

Abstract
Fibroblast growth factor (FGF) family signaling mediates cell-to-cell communication in development and organ homeostasis in adults. Of the FGF receptor (FGFR) isotypes, FGFR4 is the sole resident isotype present in mature parenchymal hepatocytes. FGFR1 that is normally associated with activated nonparenchymal cells appears ectopically in hepatoma cells. Ectopic expression and chronic activity of FGFR1 in hepatocytes accelerates diethylnitrosamine (DEN)-initiated hepatocarcinogenesis by driving unrestrained cell proliferation and tumor angiogenesis. Hepatocyte FGFR4 mediates liver’s role in systemic cholesterol/bile acid and lipid metabolism and affects proper hepatolobular restoration after damage without effect on cell proliferation. Here we ask whether FGFR4 plays a role in progression of hepatocellular carcinoma (HCC). We report that although spontaneous HCC was not detected in livers of FGFR4-deficient mice, the ablation of FGFR4 accelerated DEN-induced hepatocarcinogenesis. In contrast to FGFR1 that induced a strong mitogenic response and depressed rate of cell death in hepatoma cells, FGFR4 failed to induce a mitogenic response and increased the rate of cell death. FGFR1 but not FGFR4 induced cyclin D1 and repressed p27 expression. Analysis of activation of Erk, JNK and PI3K-related AKT signaling pathways indicated that in contrast to FGFR1, FGFR4 failed to sustain Erk activation and did not activate AKT. These differences may underlie the opposing effects of FGFR1 and FGFR4. These results suggest that in contrast to ectopic FGFR1 that is a strong promoter of hepatoma, resident FGFR4 that mediates differentiated hepatocyte metabolic functions also serves to suppress hepatoma progression.
Keywords: FGF, cholesterol metabolism, hepatocellular carcinoma, liver adenoma, metabolism, tyrosine kinase signaling
INTRODUCTION
The tripartite fibroblast growth factor (FGF) signaling system comprised of oligomeric combinations of variants of four transmembrane tyrosine kinases (FGFR), diverse oligosaccharides within heparan sulfate and 18 transmembrane receptor-activating FGFs is a ubiquitous mediator of tissue homeostasis through sensing perturbation and mediating cell-to-cell communication [1]. Cell specific expression of FGFR isotype, heparin sulfate oligosaccharide motifs and other co-factors combine to confer cell and tissue specificity of FGF signaling both in respect to activating FGF [2,3] and activation of downstream intracellular pathways [4,5]. Dependent on cellular context, specific FGF, FGFR and heparan sulfate combinations may have opposite endpoints even within the same tissue [6,7]. In addition to cell proliferation which has been most widely studied, FGF signaling impacts a wide variety of phenotypic responses related to tissue homeostasis that include cell migration, adhesion, death, differentiation and specialized functions [4,5]. Most recently FGF signaling has been implicated in organism metabolic homeostasis that includes cholesterol/bile acid, lipid, glucose and phosphate homeostasis [3,8–11].
FGFR4 is the single FGFR isotype present in mature resting hepatocytes [2]. Expression of other FGFR isotypes in liver appears to be limited to nonparenchymal cells, hepatocyte progenitors and associated with liver perturbations or abnormalities [2,12,13]. Studies using FGFR4−/− mice and mice overexpressing constitutively active FGFR4 in hepatocytes have revealed that hepatocyte FGFR4 has no impact on cellularity of normal or regenerating liver or cell proliferation rate during response to liver injury [8,14]. However, studies using the same mouse models revealed a major role of hepatocyte FGFR4 in systemic control of cholesterol/bile acid metabolism and lipid metabolism through metabolite-controlled transcription networks in the hepatocyte [8,11,15]. Exaggerated carbon tetrachloride (CCl4)-induced liver injury and fibrosis in FGFR4−/− mice also suggested a metabolic role of hepatocyte FGFR4 in limitation of internal liver injury by toxic products of liver metabolism [14]. In marked contrast to FGFR4, FGFR1 in hepatocytes has high impact on cellular homeostasis. Ectopic expression of FGFR1 and its activating FGFs is a commonly observed property of differentiated hepatoma cells [13,16,17]. Forced ectopic expression of constitutively active FGFR1 (caFGFR1) in hepatocytes increased the number of normal hepatocytes undergoing DNA synthesis in response to partial hepatectomy and accelerated diethylnitrosamine (DEN)-initiated hepatocarcinogenesis through driving cell cycling and VEGF-induced angiogenesis [18].
The role of resident hepatocyte FGFR4 in development and progression of hepatoma remains to be clarified. In addition to the lack of effect of ablation of FGFR4 on hepatocyte proliferation relative to ectopic FGFR1, other lines of evidence suggest that the resident hepatocyte FGFR4 is a candidate for limiting hepatoma progression rather than promoting it. FGF1 which binds to all four FGFR isotypes present on the differentiated hepatoma cell line HepG2 at low doses exhibits a positive and at higher doses a net negative effect on HepG2 cell growth [19]. In contrast, FGF2 that is limited from binding to FGFR4 by hepatocyte heparan sulfate [2] exhibits only the low dose positive mitogenic effect. The overexpression of FGF21 in hepatocytes delayed onset of DEN-initiated tumors, but failed to further delay rate of progression to HCC once tumors were established [20]. This switch from repressor to promoter correlated with alterations in FGFR4 and the ectopic expression of FGFR1 during progression of hepatomas. FGF21, a relatively hepatocyte-specific FGF, is a candidate autocrine ligand for FGFR4 [3]. In parenchymal cells of the prostate [6], bladder [21], salivary gland [22] and colon [23,24], the resident FGFR isotype promotes tissue homeostasis and suppresses tumor progression. This is in dramatic contrast to normally non-parenchymal FGFR1 that drives malignant progression when it appears in the parenchymal cells of these tissues. However, there are suggestions in other tissue contexts that FGFR4 may be tumor-promoting [25–28] Although it remains to be determined whether the putative mouse ortholog, FGF15, acts similarly, chronic administration of human FGF19, a candidate ligand for hepatocyte FGFR4 [29,30], has been reported to cause hepatomas in mice.
In this study, we show that similar to mice expressing ectopic hepatocyte FGFR1 [18] DEN-initiated hepatocarcinogenesis is accelerated in mice devoid of resident hepatocyte FGFR4. When restored in mouse hepatoma cells devoid of FGFR4 and FGFR1, FGFR4 had no mitogenic effect, but increased the rate of apoptosis while FGFR1 was strongly mitogenic with an opposite effect on apoptosis. The opposite endpoints of the two FGFR isotypes correlated with specific differences in elicited downstream signaling pathways. These results indicate that in contrast to ectopic FGFR1 that is a strong tumor promoter, the resident hepatocyte FGFR4 that normally mediates multiple functional roles of differentiated hepatocytes is a hepatoma suppressor.
MATERIALS AND METHODS
Mouse Models
FGFR4−/− mice were as previously[31]. FGFR4−/− and WT control mice used in this study were produced from the mating of FGFR4+/− mice. Only male mice were used due to the higher incidence of DEN-induced HCC in males. All mouse work was performed in accordance with the Institutional Animal Care and Use Committee at the Institute of Biosciences and Technology, Texas A&M Health Science Center.
Analysis of Liver Phenotype
Analysis of livers from DEN-treated mice was described previously [18]. Briefly, cohorts of FGFR4−/− and WT littermates were subjected to a single intraperitoneal injection of 10 µg/g body weight of DEN (Sigma, St. Louis, MO) 15 days after birth. Mice were sacrificed at 4, 6, 8, 10 and 12 months after birth. The body and liver weights, the number of visible tumor and the size of maximum visible tumor were recorded. Representative liver samples were taken from each lobe, excised and fixed overnight in Histochoice Tissue Fixative MB (Amresco, Solon, OH). They were then dehydrated through a series of ethanol treatments, embedded in paraffin, sectioned serially at 5 µm and stained with hematoxylin/eosin (H&E) for histopathological analysis according to standard methods.
Hepatoma Cell Culture and Forced Expression by Transfection
Human hepatoma cell lines were maintained and primary mouse hepatoma cell cultures were established and maintained as described previously [18]. Individual colonies of cells in primary culture were isolated and expanded to establish clonal secondary cultures. The clonal cultures were continuously cultured over a period of 3 months until they stabilized in respect to rate of expansion to confluence at which time they were utilized for stable transfections. Stock cultures were banked at every fifth passage.
Plasmids containing the full length cDNA coding for the human FGFR1 and FGFR4 have been described [2,6]. Expression vectors were prepared by excision of FGFR1 and FGFR4 cDNAs from the plasmids by treatment with BamH I, and Hind III and Xho I, respectively. Excised cDNAs were then inserted into eukaryotic expression vector pcDNA3.1/zeo using the same restriction sites used in excision of each construct. For stable transfections, 2 × 105 hepatoma cells were seeded in a 6-well tissue culture plate and transfected the next day with 2 µg pcDNA3.1/zeo-hFGFR1, pcDNA3.1/zeo-hFGFR4 or vector only with lipofecAMINE (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Two days after transfection, cells were transferred to 100 mm dishes and selected in medium containing zeocin (Invitrogen, Carlsbad, CA) at a final concentration of 750 µg/ml. Two weeks later, individual zeocin-resistant colonies were isolated, expanded and screened for FGFR1 and FGFR4 expression by RNA protection assays (RPA) and Western blot.
Gene Expression Analyses
For immunochemical analyses, hepatoma cell lysates were prepared as described previously [18]. Protein concentrations were determined using the BCA Protein Assay reagent (Pierce, Rockford, IL). For immunoblots, 30 µg protein was applied to 8% SDS-PAGE, transferred to Hybond-P membrane (Amersham, Piscataway, NJ) which was incubated with the primary antibody, washed 3 times and then incubated with the second anti-mouse or rabbit IgG conjugated to horseradish peroxidase (Bio-Rad, Hercules, CA). Bands were visualized by development with the ECL-Plus detection reagents (Amersham, Piscataway, NJ). Antibodies used for Western blot were as follows: Antibodies against FGFR1, FGFR4, p21, p27 and cyclin D1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against Erk1/2, phospho-Erk1/2, JNK, phospho-JNK, AKT and phospho-AKT were from Cell Signaling Technology (Danvers, MA).
For analysis of expression at the mRNA level, total RNA was isolated from cells with Ultraspec RNA isolation system (Biotecx Laboratories, Houston, TX). Specific mRNAs were measured by RPA using the HybSpeed RPA kit (Ambion, Austin, TX). For RPA about 40 µg of total RNA was hybridized with 1 × 105 cpm of [P32]–labeled specific and β-actin antisense riboprobes in the same reaction mixture. After treatment with ribonuclease, protected products were analyzed on 5% polyacrylamide sequencing gels, followed by autoradiography. Reverse transcription was carried out with Superscript II (Life Technologies, Grand Island, NY) and random primers according to protocols provided by the manufacture. The PCR was carried out for 40 cycles at 94°C for 1 minute, 55 or 60°C for 1 minute, and 72°C for 1 minute using TaqDNA Polymerase (Promega, Madison, WI) and specific primers. Murine connexin 32 was amplified by RT-PCR from mouse liver using sense primer 5’-ACATCTCAGGGACACTGTGGTGGA and antisense primer 5’-AGCATAAAGACAGTGAAGACGGTT. Murine transferrin was amplified by RT-PCR from mouse liver using sense primer 5’-AAGCTCCAAACCATGTTGTGGTCTC and antisense primer 5’- TTTCAGGTGTGGTACCCTCTGGAAG. Murine FGFR2 was amplified by RT-PCR from mouse liver using sense primer 5’-ATCCTTTCACTCTGCATGGTT and antisense primer 5’-GCGCTTGGTCAGCTTGTGCAC. Murine FGFR3 was amplified by RT-PCR from mouse liver using sense primer 5’-CTGCAAGTGCTAAATGCCTCCCACG and antisense primer 5’- TGCCCTCGGAATTCTTTGCCATTCT. Murine β-actin, albumin, FGFR1 and FGFR4 cDNAs were described previously [8,15,18]. The products of RT-PCR were verified by sequencing. Riboprobes complementary to part of the cDNAs described above which had been subcloned into pBluescript-SK and were transcribed into [P32]–labeled antisense riboprobes by T3 or T7 RNA polymerase using the MAXiscript kit (Ambion, Austin, TX). The size of probes and the predicted protected fragments were as follows: murine albumin, 365 and 287 nucleotides; murine connexin 32, 274 and 208; murine transferrin, 251 and 185; murine FGFR1, 315 and 239 nucleotides; murine FGFR2, 279 and 213 nucleotides; murine FGFR3, 377 and 301 nucleotides; murine FGFR4, 267 and 198 nucleotides; β-actin, 197 and 139 nucleotides.
Assessment of DNA Synthesis and Cell Death
The growth properties of FGFR4−/−/hFGFR1 and FGFR4−/−/hFGFR4 hepatoma cells were assessed by [3H]thymidine incorporation in 3 × 104 cells/well in 24-well plates as follows. Cells were allowed to attach overnight in 0.5 ml RD medium (1:1 RPMI and DMEM) containing 10% fetal bovine serum (FBS). After starved for 24 hr, cells were incubated for an additional 24 hr in serum-free medium with or without 20 ng/ml FGF1. For the last 6 hr of incubation, 0.25 µCi/well [3H]thymidine was added. Cells were then washed with PBS, fixed in cold 10% trichloracetic acid and lysed in 0.4 N NaOH. Thymidine incorporation was determined using a scintillation counter. For apoptosis assays, 2 × 104 cells per well were seeded in 96-well plates. The next day, cells were incubated in a serum-free medium with or without 20 ng/ml FGF1 for 72 hr. For the last 30 mins of incubation, RD medium containing APOPercentage Dye Label (Biocolor Ltd., Belfast) was added. After 3 washes with PBS to remove unbound dye, cell bound dye was released into solution by adding 100 µl/well APOPercentage Dye release reagent. Data from plates were collected using a VERSAmax microplate reader at 550 nm. Three independent experiments of four wells each were done for each variable with two independent cell lines.
Statistical Analysis
Values were expressed as the mean ± standard deviation (SD) from the numbers of replicates described in legends to figures. Statistical significance was determined by Student’s t-test or Fisher’s exact test with a significance threshold of P<0.05.
RESULTS
Ablation of FGFR4 Accelerates DEN-induced Hepatocarcinogenesis
FGFR4−/− mice exhibit no signs of neoplasia, preneoplasia or other abnormalities in liver [8,31] coincident with the otherwise dramatic effects on metabolic homeostasis [8,9,11]. To test for a potential role of FGFR4 in hepatocarcinogenesis, mice were subjected to a single exposure of 10 µg DEN per g body weight at 15 days of age to initiate hepatoma as described previously [18,20]. Strain-specific effects were minimized by use of littermates from heterozygous breeding. Livers of both wild-type (WT) and FGFR4−/− mice were examined at 4, 6, 8, 10 and 12 months after birth. Visible surface tumor nodules appeared in the FGFR4−/− mouse livers as early as 4 months (Figure 1, Table 1) with 60% of ten animals exhibiting tumors. This was in marked contrast to the WT mice where only 11% (1 of 9 animals) showed a tumor in the same 4 month period. Only adenomas were observed in the DEN-treated WT livers at a frequency of 71 and 100 percent, respectively, at 8 and 10 months (Table 1). No overt HCC was apparent in this period. In contrast, lesions characteristic of HCC were apparent in 22 and 50 percent of the FGFR4−/− livers, respectively, at 8 and 10 months (Figure 1, Table 1). The accelerated liver tumor development in FGFR4−/− mice was further reflected by increased tumor burden, tumor multiplicity and maximum tumor size compared with age-matched WT mice (Table 1). The tumor burden indicated by both ratio of liver weight to body weight and maximum tumor size was over 2-fold greater at 12 months and tumor multiplicity was 3-fold higher at 10 months in FGFR4−/− mice compared to WT controls. Thus, in striking contrast to FGFR1 that is a strong promoter of carcinogenesis in the hepatocyte context [18], the resident hepatocyte FGFR4 plays an opposing tumor suppressive role in both initiation and progression of HCC induced by carcinogen.
Figure 1.
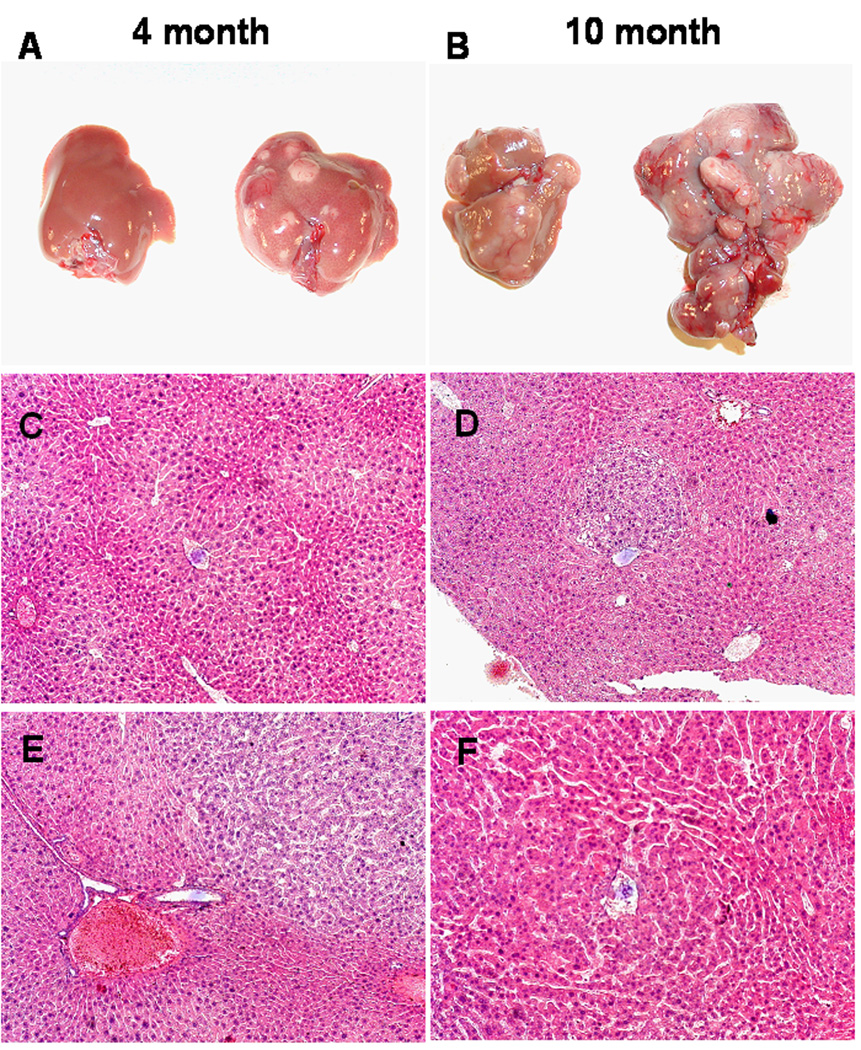
Ablation of FGFR4 accelerates DEN-induced liver tumors. Representative livers from WT and FGFR4−/− mice exposed to DEN and harvested as described in Materials and Methods. (A,B) Representative livers from WT (left) and FGFR4−/− (right) mice at 4 and 10 months after birth are shown. (C,D), Normal morphology in WT liver (C) and adenoma in a FGFR4−/− liver (D) at 4 months after birth. (E, F) Adenoma in a WT liver (E) and HCC in a FGFR4−/− liver (F) at 10 months after birth (100X magnification).
Table 1.
Hepatic Tumors in DEN-treated Wildtype (WT) and FGFR4−/− micea
| Mouse genotype | Month after birth | Tumor incidence Mice (%) | HCC incidence Mice (%) | LW/BW x 100 | Number of tumors | Max tumor size |
|---|---|---|---|---|---|---|
| WT | 4 | 1/9 (11) | 0/9 (0) | 4.02 ± 0.44 | 0 | 0 |
| 6 | 2/7 (29) | 0/7 (0) | 4.11 ± 0.46 | 0.57 ± 0.28 | 0.04 ± 0.1 | |
| 8 | 5/7 (71) | 0/7 (0) | 4.45 ± 0.47 | 3.28 ± 0.53 | 0.21 ± 0.14 | |
| 10 | 7/7 (100) | 0/7 (0) | 4.86 ± 0.54 | 9.0 ± 2.89 | 0.43 ± 0.18 | |
| 12 | 7/7 (100) | 2/7 (29) | 5.46 ± 1.37 | 13.8 ± 4.59 | 0.54 ± 0.23 | |
| FGFR4 −/− | 4 | 6/10 (60)* | 0/10 (0) | 5.04 ± 0.52* | 6.29 ± 2.47* | 0.15 ± 0.06 |
| 6 | 6/7 (86) | 0/7 (0) | 5.73 ± 0.43† | 22.57 ± 4.8† | 0.29 ± 0.17* | |
| 8 | 9/9 (100) | 2/9 (22) | 7.94 ± 2.64† | 28.17 ± 6.36* | 0.64 ± 0.26* | |
| 10 | 8/8 (100) | 4/8 (50) | 7.05 ± 1.39† | 32.16 ± 8.85* | 0.8 ± 0.24† | |
| 12 | 8/8 (100) | 7/8 (87.5)* | 12.64 ± 2.63** | NC | 1.23 ± 0.21† | |
A single ip injection of DEN (10 µg/g body weight) was administered to mice 15 d after birth. Incidence/total animals examined was scored as described in Materials and Methods. Percentages are indicated in parentheses. The numbers of visible tumors on the liver surface and maximum tumor size are also indicated. Tumor burden was estimated by liver weight (LW) divided by total body weight (BW) ± SD (n = no. animals indicated under incidence). Significance of difference in the knockout mice from WT mice at the same time determined by Fisher’s Exact Test (Tumor and HCC incidence) or Student’s t-test (LW/BW X 100, Number of tumor and Max tumor size) is indicated by
p<0.05
p<0.01
p<0.001.
NC, Not counted.
Aberrant Expression of FGFR4 in Hepatoma Cell Lines
Aberrant isoforms of FGFR4 have been reported in several types of tumors, but not hepatomas [32,33]. We examined the profiles of FGFR4 transcripts and antigens in several human hepatoma cell lines by Northern blot mRNA analysis and immunochemical analysis with an antibody against the C-terminal domain. Although no truncation of FGFR4 mRNAs was observed in all four human hepatoma cell lines (data not shown), two of them exhibited abnormal species of FGFR4 in addition to the expected 110 KDa full-length species (Figure 2). One of three hepatoma cell lines derived from DEN-induced hepatomas in FGFR4−/− mice produced an abnormal 56 KDa species from transfected human FGFR4 (Figure 3C). The 56 KDa species was observed for native FGFR4 in human hepatoma HepG2 cells (Figure 2). These results suggest that FGFR4 is altered in hepatoma cells although it remains to be determined whether the alteration has functional impact on hepatocarcinogenesis.
Figure 2.

Aberrant expression of FGFR4 in hepatoma cells. The expression of pattern of FGFR4 protein was analyzed by immunoblot (Materials and Methods) in the indicated human hepatoma cell lines.
Figure 3.

Establishment of stable FGFR1 and FGFR4 expressing cell lines in FGFR4−/− hepatoma cells. FGFR4−/− hepatoma cells were established from liver tumors in FGFR4−/− mice at 4 month. (A) Expression of albumin, connexin32 and transferrin mRNA. Three independent clonal lines 28, 38, and 50 out of 60 clonal cultures are shown. (B) Expression of FGFR1-4 mRNA. Representative clonal lines 38 and 50 exhibiting low or absent level of all four FGFR (38,50) are shown. (C) Expression of transfected human (h) FGFR1 and FGFR4 in clone 50. Expression of mRNA and protein assessed by RPA and immunoblot in three independent stably transfected clones selected for analysis are shown. FGFR4−/−/hFGFR1 clone A4 and A28 and FGFR4−/−/hFGFR4 clone B1 and C8 that expressed comparable levels of each receptor were used in subsequent analyses.
FGFR1 and FGFR4 Have Opposing Effects on Differentiated Hepatoma Cell Proliferation and Death
Stable clonal lines of cells were derived from DEN-induced hepatomas in FGFR4-deficient mice after 4 month by methods described previously [18]. Clonal lines expressed sustained levels of mouse albumin, connexin32 and transferrin mRNA, suggesting that they were well differentiated hepatoma cells (Figure 3A). Several clonal lines exhibited no or very low levels of FGFR isoypes in addition to FGFR4 (Figure 3B) and were used to compare the activities of FGFR1 and FGFR4 in the same cell type. Cells were transfected with mammalian expression vectors containing full length human FGFR1 and FGFR4 cDNA driven by the cytomegalovirus (CMV) promoter. Vector without insert was used as control. Positively expressing clones of FGFR1 and FGFR4 were screened by ribonuclease protection assay (RPA) (Figure 3C, upper panel) and Western blot (Figure 3C, lower panel). Two clones exhibiting similar expression levels of mRNA and intact protein were picked for further analysis.
The effects of FGFR1 and FGFR4 on cell proliferation were assessed by [3H]-thymidine incorporation. In the absence of FGF1 which was used as the common FGF agonist among all FGFR isotypes, no difference in basal level of DNA synthesis was observed between controls, FGFR1- or FGFR4-expressing cells. FGF1 stimulated DNA synthesis in the FGFR1-expressing hepatoma cells 2.5 times the basal levels observed in control cells. FGF1 failed to increase basal levels of DNA synthesis in FGFR4-expressing cells (Figure 4A).
Figure 4.

Differential effects of FGFR1 and FGFR4 on DNA synthesis and cell death of hepatoma cells. (A) DNA synthesis. Cells were cultured in serum-free medium with and without FGF1 (20 ng/ml) for 24 hr prior to addition of [3H]thymidine. (B) Cell death. Cells were cultured in serum-free medium with and without FGF1 (20 ng/ml) for 72 hr prior to addition of APOPercentage Dye. The two bars represents an independent transfected cell line described in Figure 4. The data is the mean ± SD of 12 wells from three independent experiments of four wells each for both DNA synthesis and cell death assays. * P<0.05, ** P<0.01, *** P<0.001 (paired tests with no FGFR transfected).
We then investigated potential differential effects of the two FGFR isotypes on cell death assessed with the APOPercentage Dye Label. Little apoptosis was observed in FGFR1 or FGFR4 expressing cells in standard growth medium (data not shown). In the absence of FGF1, serum deprivation induced a similar rate of apoptosis in cells devoid of FGFR and those expressing FGFR1 or FGFR4 (Figure 4B). Addition of FGF1 induced a 30% decrease in apoptosis (p<0.05) in cells expressing FGFR1, but a 1.9-fold increase (p<0.005) in hepatoma cells expressing FGFR4 (Figure 4B). These results indicate that FGFR1 drives hepatoma cell proliferation and contributes to inhibition of stress-induced apoptosis. Conversely, FGFR4 has no effect on hepatoma cell proliferation, but protects against stress-induced apoptosis.
Differential Impact of FGFR1 and FGFR4 on Signaling Pathways
To test for a differential effect of FGFR1 and FGFR4 on downstream signaling in the differentiated hepatoma cells, we compared effect on activation of Erk, JNK and PI3K-related AKT that are affected by FGF signaling in diverse contexts. Addition of FGF1 to both FGFR1- and FGFR4-expressing cells increased phosphorylation of Erk about equally in 15 min (Figure 5A). However, FGFR4 failed to sustain the activation which dropped markedly by 30 min relative to Erk in FGFR1-expressing cells which lasted for 1 hr. In contrast to Erk, both cells expressing FGFR1 and FGFR4 sustained the activation of JNK. Relative to FGFR1, FGFR4 failed to increase phosphorylation of AKT (Figure 5A).
Figure 5.
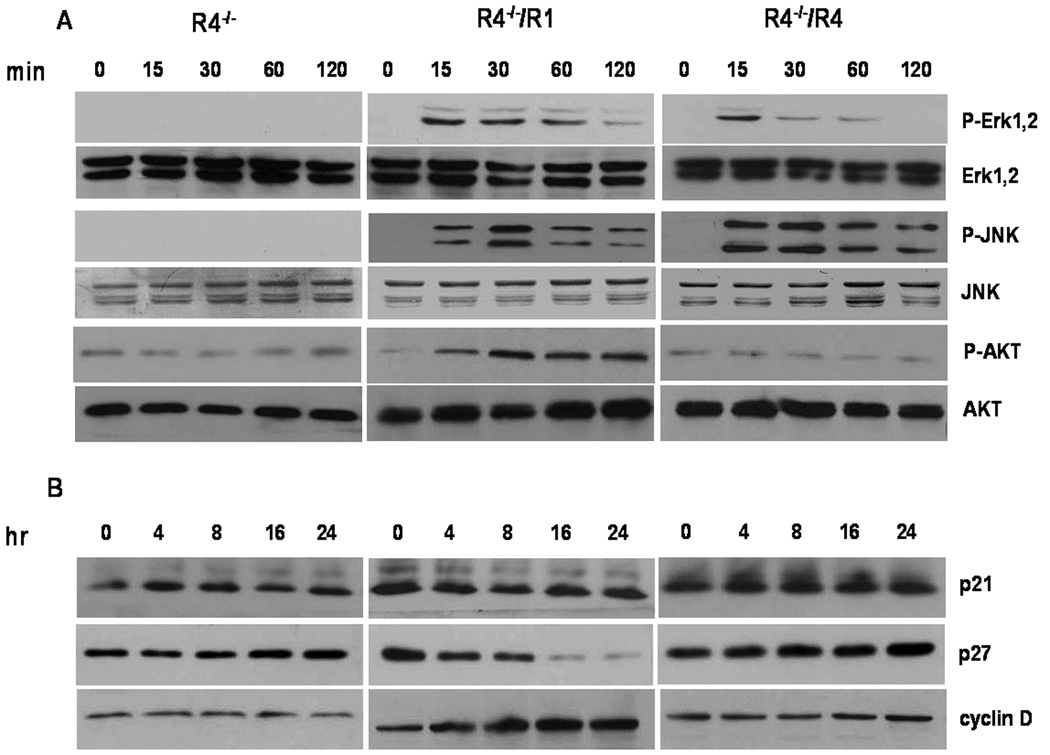
Differential activation of signaling pathways and G1/S control factors by FGFR1 and FGFR4 in hepatoma cells. Hepatoma cells were maintained in serum-free medium overnight and then treated with 20 ng/ml FGF1 for the indicated times prior to preparation of cell lysates. (A) Effect on phosphorylated Erk, JNK and AKT. Cell lysates were analyzed by immunoblotting with antibodies against phospho-Erk, phospho-JNK and phospho-AKT. The membranes were reprobed with antibodies detecting total Erk, JNK and AKT as indicated. (B) Analysis of G1/S regulators. Cell lysates were analyzed by immunoblotting with antibodies against p21, p27 and cyclin D1.
Ectopic FGFR1 in hepatocytes and hepatoma cells in vivo and in vitro [18] strongly correlates with and drives increased rates of cell cycling in contrast to FGFR4. This predicts that FGFR1 might selectively downregulate cell cycle inhibitors and upregulate cell cycle promoters relative to FGFR4. We examined the consequences of FGFR1 and FGFR4 activation on expression of G1/S cell cycle regulators, p21, p27 and cyclin D1 in the mouse hepatoma cells. FGF1 caused downregulation of CDK inhibitor p27 in FGFR1-expressing cells, but had no effect in FGFR4-expressing cells (Figure 5b). A comparable difference of the two FGFR on p21 was not apparent. FGFR1 caused an upregulation of cyclin D1 expression in FGFR1-expressing cells with no comparable effect in FGFR4-expresssing cells. Taken together, these results are consistent with the idea that resident hepatocyte FGFR4 and ectopic FGFR1 have opposing effects on cell population growth rates in differentiated hepatoma cells and hepatocytes due to differential impact on downstream cell signaling pathways.
DISCUSSION
FGFR4 is the resident FGFR isotype in mature hepatocytes. It plays key roles in hepatocyte functions that includes cholesterol to bile acid synthesis, lipid and xenobiotic metabolism {Yu, 2000 #32; Yu, 2002 #53; Huang, 2007 #279}. It plays no direct role in hepatocyte proliferation in response to loss of liver mass or injury although it plays a role in restoration of overall hepatolobular architecture [14]. In contrast to FGFR4, the homologous FGFR family member, FGFR1 that is most widely associated with nonparenchymal or mesenchymal cells only appears ectopically in differentiated hepatoma cells where it drives abnormal rates of cell cycling and in later stages angiogenesis [18]. Unlike resident FGFR4, ectopic FGFR1 in normal hepatocytes accelerates rate of hepatocyte division in response to injury. The role of resident hepatocyte FGFR4 in liver tumorigenesis is unclear.
Several studies have triggered suggestions that FGFR4 may be a positive contributor to tumorigenesis. An FGFR4 germline polymorphism with a substitution of glycine by arginine at codon 388 in the transmembrane domain correlates with advanced progression of tumors of breast, colon and prostate [25,26]. Overexpression of FGFR4 has been observed in breast, gynecological and thyroid cancers [27,28]. Variants of FGFR4 that have been observed in pituitary [33,34] and lung [35] tumors have been proposed to be tumor-promoting. Forced expression or systemic administration of FGF19 that is a candidate activating FGF for the metabolic functions of hepatocyte FGFR4 [9] has been reported to cause hepatoma-like liver lesions [29]. We show that some human hepatoma cells exhibit variants in FGFR4 that most likely occur by posttranslation proteolytic modification. The appearance of a 56 kDa truncate of FGFR4 expressed from an external transfection and detected by antibody against a C-terminal epitope indicates a posttranslational truncation somewhere near the transmembrane domain. However, the impact of the truncation on net FGFR4 activity remains to be established.
In this study we show that although a germline deficiency in FGFR4 alone has no apparent effect on liver neoplasia, DEN-initiated hepatocarcinogenesis is dramatically accelerated in the absence of FGFR4. This suggests that the resident hepatocyte FGFR4 acts as a suppressor of hepatoma progression. This is in marked contrast to FGFR1 that is a strong promoter of hepatoma progression in the same context. The concurrent evolution of a tumor suppressive effect of the resident FGFR in mature parenchymal cells along with a role in maintenance of tissue homeostasis and specialized organ function may represent a general protective mechanism against development of parenchymal cell tumors. Similar differences in the resident mature parenchymal cell FGFR and ectopic FGFR1 have been observed in prostate [6], bladder [21], salivary gland [22] and colorectal tissue [23,24]. Although our results indicate a tumor suppressive effect of FGFR4 within hepatocytes in vivo, we cannot eliminate additional contributions to promotion of hepatomas due to systemic metabolic alterations caused by the FGFR4 deficiency that include hyperlipidemia, insulin resistance, hypercholesterolemia and elevated bile acids [8,11]. Recently spontaneous hepatomas have been reported in mice deficient in the Farnesoid X receptor (FXR, NR1H4) coincident with elevated systemic bile acids similar to that observed in FGFR4−/− mice [36]. However, no spontaneous liver lesions have been observed to date in FGFR4-deficient mice. Lesions require a strong tumor initiator as DEN. FGFR1 expression was neither upregulated in normal livers nor in DEN-induced hepatomas in FGFR4−/− mice compared to WT mice (data not shown). This indicated that FGFR4 was not a suppressor of expression of tumor-promoting ectopic FGFR1 in hepatocytes or hepatoma cells. We cannot eliminate a contribution to suppression of the tumor promoting activity of ectopic FGFR1 by resident FGFR4 through a “dominant negative” mechanism as a result of heterodimerization in cells co-expressing the two FGFR isotypes. However, there is currently no evidence to suggest that FGFR4 and FGFR1 heterodimerize in the hepatocyte or hepatoma cell context.
To further clarify these issues and provide direct evidence that resident FGFR4 itself delays hepatocarcinogenesis, we determined how FGFR4 impacts growth rates of hepatoma cells in vitro. By screening multiple primary culture clones, we established hepatoma cell lines with little or no expression of any FGFR isotypes derived from DEN-induced tumors from FGFR4−/− mice. Indeed, reinstatement of FGFR4 in these FGFR-deficient cells exhibited no positive effect on rate of proliferation. Instead the presence of FGFR4 under conditions of environmental stress mimicked by deficient culture medium promoted a high rate of apoptotic cell death. This was in marked contrast to cells expressing FGFR1 that exhibited accelerated cell growth and reduced rates of apoptosis under similar conditions. This confirmed that FGFR4 and FGFR1 when acting alone in isolated hepatoma cells have opposing endpoints at the cellular level that are consistent with their opposing tumor suppression and tumor promotion effects, respectively, observed in animals. These results from isolated hepatoma cells argue that in addition to its role in the hepatocyte’s contribution to metabolic homeostasis, FGFR4 guards against hepatoma development by supporting phenotypic parameters associated with tumor suppression.
Although it has not been confirmed in intact tissue context, it has been proposed that FGF19 is a preferred activating FGF for FGFR4 and thus responsible for its metabolic activities in liver [37]. Liver neoplasias as a result of systemic elevation of FGF19 [29] and reduction of experimental tumors by an immunoblockade of FGF19 has been reported in mice [30]. However, systemic FGF19 exerts a tumor suppressive effect on xenografts of human and mouse tumors in immunosuppressed mice [38]. Our results showing that FGFR4 appears to exert a hepatoma-suppressive effect suggest that FGFR4 unlikely mediates the tumor-promoting effects of FGF19. We failed to observe a single spontaneous hepatoma in transgenic mice expressing FGF-independent, hyperactive FGFR4 in hepatocytes [15] for up to two years (unpublished observation). This indicates that chronic systemic levels of FGF19 may act on other targets than hepatocyte FGFR4 to result in induction of hepatomas. This is consistent with the report that mice with an FGFR4 gene deletion are still responsive to systemic FGF19 in respect to some metabolic monitors [39]. It should be noted that putative orthologs human FGF19 and mouse FGF15 are as divergent in sequence from each other as they are to other members of the FGF family within the same species. FGF19 particularly at high levels may activate ectopic tumor-promoting signaling pathways in mice that are not associated with activities of endogenous mouse FGF15.
Numerous studies indicate that FGFR4 and FGFR1 exhibit context-dependent qualitative and quantitative differences in tyrosine kinase activity, tyrosine phosphorylation and impact on downstream signaling mediators [40–44]. FGFR4 exhibited tyrosine phosphorylation, but was non-mitogenic and failed to exhibit responses typical of FGFR1 except for weak tyrosine phosphorylation of phospholipase C gamma in BAF3 murine lymphoid cells. Rat L6 myoblasts and mouse 3T3 fibroblasts exhibited DNA synthesis in response to both FGFR1 and FGFR4, but FGFR4 failed to activate a variety of downstream phosphoproteins characteristic of FGFR1 [41]. FGFR4-stimulated phosphorylation of mitogen activated kinases (MAPKs) was detected in L6E9 myoblasts at a fraction of that elicited by FGFR1 coincident with little effect on growth, survival and morphology relative to FGFR1 [42]. Using artificial constructs of FGFR1 and FGFR4 with the same ectodomain, the intracellular kinase domains of the two FGFR activated overlapping pathways [43]. In comparisons of drug-induced forced dimerization of the FGFR1 and FGFR4 intracellular domains in embryonic skeletal muscle cell lines, no phosphorylation of FGFR4 relative to FGFR1 was detectable [44].
Here we show that in a mouse hepatoma cell context both FGFR1 and FGFR4 initially activate the Erk pathway in similar fashion, but only FGFR1 can sustain the activation. Both FGFR1 and FGFR4 sustain the activation of JNK to about equal extent. Only FGFR1, not FGFR4, was capable of activating AKT in the hepatoma cell context. The FGFR1-associated sustained Erk and AKT activation might explain the increased rate of cell proliferation and modest rate of stress-induced apoptosis. The elevated rate of stress-induced apoptosis observed specifically in FGFR4-expressing cells is likely mediated by JNK signaling in absence of sustained Erk and AKT pathway activation. JNK signaling mediates in part the metabolic activities of FGFR4 in normal hepatocytes [15]. The depression of bile acid metabolism by ectopic FGFR1 in hepatocytes without the accompanying cell death may be due to its strong effects on JNK signaling concurrent with sustained Erk signaling [18]. The depression of p27 and stimulation of cyclin D1 by specifically FGFR1 in hepatoma cells further support the observation. Consistent with the absence of a direct role of resident hepatocyte FGFR4 on hepatocyte cell cycling in vivo [8] or in vitro (this report Fig. 4), our results indicated a complete absence of effect of FGFR4 signaling on these two regulators of cell cycling at the G1 to S boundary [45,46].
In summary, our study reveals that resident hepatocyte FGFR4 plays an additional role as a tumor suppressor in addition to mediating liver’s contribution to systemic lipid and glucose metabolism. This is the opposite of normally non-parenchymal FGFR1 that is a strong promoter of hepatic carcinogenesis when it appears in hepatocytes. The tumor suppressive effects of a parenchymal cell FGFR isotype that promotes tissue cellular homeostasis or specialized parenchymal cell function relative to an ectopic FGFR isotype may be a general paradigm within the FGF signaling family [6,21,22,24,47]. In numerous adenocarcinomas, the ectopic expression of normally mesenchymal- or stromal-derived FGFR1 in the adenocarcinoma cells seems most common and has been suggested as a marker of tumor-associated epithelial to mesenchymal cell transition (EMT) [48,49].
Hepatocyte FGFR4 is a potential target for intervention in systemic cholesterol/bile acid [8,50], lipid [11] and xenobiotic metabolism related to liver fibrosis [14]. Effects of FGFR4 at a yet to be identified non-hepatic site is also a target for intervention in glucose metabolism and diabetes [11]. The specific activating FGF ligands, control of their access to FGFR4 and mechanism of assembly and activation of the FGFR signaling complex for both its tumor suppression and metabolic activities remains to be determined. Whether the downstream pathways that target the two domains are similar or convergent is also of interest. Our results indicate that agonists applied to hepatocyte FGFR4 to modulate its metabolic functions will have an added benefit in suppression of hepatomas.
ACKNOWLEDGMENTS
This work was supported by Public Health Service Grants R01DK35310 from NIDDK and R01CA59971 from NCI.
Abbreviations
- DEN
diethylnitrosamine
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- HCC
hepatocellular carcinoma
- RPA
ribonuclease protection assay
- WT
wild type.
REFERENCES
- 1.McKeehan WL, Wang F, Kan M. The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog Nucleic Acid Res Mol Biol. 1998;59:135–176. doi: 10.1016/s0079-6603(08)61031-4. [DOI] [PubMed] [Google Scholar]
- 2.Kan M, Wu X, Wang F, McKeehan WL. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J Biol Chem. 1999;274(22):15947–15952. doi: 10.1074/jbc.274.22.15947. [DOI] [PubMed] [Google Scholar]
- 3.Kurosu H, Choi M, Ogawa Y, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282(37):26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci. 1999;4:D165–D177. doi: 10.2741/klint. [DOI] [PubMed] [Google Scholar]
- 5.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 6.Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997;57(23):5369–5378. [PubMed] [Google Scholar]
- 7.Jin C, McKeehan K, Guo W, et al. Cooperation between ectopic FGFR1 and depression of FGFR2 in induction of prostatic intraepithelial neoplasia in the mouse prostate. Cancer Res. 2003;63(24):8784–8790. [PubMed] [Google Scholar]
- 8.Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275(20):15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 9.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Yang C, Luo Y, Jin C, Wang F, McKeehan WL. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes. 2007;56(10):2501–2510. doi: 10.2337/db07-0648. [DOI] [PubMed] [Google Scholar]
- 12.Hu Z, Evarts RP, Fujio K, Marsden ER, Thorgeirsson SS. Expression of fibroblast growth factor receptors flg and bek during hepatic ontogenesis and regeneration in the rat. Cell Growth Differ. 1995;6(8):1019–1025. [PubMed] [Google Scholar]
- 13.Hu Z, Evarts RP, Fujio K, et al. Expression of transforming growth factor alpha/epidermal growth factor receptor, hepatocyte growth factor/c-met and acidic fibroblast growth factor/fibroblast growth factor receptors during hepatocarcinogenesis. Carcinogenesis. 1996;17(5):931–938. doi: 10.1093/carcin/17.5.931. [DOI] [PubMed] [Google Scholar]
- 14.Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Increased carbon tetrachloride-induced liver injury and fibrosis in FGFR4-deficient mice. Am J Pathol. 2002;161(6):2003–2010. doi: 10.1016/S0002-9440(10)64478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu C, Wang F, Jin C, Huang X, McKeehan WL. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem. 2005;280(18):17707–17714. doi: 10.1074/jbc.M411771200. [DOI] [PubMed] [Google Scholar]
- 16.Kin M, Sata M, Ueno T, et al. Basic fibroblast growth factor regulates proliferation and motility of human hepatoma cells by an autocrine mechanism. J Hepatol. 1997;27(4):677–687. doi: 10.1016/s0168-8278(97)80085-2. [DOI] [PubMed] [Google Scholar]
- 17.Ogasawara S, Yano H, Iemura A, Hisaka T, Kojiro M. Expressions of basic fibroblast growth factor and its receptors and their relationship to proliferation of human hepatocellular carcinoma cell lines. Hepatology. 1996;24(1):198–205. doi: 10.1053/jhep.1996.v24.pm0008707262. [DOI] [PubMed] [Google Scholar]
- 18.Huang X, Yu C, Jin C, et al. Ectopic activity of fibroblast growth factor receptor 1 in hepatocytes accelerates hepatocarcinogenesis by driving proliferation and vascular endothelial growth factor-induced angiogenesis. Cancer Res. 2006;66(3):1481–1490. doi: 10.1158/0008-5472.CAN-05-2412. [DOI] [PubMed] [Google Scholar]
- 19.Kan M, DiSorbo D, Hou JZ, Hoshi H, Mansson PE, McKeehan WL. High and low affinity binding of heparin-binding growth factor to a 130-kDa receptor correlates with stimulation and inhibition of growth of a differentiated human hepatoma cell. J Biol Chem. 1988;263(23):11306–11313. [PubMed] [Google Scholar]
- 20.Huang X, Yu C, Jin C, et al. Forced expression of hepatocyte-specific fibroblast growth factor 21 delays initiation of chemically induced hepatocarcinogenesis. Mol Carcinog. 2006;45(12):934–942. doi: 10.1002/mc.20241. [DOI] [PubMed] [Google Scholar]
- 21.Ricol D, Cappellen D, El Marjou A, et al. Tumour suppressive properties of fibroblast growth factor receptor 2-IIIb in human bladder cancer. Oncogene. 1999;18(51):7234–7243. doi: 10.1038/sj.onc.1203186. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Wang H, Toratani S, et al. Growth inhibition by keratinocyte growth factor receptor of human salivary adenocarcinoma cells through induction of differentiation and apoptosis. Proc Natl Acad Sci U S A. 2001;98(20):11336–11340. doi: 10.1073/pnas.191377098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang JH, Shin KH, Park YJ, Lee RJ, McKeehan WL, Park JG. Novel transcripts of fibroblast growth factor receptor 3 reveal aberrant splicing and activation of cryptic splice sequences in colorectal cancer. Cancer Res. 2000;60(15):4049–4052. [PubMed] [Google Scholar]
- 24.Jang JH. Reciprocal relationship in gene expression between FGFR1 and FGFR3: implication for tumorigenesis. Oncogene. 2005;24(5):945–948. doi: 10.1038/sj.onc.1208254. [DOI] [PubMed] [Google Scholar]
- 25.Bange J, Prechtl D, Cheburkin Y, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62(3):840–847. [PubMed] [Google Scholar]
- 26.Wang J, Stockton DW, Ittmann M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin Cancer Res. 2004;10(18 Pt 1):6169–6178. doi: 10.1158/1078-0432.CCR-04-0408. [DOI] [PubMed] [Google Scholar]
- 27.Jaakkola S, Salmikangas P, Nylund S, et al. Amplification of fgfr4 gene in human breast and gynecological cancers. Int J Cancer. 1993;54(3):378–382. doi: 10.1002/ijc.2910540305. [DOI] [PubMed] [Google Scholar]
- 28.St Bernard R, Zheng L, Liu W, Winer D, Asa SL, Ezzat S. Fibroblast growth factor receptors as molecular targets in thyroid carcinoma. Endocrinology. 2005;146(3):1145–1153. doi: 10.1210/en.2004-1134. [DOI] [PubMed] [Google Scholar]
- 29.Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160(6):2295–2307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desnoyers LR, Pai R, Ferrando RE, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27(1):85–97. doi: 10.1038/sj.onc.1210623. [DOI] [PubMed] [Google Scholar]
- 31.Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125(18):3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 32.Takaishi S, Sawada M, Morita Y, Seno H, Fukuzawa H, Chiba T. Identification of a novel alternative splicing of human FGF receptor 4: soluble-form splice variant expressed in human gastrointestinal epithelial cells. Biochem Biophys Res Commun. 2000;267(2):658–662. doi: 10.1006/bbrc.1999.2010. [DOI] [PubMed] [Google Scholar]
- 33.Ezzat S, Zheng L, Yu S, Asa SL. A soluble dominant negative fibroblast growth factor receptor 4 isoform in human MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2001;287(1):60–65. doi: 10.1006/bbrc.2001.5546. [DOI] [PubMed] [Google Scholar]
- 34.Morita K, Takano K, Yasufuku-Takano J, et al. Expression of pituitary tumour-derived, N-terminally truncated isoform of fibroblast growth factor receptor 4 (ptd-FGFR4) correlates with tumour invasiveness but not with G-protein alpha subunit (gsp) mutation in human GH-secreting pituitary adenomas. Clin Endocrinol (Oxf) 2008;68(3):435–441. doi: 10.1111/j.1365-2265.2007.03062.x. [DOI] [PubMed] [Google Scholar]
- 35.Marks JL, McLellan MD, Zakowski MF, et al. Mutational analysis of EGFR and related signaling pathway genes in lung Adenocarcinomas identifies a novel somatic kinase domain mutation in FGFR4. PLoS ONE. 2007;2(5):e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67(3):863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 37.Xie MH, Holcomb I, Deuel B, et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11(10):729–735. doi: 10.1006/cyto.1999.0485. [DOI] [PubMed] [Google Scholar]
- 38.Molina R, Chen B, Rose B, et al. Inhibition of experimental tumor models achieved through systemic elevation of FGF19 in vivo. Proc Amer Assoc Cancer Res. 2004;45 [Google Scholar]
- 39.Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145(6):2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 40.Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14(1):181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vainikka S, Joukov V, Wennstrom S, Bergman M, Pelicci PG, Alitalo K. Signal transduction by fibroblast growth factor receptor-4 (FGFR-4). Comparison with FGFR-1. J Biol Chem. 1994;269(28):18320–18326. [PubMed] [Google Scholar]
- 42.Shaoul E, Reich-Slotky R, Berman B, Ron D. Fibroblast growth factor receptors display both common and distinct signaling pathways. Oncogene. 1995;10(8):1553–1561. [PubMed] [Google Scholar]
- 43.Raffioni S, Thomas D, Foehr ED, Thompson LM, Bradshaw RA. Comparison of the intracellular signaling responses by three chimeric fibroblast growth factor receptors in PC12 cells. Proc Natl Acad Sci U S A. 1999;96(13):7178–7183. doi: 10.1073/pnas.96.13.7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwiatkowski BA, Kirillova I, Richard RE, Israeli D, Yablonka-Reuveni Z. FGFR4 and its novel splice form in myogenic cells: Interplay of glycosylation and tyrosine phosphorylation. J Cell Physiol. 2008;215(3):803–817. doi: 10.1002/jcp.21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moller MB. P27 in cell cycle control and cancer. Leuk Lymphoma. 2000;39(1–2):19–27. doi: 10.3109/10428190009053535. [DOI] [PubMed] [Google Scholar]
- 46.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272(5263):877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- 47.Freeman KW, Gangula RD, Welm BE, et al. Conditional activation of fibroblast growth factor receptor (FGFR) 1, but not FGFR2, in prostate cancer cells leads to increased osteopontin induction, extracellular signal-regulated kinase activation, and in vivo proliferation. Cancer Res. 2003;63(19):6237–6243. [PubMed] [Google Scholar]
- 48.Xian W, Schwertfeger KL, Rosen JM. Distinct roles of fibroblast growth factor receptor 1 and 2 in regulating cell survival and epithelial-mesenchymal transition. Mol Endocrinol. 2007;21(4):987–1000. doi: 10.1210/me.2006-0518. [DOI] [PubMed] [Google Scholar]
- 49.Acevedo VD, Gangula RD, Freeman KW, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Ishikawa T, Fidge N. Changes in the concentration of plasma lipoproteins and apoproteins following the administration of Triton WR 1339 to rats. J Lipid Res. 1979;20(2):254–264. [PubMed] [Google Scholar]