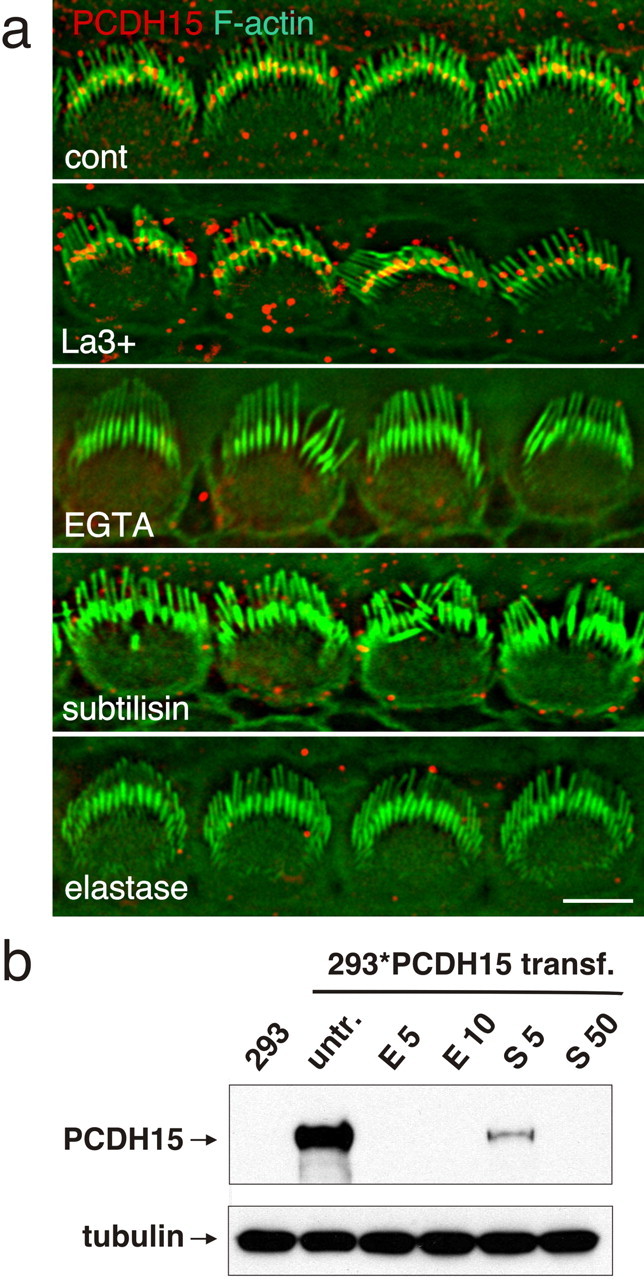
Biochemical properties of PCDH15. a, Cochlear whole mounts of P5 animals were treated as indicated and stained for PCDH15 (red) and F-actin (green). PCDH15 was sensitive to treatment with EGTA, subtilisin, and elastase. b, HEK293 cells that were transfected to express PCDH15 were treated with elastase (E, 5 or 10 μg/ml) or subtilisin (S, 5 or 50 μg/ml); extracts were prepared and analyzed by Western blotting. PCDH15 was sensitive to treatment with elastase and subtilisin. Size bar, 5 μm.
To further address whether PCDH15 is an essential component of the linkages that connect stereocilia, we analyzed the hair bundles from P5 Ames waltzerav3J mice by SEM (Fig. 3). Linkages at the base of stereocilia were clearly visible in wild-type and mutant animals (Fig. 3a–c, e–g, arrows). Filaments that connected the stereocilia along their length were also present (Fig. 3b,g, arrowheads) but were more clearly visualized by TEM (Fig. 3d,h). Together, our findings show that linkages at the base of stereocilia and along their length form in the absence of PCDH15. These results suggest that PCDH15 is not essential for the formation of at least some linkages. Indeed, it may not be part of any linkage. Alternatively, the linkage systems may be more complex and composed of several types of filaments that differ in their molecular composition; only a subset may be missing in the Ames waltzerav3J mice, or removal of PCDH15 from linkages may not cause complete disassembly of the linkages.
Figure 3.

Analysis of cochlear hair cells by SEM and TEM. P5 cochlear whole mounts from wild-type (wt) (a–c) and Ames waltzerav3J (av3J) (e–g) mice were analyzed by SEM. Links at the base of stereocilia (arrows) were visible in hair cells from wild-type and mutant animals. Links along the length of stereocilia were also present (arrowheads) but could be visualized much more clearly by TEM (d, h). Scale bars: a–c, e–g, 200 nm; d, h, 300 nm.
PCDH15 is required for hair bundle development in the cochlea and vestibule
Previous studies have reported that, in Ames waltzerav3J mice, hair bundles in cochlear hair cells are disrupted shortly after birth, whereas vestibular hair cells are affected several weeks later (Alagramam et al., 2000, 2001b, 2005). To distinguish functions of PCDH15 in hair bundle development and maintenance and to determine more precisely the morphological changes, we analyzed cochlear and vestibular hair bundles at different developmental ages. At E18, both outer and inner cochlear hair cells of wild-type mice contained stereocilia that were assembled into a bundle with a kinocilium in the center. The bundles were aligned parallel to the long axis of the cochlear duct (Fig. 4a,b,e,f). In Ames waltzerav3J mice, hair cells had formed stereociliary bundles and contained a kinocilium (Fig. 4c,d,g,h). The bundles were difficult to visualize in outer hair cells because stereocilia appeared short and were not strictly aligned along the length of the cochlear duct (Fig. 4c,d). The bundles were more clearly visible in inner hair cells but still appeared disorganized (Fig. 4g,h). Unlike in wild-type hair cells, the kinocilium of a hair cell in the mutants was frequently located toward the edge of the cell and flanked by the longest stereocilia, which were in close apposition to the kinocilium (Fig. 4g,h). Defects were more pronounced at P0. Whereas wild-type hair cells at the basal end of the cochlea had formed nice bundles that were polarized in the apical hair cell surface and contained a centrally located kinocilium (Fig. 4i,j), bundles in the mutants appeared shorter and less well organized, and the kinocilia were frequently shifted to ectopic positions; the bundles also failed to develop a clear polarity in the apical hair cell surface. By P5, some bundles in the mutants finally established polarity, but organized rows of stereocilia could rarely be detected (Fig. 5a–e). Although some stereocilia in the mutants appeared to have reached a similar length as in wild-type mice, many of the stereocilia were short and malformed, with phalloidin-labeled swellings toward the tips (Fig. 5c,e). These findings demonstrate that kinocilia and stereocilia form in the absence of PCDH15 and form a rudimentary bundle that shows defects in stereociliary morphology and cohesion and in the polarization of the hair bundle in the apical hair cell surface. Lower-power images of the sensory epithelium and quantification of hair bundle orientation confirmed that the polarity defect extended throughout the cochlear duct (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In contrast to previous findings, high-resolution images revealed stereociliary defects also in the utricle and saccule of P5 animals. Whereas vestibular hair cells in wild-type mice had formed hair bundles with a typical staircase organization, stereocilia bundles in the mutant were disorganized (Fig. 5f,g). The number of stereocilia per hair cell appeared reduced, a staircase arrangement was less apparent, and hair cells frequently contained one or several stereocilia that were abnormally long and thick (Fig. 5h–j). Together, our findings extend previous studies and demonstrate that PCDH15 is essential for the normal development of stereociliary bundles in cochlear and vestibular hair cells. We also demonstrate that, in PCDH15-deficient Ames waltzerav3J mice, several parameters of hair bundle development are affected, including bundle cohesion and polarity.
Figure 4.
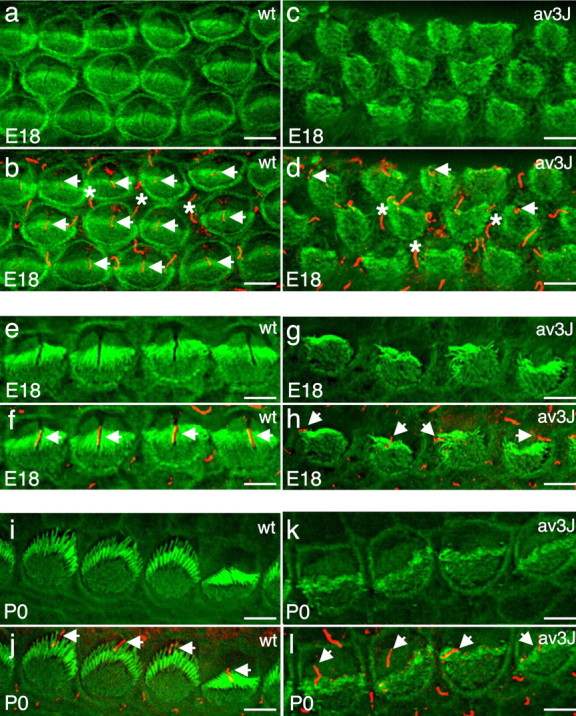
Hair bundle development in wild-type (wt) and Ames waltzerav3J (av3J) mice. Cochlear whole mounts were stained with phalloidin (green) to reveal F-actin in stereocilia and an antibody against acetylated tubulin (red) to reveal kinocilia. a–d, Outer hair cells in wild-type and mutant animals at E18. Arrows point to kinocilia of hair cells within the hair bundle. The asterisks indicate kinocilia on support cells between hair cells. e–h, Inner hair cells in wild-type and mutant animals at E18 and at P0 (i–l). Arrows point to kinocilia in hair cells. Note the symmetric organization of the hair bundles in wild-type but not mutant animals. Scale bars, 5 μm.
Figure 5.

Hair bundle development in wild-type (wt) and Ames waltzerav3J (av3J) mice. Whole mounts were stained with phalloidin (green) to reveal stereocilia. a–e, Inner hair cells of wild-type and mutant animals at P5. Note the abnormal morphology of the hair bundles in the mutants and the abnormal shape of the stereocilia with bulb-like enlargements toward the tips. Many of the stereocilia in hair cells from mutant mice also appeared to be reduced in length (b, c, e). f–j, Vestibular hair cells of mutant and wild-type animals at P5. Stereocilia in wild-type mice formed bundles with a staircase arrangement. Bundles in the mutants were abnormally shaped. Most notably, the number of stereocilia in a bundle was reduced, and abnormally thick and elongated stereocilia were visible. Scale bars: a–c, 5 μm; d, e, 1.5 μm; f, g, 12 μm; h–j, 3.5 μm.
The cytoplasmic domain of PCDH15 binds to MYO7A
Previous studies have shown that mutations in the genes encoding PCDH15, CDH23, MYO7A, sans, and harmonin lead to deafness in humans. Mice with mutations in the orthologs of these genes have defects in hair cell stereocilia (Ahmed et al., 2003a; Whitlon, 2004). To assess whether PCDH15 acts in a common pathway with any of these genes, we searched for biochemical interactions. In agreement with previous findings (Reiners et al., 2005), we demonstrated that the C-terminal PDZ binding domain of PCDH15 binds to harmonin (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We also searched for interactions between PCDH15 and MYO7A. We expressed PCDH15 and MYO7A fragments fused to GFP in HEK293 cells and performed coimmunoprecipitation experiments (Fig. 6). A fragment encompassing the tail domain of MYO7A, but lacking its motor domain, or a shorter fragment consisting of the SH3 homology domain and parts of the C-terminal MyTH4 domain could be coimmunoprecipitated with PCDH15 (Fig. 6a,b). We performed GST pull-down experiments to confirm that the interaction between PCDH15 and MYO7A was direct. Fusion proteins between GST and the SH3-MyTH4 domain or the SH3 domain of MYO7A bound to the PCDH15 cytoplasmic domain (Fig. 6a,c). These findings demonstrate that PCDH15 and MYO7A can directly interact with each other.
Figure 6.

PCDH15 binds to MYO7A. a, Diagram of the MYO7A protein and the constructs that were used for the biochemical experiments. b, Cells were transfected with PCDH15 and the GFP-tagged MYO7A constructs indicated at the top. Immunoprecipitations (IP) were performed with GFP antibodies, and proteins were resolved by SDS-PAGE. PCDH15 was visualized by Western blotting (WB). Top, PCDH15 coimmunoprecipitated with the MYO7A tail domain or a smaller construct containing the SH3 domain and C-terminal MyTH4 homology domain of MYO7A. Middle and Bottom, Extracts were directly analyzed for the expression of PCDH15 and GFP-tagged MYO7A constructs without immunoprecipitation to confirm that the transfected proteins were expressed to similar levels. c, The indicated GST-tagged MYO7A constructs were used for pull-down experiments using the in vitro translated PCDH15 cytoplasmic domain. Top, GST-MYO7A-SM and GST-MYO7A-SH3 were able to interact with PCDH15, whereas GST alone did not bind. As a control, the input PCDH15 before GST pull-down was loaded on the gel. Bottom, Coomassie blue staining revealed that all GST constructs were used in similar amounts.
We next analyzed interactions between MYO7A and the PCDH15 cytoplasmic domain in the context of cellular membranes. For this purpose, we coexpressed in cell lines a GFP-tagged MYO7A-tail construct with a fusion protein consisting of the extracellular and transmembrane domain of E-cadherin fused to the cytoplasmic domain of PCDH15 (Ecad-PCDH15cyto) (Fig. 7a). As a control, we coexpressed the GFP-tagged MYO7A-tail construct with fusion protein consisting of the extracellular and transmembrane domain of E-cadherin fused to an HA tag (Ecad-HA) (Fig. 7a). In transfected cells, Ecad-HA and Ecad-PCDH15cyto localized to cell–cell contacts that formed between transfected cells (Fig. 7b,c). MYO7A-GFP was recruited to cell–cell contacts only in cells expressing Ecad-PCDH15cyto (Fig. 7b,c). These findings extend our biochemical studies and demonstrate that the PCDH15 cytoplasmic domain can recruit MYO7A to the cell membrane. The results also raise the possibility that PCDH15 is similarly required to recruit MYO7A to the membrane of hair cell stereocilia.
Figure 7.
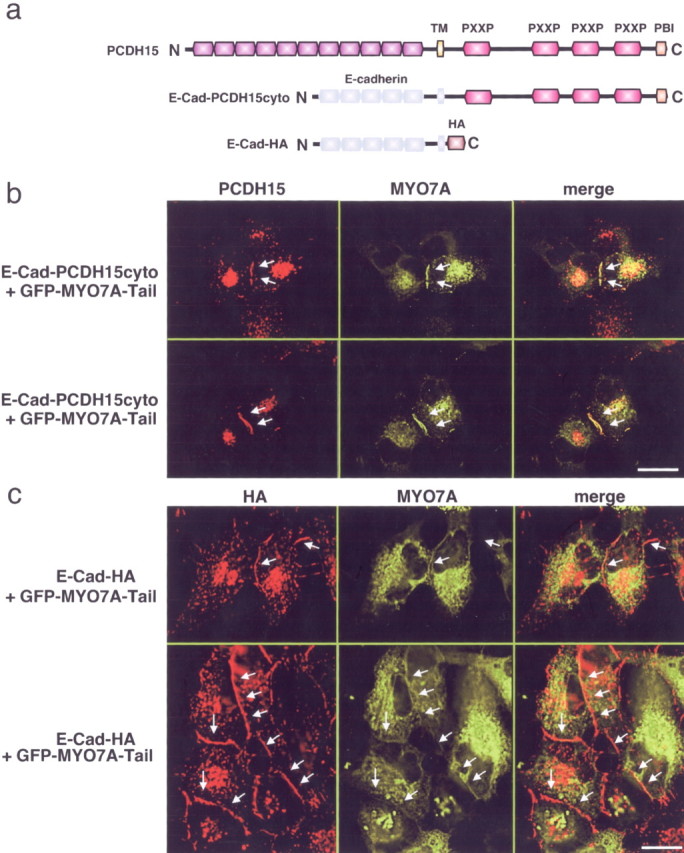
Recruitment of MYO7A by the PCDH15 cytoplasmic domain. a, Diagram of PCDH15 and chimeric proteins between the extracellular and transmembrane domain of E-cadherin fused to the PCDH15 cytoplasmic domain or a HA tag. b, c, Cells were transfected with the constructs indicated on the left. E-cadherin fusions to the PCDH15 cytoplasmic domain or an HA tag were visualized by staining with antibodies that detect the PCDH15 cytoplasmic domain or the HA tag, respectively. Myosin localization was determined by GFP fluorescence. Arrows point to cell–cell contacts between transfected cells. Note that E-cad-PCDH15cyto and E-cad-HA were recruited to cell–cell contacts, but MYO7A was only recruited in cells expressing E-cad-PCDH15-cyto. Scale bars, 12 μm.
Localization of MYO7A and PCDH15 in Ames waltzerav3J and shaker-1 mice
To test whether PCDH15 is required for MYO7A localization in hair cells, we compared the subcellular distribution of both proteins in wild-type mice, PCDH15-deficient Ames waltzerav3J mice, and MYO7A-deficient shaker-1 mice. As described above, PCDH15 was localized toward the base of stereocilia in P5 cochlear hair cells from wild-type mice (Fig. 8a). In agreement with previous findings, we observed that MYO7A was expressed more broadly throughout stereocilia (Hasson et al., 1997). High-resolution image analysis of hair bundles revealed that MYO7A staining was punctate (Fig. 8a). A significant fraction of MYO7A was localized toward the base of stereocilia in a similar region to PCDH15 (Fig. 8a, arrows). Bright MYO7A puncta were also visible in the cuticular plate (Fig. 8a, arrowheads) and along the length of the stereocilia and below their tips (Fig. 8a). In PCDH15-deficient Ames waltzerav3J mice, MYO7A distribution was perturbed. In some hair cells, MYO7A staining was absent, whereas in others MYO7A was mislocalized with high expression in the cuticular plate and at stereociliary tips (Fig. 8b). In MYO7A-deficient shaker-1 mice, PCDH15 was no longer localized to stereocilia (Fig. 8c). However, PCDH15 was still expressed in shaker-1 mice, as confirmed by Western blotting (data not shown). We next performed several control experiments. First, we analyzed PCDH15 expression in hair cells from CDH23-deficient waltzer mice, which show splayed bundles similar to MYO7A-deficient mice. PCDH15 localization was unaffected, providing strong evidence that mislocalization of PCDH15 in shaker-1 mice was not simply a consequence of stereociliary splaying (Fig. 8c). Similarly, CDH23 was still localized at the tips of stereocilia in PCDH15-deficient Ames waltzerav3J mice (Fig. 8d). In MYO7A-deficient shaker-1 mice, some of the CDH23 protein was more broadly distributed along the length of stereocilia, but it was still prominently localized toward stereociliary tips (Fig. 8d). Together, these findings show that PCDH15 and MYO7A protein are expressed in a partially overlapping pattern in stereocilia. In addition, PCDH15 expression is no longer observed in stereocilia of MYO7A-deficient shaker-1 mice, whereas MYO7A localization in stereocilia is perturbed in PCDH15-deficient Ames waltzerav3J mice. The data also demonstrate that the distribution of PCDH15 in hair cells is independent of the distribution of CDH23 and vice versa, suggesting that both proteins are part of distinct protein complexes.
Figure 8.
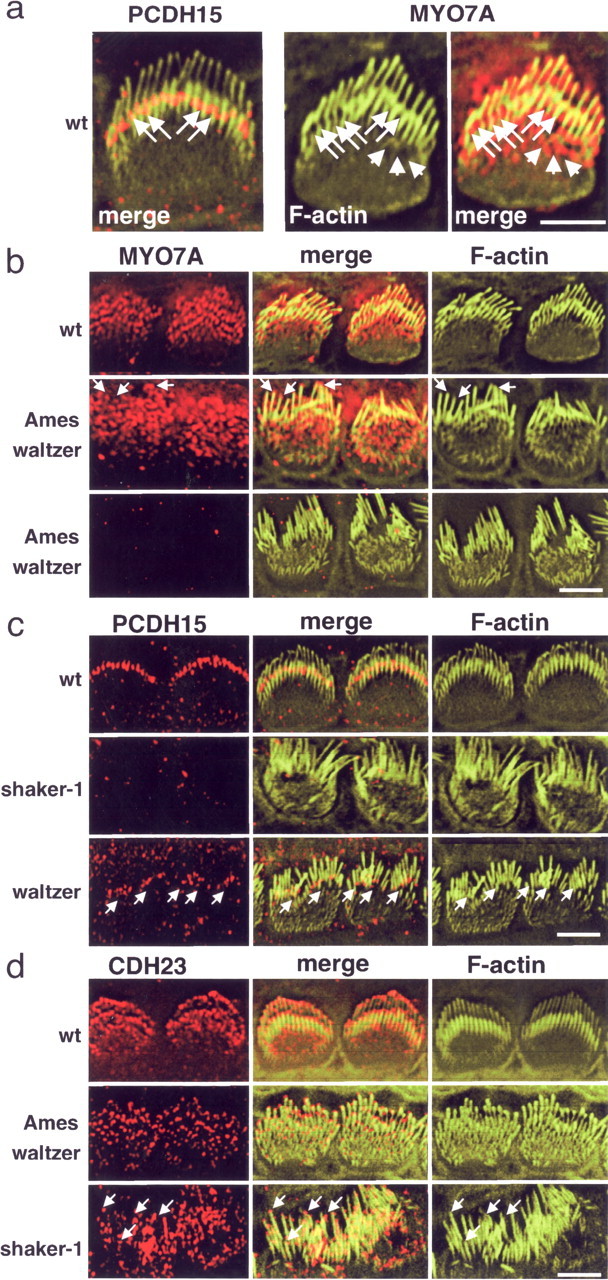
Localization of PCDH15 and MYO7A in wild-type (wt) and mutant mice. Inner hair cells of P5 animals are shown. a, The left panel shows cells from a wild-type mouse stained for PCDH15 (red) and F-actin (green; same cell as that shown in Fig. 1C). The middle and right panels show a cell stained for F-actin (green) and MYO7A (red). Note that two rows of stereocilia could be resolved. PCDH15 and MYO7A were localized toward the base of the longest stereocilia (arrows); MYO7A was also localized to the cuticular plate (arrowheads) and toward tips of stereocilia. b, MYO7A localization in PCDH15-deficient Ames waltzerav3J mice. In some hair cells, MYO7A could still be visualized, but it was mostly found at the apical hair cell surface and also at tips of stereocilia (arrows). In many hair cells, MYO7A could no longer be detected. Note that the wild-type control includes the hair cell shown at higher magnification in a. c, PCDH15 localization in MYO7A-deficient shaker-1 mice and CDH23-deficient waltzer mice. PCDH15 could no longer be detected in stereocilia in shaker-1 mice but was still confined toward the base of stereocilia in waltzer mice. Note the disorganization of the stereociliary bundles in the waltzer mice. d, CDH23 localization in PCDH15-deficient Ames waltzerav3J mice and MYO7A-deficient shaker-1 mice. CDH23 was still localized at the tips of stereocilia in the two mouse strains. Note that, in the Ames waltzerav3J mice, stereocilia were splayed but less affected than in shaker-1 mice. Because of the strong variations in stereociliary length in shaker-1 mice, it was more difficult to resolve CDH23 distribution, but it was clearly detectable at tips (arrows). Scale bars: a, 5 μm; b–d, 4 μm.
Mechanotransduction in hair cells of Ames waltzerav3J mice
Previous studies have shown that MYO7A is required not only for hair bundle morphogenesis but also for normal mechanotransduction (Kros et al., 2002). We therefore analyzed the mechanotransduction properties of hair cells in PCDH15-deficient Ames waltzerav3J mice. First, we imaged uptake of the fluorescence dye FM1-43, which can enter hair cells through open mechanotransduction channels (Gale et al., 2001; Meyers et al., 2003). The experiments were performed with whole mounts of cochlear and utricular sensory epithelia from P5–P10 mutant mice and wild-type littermates. FM1-43 rapidly entered hair cells from wild-type animals, after a 10 s exposure, and diffused into cell bodies, but hair cells from PCDH15-deficient Ames waltzerav3J mice did not take up the dye (Fig. 9a). Because dye uptake in unstimulated hair bundles depends on channels being open at rest, we wondered whether Ames waltzerav3J hair cells had functional transduction but with a stimulus–response relationship shifted such that the channels were closed at rest. Gale et al. (2001) reported that Myo7a6J mutant hair cells do not take up FM1-43 but retain the ability to transduce very large positive bundle deflections. Thus, we measured mechanically evoked transduction currents by deflecting hair bundles and recording whole-cell currents from 15 hair cells (eight from homozygous Ames waltzerav3J mice and seven from homozygous wild-type littermate controls). Representative currents are shown in Figure 9b. We were unable to evoke transduction currents in any of the five utricular type II or three cochlear outer hair cells from mutant animals. We used a variety of stimuli, including very large (∼2 μm) positive and negative deflections, off-axis deflections, and a range of holding positions, all to no avail. The mean ± SE maximal current for wild-type vestibular cells was 140 ± 28 pA (n = 4), whereas currents for wild-type cochlear cells were 103 ± 29 pA (n = 3). Together, the data demonstrate that inner and outer hair cells in the cochlea and type I and type II hair cells of the vestibule not only show morphological defects; they are also functionally impaired. These defects in hair bundle morphology and mechanotransduction likely cause deafness and balance defects in Ames waltzerav3J mice.
Figure 9.

Mechanotransduction in hair cells of Ames waltzerav3J mice. a, Cochlear and vestibular whole mounts that had been incubated for 10 s with 5 μm FM1-43 are shown. FM1-43 (red) was taken up by cochlear and vestibular hair cells from wild-type (wt), but not Ames waltzerav3J (av3J), mice. In the cochlea, hair cells were counterstained with phalloidin (green) to reveal F-actin. b, Examples of transduction currents in wild-type and Ames waltzerav3J mice. For both genotypes, a representative family of currents is shown for both a utricular type II and cochlear outer hair cell. Transduction currents were absent in all eight mutant cells examined. The bundle displacement protocol is shown at the bottom. Scale bar, 7.5 μm.
Discussion
We demonstrate here that PCDH15-deficient Ames waltzerav3J mice show defects in the integrity and polarity of hair bundles in cochlear and vestibular hair cells that manifest during development of the hair bundle and resemble those reported previously for MYO7A-deficient shaker-1 mice (Self et al., 1998). In agreement with the defects in hair bundle morphology, mechanotransduction currents can no longer be evoked in cochlear and vestibular hair cells from Ames waltzerav3J mice. To define the signaling pathways that are regulated by PCDH15, we have identified some of its downstream effectors. Consistent with a previous report (Reiners et al., 2005), we show that the C terminus of PCDH15 binds to harmonin. In addition, the cytoplasmic domain of PCDH15 mediates interactions with MYO7A. MYO7A and PCDH15 are expressed in hair cells toward the base of developing stereocilia, and PCDH15 expression is drastically reduced in stereocilia of MYO7A-deficient shaker-1 mice, whereas MYO7A ex-pression is drastically reduced in stereocilia of PCDH15-deficient Ames waltzerav3J mice. Together, the findings suggest that PCDH15 and MYO7A cooperate to regulate the development of hair bundles in cochlear and vestibular hair cells.
Previous studies have demonstrated that hair bundles of cochlear hair cells in PCDH15-deficient mice are shaped abnormally at early postnatal ages, whereas vestibular hair cells were reported to be affected several weeks later (Alagramam et al., 2000, 2001b, 2005). It therefore has remained unclear whether PCDH15 has a primary function in hair bundle development or maintenance, or different functions in cochlear and vestibular hair cells. Our findings now clarify this issue. Consistent with the expression time course of PCDH15, we demonstrate that the development of cochlear and vestibular hair bundles is affected in Ames waltzerav3J mice. The kinocilium and stereocilia start to develop in PCDH15-deficient hair cells and are assembled into a rudimentary hair bundle. However, the stereocilia in the mutants appear disorganized and the kinocilium is frequently mislocalized toward one side of the apical hair cell surface. It therefore appears that PCDH15 is required for maintaining the integrity of the developing hair bundle and possibly for the movement of the hair bundle that leads to its polarization in the apical hair cell surface. Consistent with the defects in hair bundle morphogenesis, FM1-43 uptake assays and single-cell recordings demonstrated that mechanotransduction current could not be evoked in cochlear or vestibular hair cells from Ames waltzerav3J mice. Although the defects in mechanotransduction are likely a consequence of the defects in hair bundle morphogenesis, our findings do not exclude a more direct role for PCDH15 in mechanotransduction as well.
PCDH15 is an attractive candidate for contributing to the extracellular filaments that connect hair cell stereocilia into a bundle, but it is still unclear whether it is an essential component of any of the filaments. Electron microscopic studies have revealed a variety of filaments that connect stereocilia into a bundle. In mice, these include ankle links and transient lateral links in developing hair cells and horizontal top connectors and tip links in mature hair cells. During development, the longest stereocilia are also connected to the kinocilium by kinociliary links (Goodyear et al., 2005 and references therein). Recent studies have provided evidence that CDH23 is a component of kinociliary links, transient lateral links, and tip links (Boeda et al., 2002; Siemens et al., 2002, 2004; Sollner et al., 2004; Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). The phosphatidylinositol lipid phosphatase PTPRQ has been shown to be required for the formation of lateral links that connect stereocilia and likely is a component of these linkages (Goodyear et al., 2003). We show here that PCDH15 shares many features with ankle links. It is transiently expressed toward the base of stereocilia, in which ankle links are transiently present. Agents that disrupt ankle links, such as EGTA and subtilisin, affect the localization of PCDH15 in hair cells, whereas La3+, which does not affect ankle links, also does not affect PCDH15 localization. These biochemical features clearly distinguish PCDH15 from other linkages such as transient lateral links and top connectors (not sensitive to any of the treatments) or the tip link (not sensitive to subtilisin) (Osborne and Comis, 1990; Assad et al., 1991; Goodyear and Richardson, 1999; Kachar et al., 2000; Goodyear et al., 2005). However, unlike the ankle link, PCDH15 localization is sensitive to elastase. Furthermore, filaments that connect stereocilia at the ankle region and along their length are detectable in PCDH15-deficient Ames waltzerav3J mice. How can these findings be reconciled? As one possibility, PCDH15 may not be a part of any of the linkage systems. Although members of the cadherin superfamily such as classical cadherins function as homophilic cell adhesion molecules, there is little evidence that protocadherins mediate adhesive interactions important for their function (Frank and Kemler, 2002). Alternatively, linkages that connect stereocilia may be molecularly more heterogenous than indicated by the names given to individual linkages. Consistent with this model, studies in several species have shown that stereocilia are coated with a dense network of filaments during development, which becomes refined during hair cell maturation (Goodyear et al., 2005 and references therein). Some of the filaments may contain PCDH15, and absence of a subset of filaments would be difficult to detect by electron microscopy. This issue can be resolved once antibodies to the extracellular domain of PCDH15 are available that are suitable for immunogold labeling.
Pcdh15 belongs to a small group of genes that have been linked to deaf blindness [Usher syndrome type I (USH1)] and several nonsyndromic forms of deafness. Besides PCDH15, these genes encode CDH23, harmonin, MYO7A, and sans. Mutations in the murine orthologs of any of the USH1 proteins lead to defects in the structure of hair bundles (Ahmed et al., 2003a; Whitlon, 2004). Biochemical studies have furthermore demonstrated that harmonin can form homomeric complexes and bind to CDH23, PCDH15, MYO7A, and sans (Boeda et al., 2002; Siemens et al., 2002; Adato et al., 2005). It therefore has been suggested that the proteins form a network that regulates hair bundle morphogenesis (Adato et al., 2005). However, several lines of evidence suggest that the situation is more complex. First, USH1 proteins are likely not present simultaneously in the same protein complexes because the binding sites for several proteins overlap. For example, PCDH15 and CDH23 bind to the PDZ2 domain of harmonin; CDH23, MYO7A, sans, and the C terminus of harmonin bind to the PDZ1 domain of harmonin (this study) (Boeda et al., 2002; Siemens et al., 2002; Adato et al., 2005; Reiners et al., 2005). Second, we demonstrate here that PCDH15 and CDH23 have a distinct subcellular distribution in developing hair cells. Although CDH23 is localized toward the tips of stereocilia, PCDH15 is localized toward the base. Third, we show that the localization of CDH23 is not perturbed in PCDH15-deficient mice and vice versa, suggesting that the two proteins are appropriately localized in hair cells independent of each other. These data are consistent with a model in which the transmembrane receptors PCDH15 and CDH23 assemble distinct protein complexes in different locations in hair cells. The precise composition of these protein complexes in vivo as well as their function in hair cells still need to be defined. In the current study, we have taken a first step toward this goal. We demonstrate that the PCDH15 cytoplasmic domain can bind to the tail domain and MYO7A and is sufficient to recruit MYO7A to the cell membrane of cells transfected to express these proteins. Because MYO7A binds F-actin (Udovichenko et al., 2002), the findings suggest that it links PCDH15 to the actin cytoskeleton. Consistent with the in vitro findings, the expression of PCDH15 is drastically reduced in stereocilia of MYO7A-deficient shaker-1 mice, whereas MYO7A expression is drastically reduced in stereocilia of PCDH15-deficient Ames waltzerav3J mice. Although additional experiments are necessary to address this issue, our findings raise the possibility that complex formation between PCDH15 and MYO7A is important for protein targeting and/or maintenance of the protein complex at target sites. Because MYO7A is an actin-based molecular motor, it is tempting to speculate that the protein complex serves to adjust tension force within the stereociliary cytoskeleton. Importantly, PCDH15 also binds to harmonin, which can bind MYO7A and F-actin (this study) (Boeda et al., 2002; Siemens et al., 2002; Adato et al., 2005; Reiners et al., 2005), suggesting that PCDH15 may be linked to the stereociliary cytoskeleton by multiple mechanisms. Defects in the hair bundle in Ames waltzerav3J mice [as well as shaker-1 mice (Self et al., 1998)] could be, at least in part, a consequence of defects in tension that is required to stabilize the base of stereocilia and provide cohesion between rows of stereocilia during rearrangements that lead to the development of a polarized bundle.
Footnotes
This work was supported by National Institutes of Health/National Institute on Deafness and Other Communication Disorders Grants DC005965 and DC007704 (U.M.), DC005439 and DC003279 (J.R.H.), DC006183 (G.S.G.G.), DC002368 and DC003279 (P.G.G.), and EY07042 and Core Grant EY12598 (D.W.). We thank members of the Müller laboratory for helpful discussions and Evelyn York (University of California at San Diego) and Dr. J. Pickles (University of Queensland, Brisbane, Queensland, Australia) for technical advice.
References
- Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C (2005). Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet 14:347–356. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Wilcox ER (2003a). The molecular genetics of Usher syndrome. Clin Genet 63:431–444. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER (2003b). PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12:3215–3223. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Zahorsky-Reeves J, Wright CG, Pawlowski KS, Erway LC, Stubbs L, Woychik RP (2000). Neuroepithelial defects of the inner ear in a new allele of the mouse mutation Ames waltzer. Hear Res 148:181–191. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP (2001a). The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet 27:99–102. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, Schwartz S, Lee C, Morton CC, Mullins RF, Ramesh A, Van Camp G, Hageman GS, Woychik RP, Smith RJ, Hagemen GS (2001b). Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 10:1709–1718. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Stahl JS, Jones SM, Pawlowski KS, Wright CG (2005). Characterization of vestibular dysfunction in the mouse model for Usher syndrome 1F. J Assoc Res Otolaryngol 6:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assad JA, Shepherd GM, Corey DP (1991). Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron 7:985–994. [DOI] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB (2005). Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7:148–156. [DOI] [PubMed] [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C (2002). Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J 21:6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS (2004). TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature 432:723–730. [DOI] [PubMed] [Google Scholar]
- Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, Perfettini I, Legrain P, Richardson G, Hardelin JP, Petit C (2005). Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet 14:401–410. [DOI] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K (2001). Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 27:103–107. [DOI] [PubMed] [Google Scholar]
- Frank M, Kemler R (2002). Protocadherins. Curr Opin Cell Biol 14:557–562. [DOI] [PubMed] [Google Scholar]
- Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP (2001). FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J Neurosci 21:7013–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Walker RG (2001). Molecular basis of mechanosensory transduction. Nature 413:194–202. [DOI] [PubMed] [Google Scholar]
- Goodyear R, Richardson G (1999). The ankle-link antigen: an epitope sensitive to calcium chelation associated with the hair-cell surface and the calycal processes of photoreceptors. J Neurosci 19:3761–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear RJ, Legan PK, Wright MB, Marcotti W, Oganesian A, Coats SA, Booth CJ, Kros CJ, Seifert RA, Bowen-Pope DF, Richardson GP (2003). A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci 23:9208–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear RJ, Marcotti W, Kros CJ, Richardson GP (2005). Development and properties of stereociliary link types in hair cells of the mouse cochlea. J Comp Neurol 485:75–85. [DOI] [PubMed] [Google Scholar]
- Hampton LL, Wright CG, Alagramam KN, Battey JF, Noben-Trauth K (2003). A new spontaneous mutation in the mouse Ames waltzer gene, Pcdh15. Hear Res 180:67–75. [DOI] [PubMed] [Google Scholar]
- Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP (1997). Unconventional myosins in inner-ear sensory epithelia. J Cell Biol 137:1287–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG (2002). A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 108:371–381. [DOI] [PubMed] [Google Scholar]
- Hudspeth AJ (1997). How hearing happens. Neuron 19:947–950. [DOI] [PubMed] [Google Scholar]
- Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG (2000). High-resolution structure of hair-cell tip links. Proc Natl Acad Sci USA 97:13336–13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa Y, Mburu P, Morse S, Kominami R, Townsend S, Brown SD (2005). Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum Mol Genet 14:391–400. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP (2002). Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci 5:41–47. [DOI] [PubMed] [Google Scholar]
- Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB (2005). Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol 280:295–306. [DOI] [PubMed] [Google Scholar]
- Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS (1999). Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci 19:6267–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon AM, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ (2003). Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet 34:421–428. [DOI] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP (2003). Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci 23:4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, Kros CJ, Richardson GP, Petit C (2005). Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol 280:281–294. [DOI] [PubMed] [Google Scholar]
- Müller U, Evans AL (2001). Mechanisms that regulate mechanosensory hair cell differentiation. Trends Cell Biol 11:334–342. [DOI] [PubMed] [Google Scholar]
- Osborne MP, Comis SD (1990). Action of elastase, collagenase and other enzymes upon linkages between stereocilia in the guinea-pig cochlea. Acta Otolaryngol 110:34–45. [DOI] [PubMed] [Google Scholar]
- Osborne MP, Comis SD, Pickles JO (1984). Morphology and cross-linkage of stereocilia in the guinea-pig labyrinth examined without the use of osmium as a fixative. Cell Tissue Res 237:43–48. [DOI] [PubMed] [Google Scholar]
- Raphael Y, Kobayashi KN, Dootz GA, Beyer LA, Dolan DF, Burmeister M (2001). Severe vestibular and auditory impairment in three alleles of Ames waltzer (av) mice. Hear Res 151:237–249. [DOI] [PubMed] [Google Scholar]
- Reiners J, Marker T, Jurgens K, Reidel B, Wolfrum U (2005). Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). Mol Vis 11:347–355. [PubMed] [Google Scholar]
- Rzadzinska AK, Derr A, Kachar B, Noben-Trauth K (2005). Sustained cadherin 23 expression in young and adult cochlea of normal and hearing-impaired mice. Hear Res 208:114–121. [DOI] [PubMed] [Google Scholar]
- Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP (1998). Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125:557–566. [DOI] [PubMed] [Google Scholar]
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Muller U (2002). The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci USA 99:14946–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Muller U (2004). Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 428:950–955. [DOI] [PubMed] [Google Scholar]
- Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, Nicolson T (2004). Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428:955–959. [DOI] [PubMed] [Google Scholar]
- Stauffer EA, Scarborough JD, Hirono M, Miller ED, Shah K, Mercer JA, Holt JR, Gillespie PG (2005). Fast adaptation in vestibular hair cells requires Myosin-1c activity. Neuron 47:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel KP, Kros CJ (2001). A genetic approach to understanding auditory function. Nat Genet 27:143–149. [DOI] [PubMed] [Google Scholar]
- Udovichenko IP, Gibbs D, Williams DS (2002). Actin-based motor properties of native myosin VIIa. J Cell Sci 115:445–450. [DOI] [PubMed] [Google Scholar]
- Whitlon DS (2004). Cochlear development: hair cells don their wigs and get wired. Curr Opin Otolaryngol Head Neck Surg 12:449–454. [DOI] [PubMed] [Google Scholar]
- Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR, Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA (2000). The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]