

Abstract
Although millions of cells are shed from a tumour every day, haematogenous metastasis is believed to be very inefficient. This inefficiency is widely assumed to be a result of the destruction of cells in the bloodstream by shear stress and the immune system and a slow rate of extravasation and proliferation in the stroma at a secondary site. Here, we propose that, whereas active intravasation of cells into the circulation is important in some tumours, others might shed cells passively into the blood or lymphatic vessels without the involvement of active cell migration. We discuss the evidence for and against this passive-shedding hypothesis and the implications for future treatments.
Introduction
The spread of metastases to distant sites is the leading cause of death from cancer. More than half of all patients with colorectal cancer will eventually develop distant metastases, including patients who undergo an apparently curative, complete resection of the primary tumour. Consequently, much effort in cancer research focuses on the detection, prevention, and treatment of distant metastases. At present, the available group of prognostic markers is limited to, at best, genetic-based or proteomic-based diagnosis of the primary tumour and its potential for metastasis. However, these prognostic indicators are often inaccurate, and even primary tumours with supposedly low potential for metastasis sometimes metastasise. Furthermore, even if a perfect diagnostic marker for metastatic potential existed, we would still need to know which aspects of biology or physiology to target to prevent metastatic colonies.
A remaining question with metastasis is whether cancer cells need to actively adhere to and migrate out of blood vessels at the secondary site in order to colonise. On the one hand, we know that cancer cells express endothelial adhesion receptors, similar to leukocytes, which allow them to roll on and adhere to endothelium.1–3 But on the other hand, studies have found clumps of cancer cells survi ving and growing within the lumen of blood vessels at the secondary site.4 These disparate observations suggest that some manifestations of metastasis are actively enabled by cancer cells, but others might be dictated by chance. The example of intravascular colonisation pertains to the later stages of metastasis, but we propose that similar passive mechanisms operate during the initial, intravasation step as well.
Most cancer treatments deal with established metastases because surprisingly little is known about the early steps of metastases or how to prevent them. Butler and Gullino,5 and Liotta and co-workers,6 showed that millions of cells are shed from a tumour every day, even though few clinically detectable metastatic colonies are formed. The widely accepted hypothesis is that shear stress and immune cells in the circulation cause destruction of the shed cells and prevent all but the most capable cells from producing secondary colonies. The question remains whether these early intravasation steps are taken by cells that actively move toward and then into nearby blood vessels, or whether the process is passive and coincidental (figure 1 and panel). The answer to this question would have major implications for future treatments.
Figure 1. Active and passive mechanisms in initial steps of metastasis.
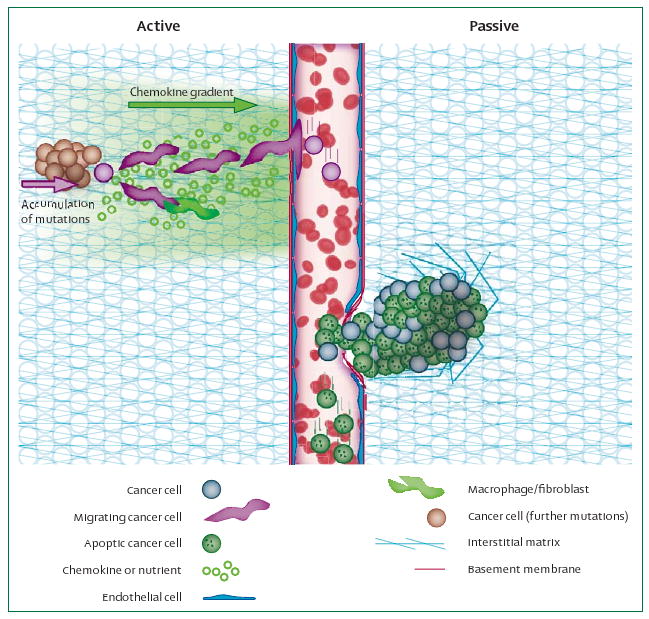
Left: reports that cancer cells accumulate mutations, upregulate migration machinery, and align and migrate up nutrient or chemokine gradients, are in support of active metastasis. Fibroblasts, macrophages, or other stromal cells are also likely to cooperate with cancer cells to actively help with the initial stage of metastasis. Right: however, there is evidence that many dead cells are shed into the vasculature, which implies a passive mechanism. It is possible that uncontrolled focal growth crushes or impinges upon fragile tumour blood vessels, leading to passive shedding. Similarly, fluid oozing from the tumour surface might help with shedding of cancer cells into lymphatic vessels.
Panel: Evidence for active and passive mechanisms in cancer cell intravasation.
Active
Passive
Requirements for haematogenous metastasis
To develop clinically detectable lesions at a distant site, metastasising cells must complete a series of sequential, interrelated steps. First, they have to move from the primary tumour mass into the bloodstream, where they must endure the haemodynamic stresses, evade the natural host immune response, and arrest at the secondary site. Once at this site they then need to adapt to the local microenvironment, proliferate, and induce angiogenesis to form a successful secondary colony. Failure to complete any step in this cascade prevents formation of metastatic colonies. Fortunately, many physiological barriers render metastasis relatively inefficient.34–36 Although the later steps of the metastasising process (ie, arrest at the secondary site, extravasation, and colonisation) have been well studied, little is known about the early intravasation step (figure 1). This initial step involves interactions between cancer cells and blood vessels.
Tumours larger than about 1 mm in diameter must recruit new blood vessels to supply sufficient oxygen and nutrients. Physiological angiogenesis is regulated by an orches trated series of proangiogenic and antiangiogenic molecules, such as vascular endothelial growth factor, basic fibroblast growth factor, angiopoietins, ephrins, thrombospondin, angiostatin, endostatin, and other matrix degradation products that are involved in angiogenesis. This exquisitely balanced process is disturbed in cancer, where many angiogenic molecules are chronically overexpressed, overwhelming the inhibitors.37 The resulting blood vessels that supply the tumour are immature, poorly-structured, and not well fortified with pericytes and basement membrane.25,38 How these structural and functional abnormalities in the vessel wall affect intravasation of cancer cells is not known, but they probably help with the process.
Is cancer-cell intravasation active or passive?
Many studies suggest that cancer cells actively crawl towards blood or lymphatic vessels by following nutrient or chemokine gradients.7 This active migration has been postulated to be a result of the ability of cancer cells to mimic some aspects of lymphocyte behaviour. Other evidence supports the hypothesis that metastasis in the early stages is a more accidental, passive process.29 Our lack of understanding of this process is largely a result of the difficulties in establishing tumour models in which tumour-cell intravasation and shedding can be monitored and characterised.
Evidence that cells are shed passively
Tumour-cell intravasation
To address this lack of understanding, Butler and Gullino5 developed a method for isolating a tumour and its vascular system in the ovarian pedicle. In this model, the vessels that feed and drain the tumour are easily identified, allowing quantitative control over what enters—or collection of what leaves—the tumour. With this approach, they noted a high rate of cancer-cell shedding (3–4×106 malignant cells/day per gram of tumour) by various tumours. Similar findings were reported by Liotta and co-workers39 in another model system. These results collectively highlighted a disparity between the millions of cells shed from a tumour every day and the relatively few clinically detectable metastases. Several explanations exist for this disparity. One possibility is that, once in the vasculature, metastatic cells encounter barriers that prevent colonisation at the secondary site—ie, they perish in the bloodstream as a result of fluid shear-forces or immune-cell attack, or else they land in a tissue with inappropriate microenvironment for growth. The question is what differentiates those cells that are able to metastasise from those that cannot.
Several years ago, we collected evidence suggesting that a simple answer to this question might be that successful cells need only be viable.20,24 Adapting the isolated tumour model to mice, we confirmed the high rate of tumour-cell shedding, and also collected and characterised the shed tumour cells. The shed cells were less clonogenic compared with their counterparts in the primary tumour.24 More viable cells were shed from the highly metastatic primary tumours, but most of the shed cells were already apoptotic or dead before they entered the vasculature.20 We postulated that uncontrolled, haphazard tumour growth causes proliferation of cells in regions of ade quate nutrient supply, but at the same time, death of cells in poorly-perfused regions. Because of their large number and the competition for space in a growing tumour, these dead or dying cells are probably shed into the bloodstream. The low viability of circulating cancer cells showed in preclinical models also extends to human tumours.21 When Mehes and co-workers40 and Larson and colleagues22 collected and characterised circulating epithelial tumour cells in patients with advanced breast cancer and prostate cancer, respectively, they noted that most of the circulating cells were apoptotic and concluded that apoptotic cells substantially contribute to the circulating-tumour-cell fraction in patients with cancer. The question of whether the initiation of apoptosis or necrosis takes place before or after intravasation is, in general, difficult to answer. In our animal model, we carefully controlled the conditions so that cells shed from the tumour passed through a short segment of vasculature, into a collecting cannula and to a test tube, where they were immediately fixed for analysis. Thus, we ensured that the cells were fixed within minutes of being shed, which kept the likelihood that changes occurred after intravasation to a minimum. In support of preintravasation cell death are reports in which apoptosis is associated with loss of cell–cell or cell–matrix adhesion, the same process that would allow cell shedding.41 Furthermore, squamous-cell carcinomas for instance—known for their aggressive ness and their high potential to metastasise—have a very high percentage of necrosis and apoptosis.42
Structural and mechanical aspects of early metastasis
The passage of large numbers of dead and apoptotic cells contrasts sharply with previous theories favouring active cell migration into the vessel. Could some tumours metastasise through a passive shedding process, without active migration? Tumours have long been known to have pockets of necrotic cells, produced by poor perfusion and insufficient nutrient and oxygen delivery.43 Additionally, blood vessels themselves have been shown to be highly abnormal, with immature, disorganised wall structure, detached endothelial cells, and an abnormal or missing basement membrane.25,38,44 These fragile vessels, combined with poor structural integrity of the tumour, could possibly help with the shedding of tumour material, including live and dead cells.
In support of passive intravasation is a seminal study by Liotta and co-workers,6 who showed that trauma or massage to the primary tumour increases the number of tumour cells released into the effluent blood. They also noted a linear relation between the proportion of vessels with diameters large enough to pass a tumour clump of a given size and the proportion of clumps of that size within the venous effluent.6 This means that once inside the vessel, clumps of cells—and debris—are limited by the blood-vessel conduit size, rather than by the ability of cells to negotiate or survive in the bloodstream. Also corroborating the passive intravasation hypothesis are studies showing that tumours can produce their own mechanical stresses through unchecked cell growth.27 Cells growing in a confined space push against each other, producing stress that can collapse blood and lymphatic vessels and, potentially, force cells to breach fragile blood and lymphatic vessels.25,28,38
These findings suggest that the early steps in metastasis can be dominated by passive events. However, other factors such as cell chemotaxis and phenotypic adaptability point toward an active metastatic process.
Evidence that the initial steps in metastasis are active
Cancer cells migrate actively and express matrix metalloproteinases
By contrast with the passive intravasation hypothesis is the finding that highly metastatic cells can change alignment and migrate toward blood vessels, and less metastatic cells cannot.8 This in vivo result supports a number of in vitro studies showing that cancer cells are able to polarise and migrate in response to growth factor and nutrient gradients.45–49
In addition to activating cytoskeletal and adhesion mechanisms necessary for directed migration, cells can also actively digest interstitial matrix and basement membrane to migrate through tissue and into blood vessels. For example, Morikawa and colleagues50 noted a strong correlation between type IV collagenase activity of human colon cancer and the incidence of liver metastases. The microenvironment of orthotopically grown tumours contained twice the levels of enzymes compared with ectopic tumours. Thus, the tumour microenvironment probably plays an important part in cell shedding and metastasis.
Cancer cells change their transcriptional profile during intravasation
Under common experimental protocols, most cells injected into the circulation die rapidly. For example, Fidler2 showed that metastases were produced from less than 0.1% of the original inoculated cells and concluded that metastasis resulted from only a few surviving cells. Experiments such as this suggest that neoplasms are heterogeneous and contain subpopulations of cells with differing angiogenic, invasive, and metastatic properties.51 Shioda and colleagues14 followed individual cells by intravital microscopy and reverse transcriptase-polymerase chain reaction as they completed the metastatic sequence. They showed that as B16F10 melanoma cells adhered to the vasculature, extravasated, migrated through the tissue and then proliferated, distinct genes were upregulated that would be necessary for each of those steps. Although this study did not directly examine the initial intravasation step, it suggests that, at least in some cases, cancer cells actively respond to their environment by regulating genes needed for metastasis. In short, there are many findings supportive of the hypothesis that metastases result from a unique subpopulation of cells that are actively equipped with mechanisms to undertake the different steps of metastases rather than fortuitous survival and growth of randomly chosen cells.
Collaboration of stromal and cancer cells in metastasis
The stroma constitutes a large part of most solid tumours and tumour–stroma interactions contribute functionally to tumour growth and progression. The infiltration of stromal cells has been well documented in animal models and human tumours.52,53 The generation of tumour stroma is triggered by tumour cells and induces the in-growth of new blood vessels and mesenchymal cells from the adjacent normal tissue.54 Host stromal cells apparently regulate angiogenesis by changing the local equilibrium between proangiogenic and antiangiogenic molecules. Nakagawa and co-workers55 showed that in patients with colorectal cancer, cancer-associated fibroblasts express distinct molecular expression profiles, supporting the notion that these fibroblasts change to provide a favourable micro-environment for cancer cells. Kitadai and co-workers56 showed that the expression of platelet-derived growth factor receptor in the stroma was higher in highly metastatic colon carcinoma than in less metastatic tumours and concluded that the expression of platelet-derived growth factor receptor in stromal cells is influenced by the organ-specific microenvironment and is associated with metastatic potential. On the basis of these and other studies, there exists an active interplay between host-stroma and tumour, which, in some circumstances, can enable metastasis.
Prediction of metastasis in gene expression profiles
Various theories have been proposed to explain the genetic basis of metastatic inefficiency. The traditional view depicts a series of events that produce a small sub population of cells able to overcome all barriers to successfully metastasise. At each stage, modulation of gene expression during metastasis is a dynamic, orchestrated process, with crucial genes being modulated at each stage,1 and only a small fraction of the tumour cells acquire full metastatic potential resulting from an accumulation of mutational events. Changes in the genome by amplification, translocation, or loss of heterozygosity all support this theory. However, Bernards and Weinberg12 have proposed that metastatic propensity is acquired very early during tumorigenesis, and Ramaswamy and co-workers13 discovered expression signatures in primary tumours that were predictive of successful metastases. They all claim that the cells in primary tumours are relatively stable, and any of these cells are capable of metastasising, provided the tumour acquired the correct genetic signature early on. However, if most cells have the active signature to metastasise, and millions of cells are shed by the primary tumour, this theory would predict a highly efficient metastatic process.
There are several microenvironmental or epigenetic factors, along with position in the tumour, that establish a given cell's likelihood to metastasise. Hunter has added another theory to reconcile the inconsistencies.57 Using his genetic efficiency model, he claims that host tissue can be classified as either a low or high metastatic genotype, and this genotype defines the ability of a tumour to flourish at the secondary site. This theory, in essence, claims that the host stromal cells, rather than the mutated cancer cells, predict metastatic ability. Several studies support this theory and have shown that gene expression can vary as a result of genetic background.51,52,55,57,58
Additionally, exciting studies have recently identified cancer stem cells—putative cells that are necessary and sufficient for primary tumour growth and dissemination.59–61 The low success rate for metastasis might exist, at least in part, because very few of the cells shed from a tumour are these fertile stem cells.
Conclusion
Several interdependent mechanisms affect the metastatic cascade, and despite the impressive rate of publication in this area, we still know little about the key steps in the process. Tumours seem to be able to use both active and passive methods to enter the vasculature, but whether the preferred mode is tumour-type dependent, organ dependent, or stage dependent, or whether specific genetic mutations are needed is not known. Furthermore, although intravasation into lymphatic vessels is not as well characterised as that into blood vessels, we can reasonably assume that both passive and active mechanisms are important in lymphatic metastasis as well.62
The key for successful treatment of metastases will probably include a combination of drugs that are not only able to target the late steps of metastasis, but also the early steps. Future treatments could be directed at blocking metastatic progression during the initial step of cell shedding—eg, by inducing even more apoptosis in the shed-cell population or by targeting blood vessels or stroma. If certain tumours do rely on passive intravasation into fragile blood vessels to metastasise, then treatments aimed at fortifying blood vessels might be successful at preventing metastases. One such approach, currently in clinical trials, focuses on normalising tumour blood vessels through prudent application of antiangiogenic treatment. The result is blood vessels that appear more mature, with lower permeability, more normal pericyte and basement membrane association, and better perfusion.38,43,44,62 Nevertheless, there are many manifestations of metastasis, with different tumours using different modes of dissemination. This variability is probably the greatest barrier to our development of a general, fundamental understanding of the process. Development of tumour models that more closely mimic the range of behaviour seen in human disease, and the ability to visualise these steps in vivo, might help overcome this difficulty.
Footnotes
Conflicts of interest: The authors declared no conflicts of interest.
References
- 1.Zetter BR. Adhesion molecules in tumor metastasis. Semin Cancer Biol. 1993;4:219–29. [PubMed] [Google Scholar]
- 2.Fidler IJ. Critical determinants of metastasis. Semin Cancer Biol. 2002;12:89–96. doi: 10.1006/scbi.2001.0416. [DOI] [PubMed] [Google Scholar]
- 3.Ruoslahti E. Cell adhesion and tumor metastasis. Princess Takamatsu Symp. 1994;24:99–105. [PubMed] [Google Scholar]
- 4.Wong CW, Song C, Grimes MM, et al. Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol. 2002;161:749–53. doi: 10.1016/S0002-9440(10)64233-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512–16. [PubMed] [Google Scholar]
- 6.Liotta LA, Saidel MG, Kleinerman J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976;36:889–94. [PubMed] [Google Scholar]
- 7.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–66. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Condeelis JS, Wyckoff J, Segall JE. Imaging of cancer invasion and metastasis using green fluorescent protein. Eur J Cancer. 2000;36:1671–80. doi: 10.1016/s0959-8049(00)00155-6. [DOI] [PubMed] [Google Scholar]
- 9.Fidler IJ. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer. 1973;9:223–27. doi: 10.1016/s0014-2964(73)80022-2. [DOI] [PubMed] [Google Scholar]
- 10.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 11.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 12.Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 13.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 14.Shioda T, Munn LL, Fenner MH, Jain RK, Isselbacher KJ. Early events of metastasis in the microcirculation involve changes in gene expression of cancer cells. Tracking mRNA levels of metastasizing cancer cells in the chick embryo chorioallantoic membrane. Am J Pathol. 1997;150:2099–112. [PMC free article] [PubMed] [Google Scholar]
- 15.Freije JM, Balbin M, Pendas AM, Sanchez LM, Puente XS, Lopez-Otin C. Matrix metalloproteinases and tumor progression. Adv Exp Med Biol. 2003;532:91–107. doi: 10.1007/978-1-4615-0081-0_9. [DOI] [PubMed] [Google Scholar]
- 16.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 17.Moses MA. The regulation of neovascularization of matrix metalloproteinases and their inhibitors. Stem Cells. 1997;15:180–89. doi: 10.1002/stem.150180. [DOI] [PubMed] [Google Scholar]
- 18.Kopfstein L, Christofori G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci. 2006;63:449–68. doi: 10.1007/s00018-005-5296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribatti D, Nico B, Vacca A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene. 2006;25:4257–66. doi: 10.1038/sj.onc.1209456. [DOI] [PubMed] [Google Scholar]
- 20.Bockhorn M, Roberge S, Sousa C, Jain RK, Munn LL. Differential gene expression in metastasizing cells shed from kidney tumors. Cancer Res. 2004;64:2469–73. doi: 10.1158/0008-5472.can-03-0256. [DOI] [PubMed] [Google Scholar]
- 21.Racila E, Euhus D, Weiss AJ, et al. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci USA. 1998;95:4589–94. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson CJ, Moreno JG, Pienta KJ, et al. Apoptosis of circulating tumor cells in prostate cancer patients. Cytometry. 2004;62:46–53. doi: 10.1002/cyto.a.20073. [DOI] [PubMed] [Google Scholar]
- 23.Mehes G, Witt A, Kubista E, Ambros PF. Circulating breast cancer cells are frequently apoptotic. Am J Pathol. 2001;159:17–20. doi: 10.1016/S0002-9440(10)61667-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swartz MA, Kristensen CA, Melder RJ, et al. Cells shed from tumours show reduced clonogenicity, resistance to apoptosis, and in vivo tumorigenicity. Br J Cancer. 1999;81:756–59. doi: 10.1038/sj.bjc.6690760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA. 2000;97:14608–13. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK. Solid stress inhibits the growth of multicellular tumor spheroids. Nat Biotechnol. 1997;15:778–83. doi: 10.1038/nbt0897-778. [DOI] [PubMed] [Google Scholar]
- 28.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 29.Cavallaro U, Christofori G. Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochim Biophys Acta. 2001;1552:39–45. doi: 10.1016/s0304-419x(01)00038-5. [DOI] [PubMed] [Google Scholar]
- 30.de Amicis F, Lanzino M, Kisslinger A, et al. Loss of proline-rich tyrosine kinase 2 function induces spreading and motility of epithelial prostate cells. J Cell Physiol. 2006;209:74–80. doi: 10.1002/jcp.20709. [DOI] [PubMed] [Google Scholar]
- 31.Koike C, McKee TD, Pluen A, et al. Solid stress facilitates spheroid formation: potential involvement of hyaluronan. Br J Cancer. 2002;86:947–53. doi: 10.1038/sj.bjc.6600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raghunand N, Gatenby RA, Gillies RJ. Microenvironmental and cellular consequences of altered blood flow in tumours. Br J Radiol. 2003;76:S11–22. doi: 10.1259/bjr/12913493. Spec No 1. [DOI] [PubMed] [Google Scholar]
- 33.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786–95. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 34.Chambers AF, MacDonald IC, Schmidt EE, Morris VL, Groom AC. Clinical targets for anti-metastasis therapy. Adv Cancer Res. 2000;79:91–121. doi: 10.1016/s0065-230x(00)79003-8. [DOI] [PubMed] [Google Scholar]
- 35.Sugarbaker PH. Metastatic inefficiency: the scientific basis for resection of liver metastases from colorectal cancer. J Surg Oncol. 1993;3(Suppl):158–60. doi: 10.1002/jso.2930530541. [DOI] [PubMed] [Google Scholar]
- 36.Weiss L. Cancer cell traffic from the lungs to the liver: an example of metastatic inefficiency. Int J Cancer. 1980;25:385–92. doi: 10.1002/ijc.2910250313. [DOI] [PubMed] [Google Scholar]
- 37.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 38.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 39.Liotta LA, Kleinerman J, Saidel GM. Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res. 1974;34:997–1004. [PubMed] [Google Scholar]
- 40.Mehes G, Witt A, Kubista E, Ambros PF. Circulating breast cancer cells are frequently apoptotic. Am J Pathol. 2001;159:17–20. doi: 10.1016/S0002-9440(10)61667-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glinsky GV. Apoptosis in metastatic cancer cells. Crit Rev Oncol Hematol. 1997;25:175–86. doi: 10.1016/s1040-8428(97)00234-5. [DOI] [PubMed] [Google Scholar]
- 42.Sarbia M, Gabbert HE. Modern pathology: prognostic parameters in squamous cell carcinoma of the esophagus. Recent Results Cancer Res. 2000;155:15–27. doi: 10.1007/978-3-642-59600-1_2. [DOI] [PubMed] [Google Scholar]
- 43.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 44.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–36. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 45.Saadi W, Wang SJ, Lin F, Jeon NL. A parallel-gradient microfluidic chamber for quantitative analysis of breast cancer cell chemotaxis. Biomed Microdevices. 2006;8:109–18. doi: 10.1007/s10544-006-7706-6. [DOI] [PubMed] [Google Scholar]
- 46.Schneider IC, Haugh JM. Mechanisms of gradient sensing and chemotaxis: conserved pathways, diverse regulation. Cell Cycle. 2006;5:1130–34. doi: 10.4161/cc.5.11.2770. [DOI] [PubMed] [Google Scholar]
- 47.Wang SJ, Saadi W, Lin F, Minh-Canh Nguyen C, Li Jeon N. Differential effects of EGF gradient profiles on MDA-MB-231 breast cancer cell chemotaxis. Exp Cell Res. 2004;300:180–89. doi: 10.1016/j.yexcr.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 48.Bartolome RA, Galvez BG, Longo N, et al. Stromal cell-derived factor-1alpha promotes melanoma cell invasion across basement membranes involving stimulation of membrane-type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 2004;64:2534–43. doi: 10.1158/0008-5472.can-03-3398. [DOI] [PubMed] [Google Scholar]
- 49.Entschladen F, Drell TL, 4th, Lang K, Joseph J, Zaenker KS. Tumour-cell migration, invasion, and metastasis: navigation by neurotransmitters. Lancet Oncol. 2004;5:254–58. doi: 10.1016/S1470-2045(04)01431-7. [DOI] [PubMed] [Google Scholar]
- 50.Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res. 1988;48:6863–71. [PubMed] [Google Scholar]
- 51.Hart IR. ‘Seed and soil’ revisited: mechanisms of site-specific metastasis. Cancer Metastasis Rev. 1982;1:5–16. doi: 10.1007/BF00049477. [DOI] [PubMed] [Google Scholar]
- 52.Fukumura D, Xavier R, Sugiura T, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–25. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 53.Hewitt RE, Powe DG, Carter GI, Turner DR. Desmoplasia and its relevance to colorectal tumour invasion. Int J Cancer. 1993;53:62–69. doi: 10.1002/ijc.2910530113. [DOI] [PubMed] [Google Scholar]
- 54.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–59. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa H, Liyanarachchi S, Davuluri RV, et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23:7366–77. doi: 10.1038/sj.onc.1208013. [DOI] [PubMed] [Google Scholar]
- 56.Kitadai Y, Sasaki T, Kuwai T, et al. Expression of activated platelet-derived growth factor receptor in stromal cells of human colon carcinomas is associated with metastatic potential. Int J Cancer. 2006;119:2567–74. doi: 10.1002/ijc.22229. [DOI] [PubMed] [Google Scholar]
- 57.Hunter KW. Allelic diversity in the host genetic background may be an important determinant in tumor metastatic dissemination. Cancer Lett. 2003;200:97–105. doi: 10.1016/s0304-3835(03)00420-8. [DOI] [PubMed] [Google Scholar]
- 58.Domann FE, Rice JC, Hendrix MJ, Futscher BW. Epigenetic silencing of maspin gene expression in human breast cancers. Int J Cancer. 2000;85:805–10. doi: 10.1002/(sici)1097-0215(20000315)85:6<805::aid-ijc12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 59.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 60.Widschwendter M, Fiegl H, Egle D, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–58. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 61.Setoguchi T, Taga T, Kondo T. Cancer stem cells persist in many cancer cell lines. Cell Cycle. 2004;3:414–15. doi: 10.4161/cc.3.4.799. [DOI] [PubMed] [Google Scholar]
- 62.Jain RK, Tong R, Munn LL. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007 doi: 10.1158/0008-5472.CAN-06-4102. published online March 15. DOI:10.1158/0008-5472. CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]