

Abstract
Protein palmitoylation is the most common posttranslational lipid modification; its reversibility mediates protein shuttling between intracellular compartments. A large family of DHHC (Asp-His-His-Cys) proteins has emerged as protein palmitoyl acyltransferases (PATs). However, mechanisms that regulate these PATs in a physiological context remain unknown. In this study, we efficiently monitored the dynamic palmitate cycling on synaptic scaffold PSD-95. We found that blocking synaptic activity rapidly induces PSD-95 palmitoylation and mediates synaptic clustering of PSD-95 and associated AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)-type glutamate receptors. A dendritically localized DHHC2 but not the Golgi-resident DHHC3 mediates this activity-sensitive palmitoylation. Upon activity blockade, DHHC2 translocates to the postsynaptic density to transduce this effect. These data demonstrate that individual DHHC members are differentially regulated and that dynamic recruitment of protein palmitoylation machinery enables compartmentalized regulation of protein trafficking in response to extracellular signals.
Introduction
Posttranslational modification, including phosphorylation, ubiquitination, and lipid modification, adds functional regulation to proteins beyond genomic information. Lipid modification increases protein hydrophobicity and plays a critical role in protein trafficking, targeting, and function. Thioester-linked palmitate modifies signaling proteins, enzymes, cytoskeletal proteins, ion channels, and scaffolding proteins and is involved in diverse aspects of cellular signaling (El-Husseini and Bredt, 2002; Resh, 2006; Linder and Deschenes, 2007). Recent global proteomic analyses have further expanded the known complement of palmitoylated proteins (Roth et al., 2006; Kang et al., 2008). Palmitoylation is unique in that it is a reversible modification and is proposed to be regulated by specific extracellular signals. Recent cell biological analyses revealed that some palmitoyl substrates such as small GTPases, Harvey Ras/neuroblastoma Ras (Rocks et al., 2005), and trimeric G proteins Gαo (Chisari et al., 2007)/Gαq (Tsutsumi et al., 2009) constitutively shuttle between the plasma membrane and the Golgi membrane by a palmitoylation/depalmitoylation cycle. This palmitate cycling generates and maintains the specific intracellular compartmentalization of substrates in nonpolarized cells (Rocks et al., 2006).
The postsynaptic scaffolding protein PSD-95 represents a major palmitoylated protein in neurons and plays critical roles in synaptogenesis and synaptic plasticity (Migaud et al., 1998; El-Husseini et al., 2000; Kennedy, 2000; Kim and Sheng, 2004; Funke et al., 2005). PSD-95 provides a platform for the postsynaptic clustering of crucial synaptic proteins, including AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) and N-methyl-d-aspartate (NMDA)-type glutamate receptors and cell adhesion molecules. The postsynaptic targeting of PSD-95 depends on protein palmitoylation (Topinka and Bredt, 1998). Importantly, palmitate cycling on PSD-95 is dynamically regulated by receptor activation (El-Husseini et al., 2002). Upon glutamate receptor stimulation, accelerated depalmitoylation of PSD-95 dissociates PSD-95 from postsynaptic sites and causes AMPA receptor (AMPAR) endocytosis. This receptor activation–induced depalmitoylation has also been reported in Gα (Wedegaertner and Bourne, 1994). Thus, agonist-dependent depalmitoylation down-regulates synaptic strength and G protein signaling. However, it is not yet clear whether addition of palmitate to proteins is accelerated in response to extracellular signals.
The dynamic regulation of palmitate cycling should be finely tuned by palmitoyl acyltransferases (PATs) and palmitoyl protein thioesterases (PPTs). Transmembrane proteins containing a DHHC (Asp-His-His-Cys) Cys–rich domain (DHHC proteins) have recently emerged as PATs in yeast (Bartels et al., 1999; Lobo et al., 2002; Roth et al., 2002; Linder and Deschenes, 2004). At least 23 mammalian DHHC proteins exist, and a systematic screening method has identified specific enzyme–substrate pairs (Fukata et al., 2004; Fang et al., 2006; Fernandez-Hernando et al., 2006; Fukata et al., 2006; Ponimaskin et al., 2008; Tsutsumi et al., 2009). The DHHC family is present in species ranging from yeast to human and to plants (Hemsley et al., 2005; Hemsley and Grierson, 2008). Several DHHC genes are associated with diseases, including cancers (Oyama et al., 2000), schizophrenia (Mukai et al., 2004, 2008), mental retardation (Mansouri et al., 2005; Raymond et al., 2007), and Huntington’s (Yanai et al., 2006). Although the large DHHC family plays essential roles in a range of physiological functions, how the DHHC PAT family is regulated and thereby dynamically controls palmitate cycling remains uncertain.
In this study, we found that suppression of neuronal activity induces palmitoylation and synaptic accumulation of PSD-95. This activity-sensitive PSD-95 palmitoylation recruits synaptic AMPARs. Dendritically localized DHHC2 mediates this rapid synaptic palmitoylation of PSD-95. In contrast, Golgi-resident DHHC3 constitutively palmitoylates PSD-95. These experiments indicate that large DHHC family members are individually regulated, which enables their participation in specific physiological processes such as synaptic plasticity.
Results
Total internal reflection fluorescence microscopy (TIRFM) reveals the synaptic accumulation of PSD-95 upon activity blockade
To follow changes in synaptic PSD-95 accumulation over time, we first performed time-lapse imaging of cultured hippocampal neurons transfected with PSD-95–GFP by TIRFM, which excites only molecules within 100 nm of the cover glass. TIRFM preferentially visualizes wild-type (WT) PSD-95–GFP as discrete punctae on dendrites, which are not seen with cytosolic palmitoylation–deficient (CS) mutant PSD-95 or GFP (Fig. 1, A and B). We confirmed comparable expression levels of PSD-95 (WT) and PSD-95 (CS) in transfected culture (Fig. 1 B). These data confirm that palmitoylation mediates membrane trafficking and synaptic clustering of PSD-95 (Topinka and Bredt, 1998). Because PSD-95 visualized by TIRFM apposes presynaptic synaptophysin and VGLUT1 and overlaps postsynaptic NR1 NMDA receptor (Fig. 1 C), TIRFM tracks synaptic PSD-95. When ionotropic glutamate receptor activity was blocked by kynurenic acid (Kyn), the intensity of PSD-95–GFP by TIRFM steadily increased over 2 h, whereas the intensity of PSD-95 (CS) did not change (Fig. 1, D and E; and Video 1). This Kyn-induced PSD-95 increase was blocked by coapplication of 2-bromopalmitate (2-BP), which is a palmitoyl acyl transfer inhibitor. PSD-95 signals did not detectably change within 2 h of 2-BP treatment alone (Fig. 1 E). These results indicate that newly occurring palmitoylation mediates this synaptic accumulation of PSD-95. Tetrodotoxin (TTX), a blocker of action potentials, also increased PSD-95 accumulation. The dynamic change of PSD-95 intensity was specific to palmitoylation as the localizations of GFP-Rac1-CLLL (Cys-Leu-Leu-Leu), a geranylgeranylated CaaL motif, and synaptophysin-GFP, a presynaptic protein, did not change upon Kyn treatment (Fig. 1 E). Synaptic PSD-95 accumulation upon activity blockade was also confirmed by antibody staining of native PSD-95 (see Fig. 4, C and D). The effect of Kyn or TTX on PSD-95 accumulation does not reflect newly synthesized PSD-95, as cycloheximide (CHX), an inhibitor of protein synthesis, did not affect the Kyn- or TTX-induced PSD-95 increase (Fig. S1, A and B; and Video 2). Thus, PSD-95 palmitoylation increases at the postsynaptic membrane upon activity blockade. These results are complementary to receptor activation–induced depalmitoylation of PSD-95 (El-Husseini et al., 2002).
Figure 1.

TIRFM imaging of activity-sensitive PSD-95 palmitoylation. (A) Compared with epifluorescent microscopy (Epi; green), TIRFM selectively reveals punctae from GFP-tagged PSD-95 (WT) (top; red) but not palmitoylation-deficient PSD-95 (CS) (bottom; red) in cultured hippocampal neurons. To define dendritic morphology, we coexpressed mCherry (Epi; blue). (B) TIRFM preferentially visualizes PSD-95 (WT)–GFP punctae as compared with PSD-95 (CS)–GFP or GFP alone. n = 10 neurons; ***, P < 0.001. Comparable expression levels of PSD-95 (WT)– and PSD-95 (CS)–GFP in transfected neuron culture were confirmed. (C) TIRFM tracks synaptic PSD-95. PSD-95 punctae (green) visualized by TIRFM apposed presynaptic synaptophysin and VGLUT1 and overlapped postsynaptic NR1. (D) PSD-95–GFP dynamics were analyzed by time-lapse TIRFM imaging. Inhibition of glutamate receptor activity with 10 mM Kyn increased PSD-95 (WT)–GFP intensity within 2 h. In contrast, the palmitoylation-deficient mutant PSD-95 (CS) did not change. Kymographs represent the changes in the intensity of PSD-95–GFP over 2 h. White lines indicate the regions used for the kymographs. (E) Synaptic accumulation of PSD-95 depends on newly occurring palmitoylation. Fluorescent intensities of PSD-95–GFP (WT and CS), GFP containing a C-terminal prenylation CaaL motif of Rac1 (GFP-CLLL), and synaptophysin-GFP (Syn-GFP) at 2 h after the indicated treatments were quantified. The intensity of PSD-95 (WT)–GFP but not other membrane-targeting proteins significantly increased upon 10 mM Kyn or 2 µM TTX treatment. Coapplication of 100 µM 2-BP with Kyn completely inhibited Kyn-induced increase of PSD-95–GFP intensity. n = 3–8 experiments; ***, P < 0.001 compared with control. (B and E) Error bars indicate SD. Bars: (A) 10 µm; (C and D) 5 µm.
Figure 4.

DHHC2 and -3 are differently involved in PSD-95 trafficking. (A and B) In the DHHC2 or -3 knocked down neurons (labeled with mCherry), the number of native PSD-95 puncta (green) was significantly decreased. n = 5 neurons; ***, P < 0.001. (C and D) Knockdown of DHHC2 but not DHHC3 prevented TTX- or Kyn-induced augmentation of endogenous PSD-95 accumulation. Dashed lines (100%) indicate the normalized control level. ***, P < 0.001. (C) Analyzed by confocal laser-scanning microscopy (CLSM). n = 10–15 neurons. (D) Analyzed by TIRFM. n = 5 neurons. miLacZ is a control miRNA targeting LacZ (β-galactosidase). (B–D) Error bars indicate SD. Bar, 5 µm.
The DHHC2/15 subfamily of PSD-95 palmitoylating enzymes is regulated by synaptic activity
To monitor PSD-95 palmitoylation biochemically, we used the acyl-biotin exchange (ABE) method (Roth et al., 2006; Kang et al., 2008). We confirmed that this method specifically identified palmitoylated proteins, including PSD-95, in heterologous cells (Fig. S2 A). As previously reported (El-Husseini et al., 2002), treating neurons for 12 h with 2-BP reduced PSD-95 palmitoylation (palmitoylated PSD-95 = 13 ± 15% of control cells; P < 0.001; Fig. 2 A). When we treated neurons for 2 h with Kyn, the amount of palmitoylated PSD-95 significantly increased (198 ± 13% of control cells; P < 0.001; Fig. 2, A and B). Blocking glutamate receptors with a combination of APV (D-[-]-2-amino-5-phosphonopentanoic acid), which blocks NMDA receptors, and CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), which blocks AMPARs, also enhanced PSD-95 palmitoylation within 2 h (palmitoylated PSD-95 = 184 ± 23% of control cells; P < 0.01). 2-BP blocked the rapid enhancement of PSD-95 palmitoylation, indicating that inhibition of depalmitoylation is not solely responsible and that newly occurring palmitoylation mediates this effect. This activity-sensitive PSD-95 palmitoylation is stoichiometric, as Kyn and APV + CNQX quantitatively shifted the PSD-95 band upward (Fig. 2, A and B; Fig. S1 C; and Fig. S2 B). This upward shift reflects palmitoylation, as β-mercaptoethanol (βME), which hydrolyses the palmitoyl thioester, leaves only the lower band (Fig. 2, A and B, bottom). Both the increased PSD-95 palmitoylation and synaptic accumulation were reversible upon washing out of Kyn, indicating that this process is activity sensitive (Fig. 2, B and C).
Figure 2.

The DHHC2/15 subfamily of PSD-95 PATs is regulated by synaptic activity. (A) Activity blockade induces quantitative palmitoylation of PSD-95 but not Gαq. Hydroxylamine (H)-sensitive palmitoylated proteins were purified from treated neurons by the ABE method. The amount of palmitoylated PSD-95 and Gαq was analyzed by Western blotting. T, Tris treatment as a control of hydroxylamine. (B and C) Kyn-induced PSD-95 palmitoylation and synaptic accumulation were reversible upon washing out Kyn. (B) Treatment of hippocampal neurons with Kyn for 2 h enhanced PSD-95 palmitoylation. After washout, PSD-95 palmitoylation level returned to the basal level within 2 h (ABE), with consistent mobility change of PSD-95 (−βME). In contrast, Gαq, GluR2, and GRIP1 palmitoylation did not change upon activity blockade. Kyn-induced palmitoylation changes were quantified. n = 3 each; ***, P < 0.001. Error bars indicate SD. The dashed line (100%) indicates the normalized control level. IB, immunoblot. (A and B) Closed and open arrows indicate the positions of palmitoylated and nonpalmitoylated PSD-95, respectively. (C) The increased accumulation of PSD-95–GFP upon Kyn treatment returned to the basal level at 2 h after Kyn washout. (D) Cultured hippocampal neurons expressing a DN mutant of the DHHC2 and -15 subfamily (DN-DH2/15) were treated with 3 mCi/ml [3H]palmitate for 2 h in the presence or absence of Kyn. Immunoprecipitated PSD-95 was resolved by SDS-PAGE, followed by fluorography ([3H]palm) and Coomassie staining (CBB). Inhibition of glutamate receptor activity with Kyn greatly enhanced PSD-95 palmitoylation. This enhancement was decreased by DN-DH2/15. IP, immunoprecipitation. Bar, 5 µm.
This activity-sensitive palmitoylation is specific for PSD-95, as Gαq, GluR2, and GRIP1 palmitoylation did not change upon activity blockade (Fig. 2, A and B). Our previous study demonstrated that PSD-95 PATs are DHHC2, -3, -7, and -15, which are phylogenetically divided into two subfamilies, DHHC3/7 and DHHC2/15 (Fukata et al., 2004). Gαq PATs are DHHC3 and -7 (Tsutsumi et al., 2009), and GluR2 PAT is DHHC3 (Fig. S2 C; Hayashi et al., 2005). These substrate selectivities allowed us to ask whether synaptic activity regulates a specific PAT subfamily (i.e., DHHC2/15). We metabolically labeled hippocampal neurons with [3H]palmitic acid for 2 h in the presence or absence of Kyn. We found that Kyn-enhanced PSD-95 palmitoylation was partially blocked by a dominant-negative (DN) mutant, DN-DH2/15, which specifically inhibits the DHHC2/15 subfamily (palmitoylated PSD-95 = 61 ± 15% of Kyn-treated control cells; P < 0.01; Fig. 2 D; Fukata et al., 2004). The partial effect of DN-DH2/15 is probably caused by the infection efficiency of DN-DH2/15. Under our conditions, ∼50% of neurons were expressing DN-DH2/15, which correlates with the extent of inhibition (∼40% inhibition). Although the involvement of other PATs cannot be completely ruled out, our results strongly suggest that the DHHC2/15 subfamily plays a major role in activity-sensitive PSD-95 palmitoylation.
Differential regulation of PSD-95 palmitoylating enzymes in neurons
We next examined the cellular locus for PSD-95 palmitoylation. We focused on DHHC2 and -3, as hippocampal neurons express these PATs but much less DHHC7 and -15 (Fig. S3 A). Immunoblotting with specific antibodies (Fig. 3 A) showed that DHHC2 occurred in the postsynaptic density fraction, whereas DHHC3 was present only in the P3 fraction, which contains Golgi proteins (Fig. 3 B). Consistent with this finding, DHHC3 specifically localizes to the somatic Golgi apparatus (Keller et al., 2004; Tsutsumi et al., 2009), whereas DHHC2 distributes in the dendrites and cell body as small vesicular-like structures (Fig. 3 C). These signals are specific, as the staining completely disappeared in the validated knockdown vector–transfected neuron (Fig. 3 D).
Figure 3.

Differential subcellular distribution of PSD-95 palmitoylating enzymes. (A) Specificity of antibodies to DHHC2 and -3. The bands detected by anti-DHHC2 (closed arrowheads) and anti-DHHC3 (open arrowhead) antibodies disappeared when protein expression was knocked down by siRNAs. IB, immunoblot; scr, scramble. (B) DHHC2 was enriched in the postsynaptic density (PSD) fractions (Triton X-100–insoluble postsynaptic; closed arrowheads), whereas DHHC3 was detected in only the P3 fraction (open arrowhead). H, homogenate; S, supernatant; P, precipitate; Syn, synaptosome; Sol, Triton X-100 soluble; Ins, Triton X-100–insoluble postsynaptic density fractions. (C) DHHC2 localized in dendrites and the cell body as small vesicular structures, whereas DHHC3 specifically localized at the Golgi apparatus in 18-DIV hippocampal neurons. (D) Effective knockdown of endogenous DHHC2 and -3. Cultured hippocampal neurons were transfected with mCherry-miR RNAi (miDHHC2 and -3) expression vectors at 10 DIV. 18-DIV neurons were stained by DHHC2 or -3 antibody. Note that somatodendritic DHHC2 vesicles and Golgi DHHC3 (arrows) were not stained in mCherry-expressing knocked down neurons (red). Bars: (C [left] and D) 20 µm; (C [right]) 5 µm.
DHHC2 or -3 knockdown by microRNA (miRNA; miDHHCs) greatly reduced the number of PSD-95 punctae (Fig. 4, A and B). Importantly, knockdown of DHHC2 but not DHHC3 prevented Kyn- or TTX-induced increase of endogenous PSD-95 accumulation at synaptic sites (Fig. 4, A, C and D) and Kyn-induced augmentation of PSD-95–GFP accumulation (Fig. 5 and Video 3). The inhibitory effect of miDHHC2 was rescued by miDHHC2-resistant WT DHHC2 (WT) but not by PAT-inactive DHHC2 (CS) (Fig. 5 and Fig. S4 C). These results indicate that DHHC3 localizes to the Golgi apparatus and mediates constitutive palmitoylation of various substrates, including PSD-95, Gαq, and GABAA receptor-γ subunit (Fukata et al., 2004; Keller et al., 2004; Tsutsumi et al., 2009). In contrast, dendritic DHHC2 mediates activity-sensitive PSD-95 palmitoylation.
Figure 5.
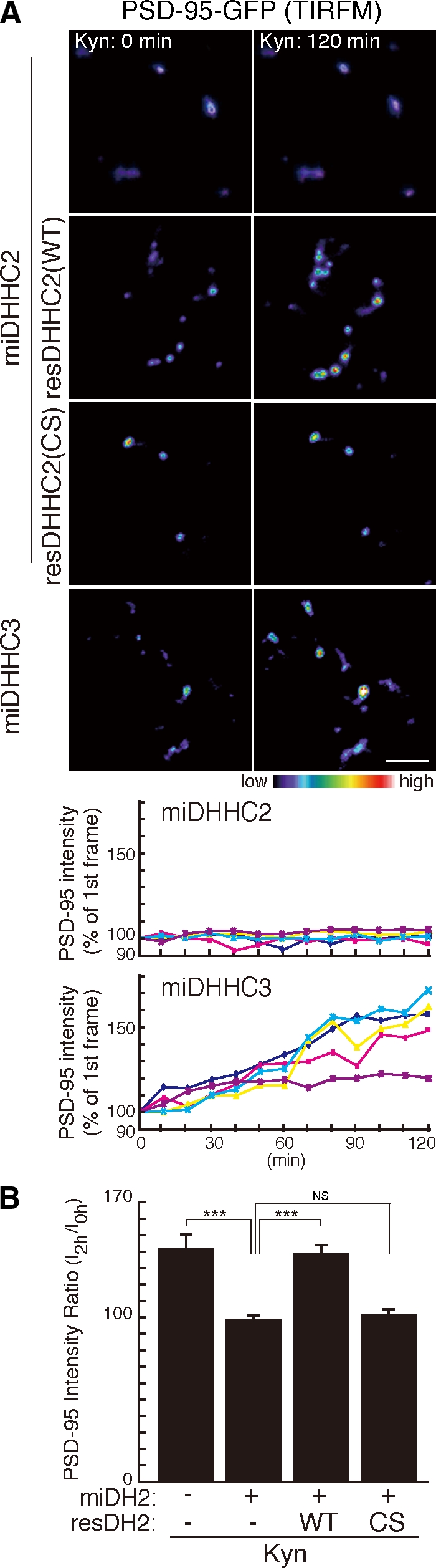
DHHC2 is essential for activity-sensitive PSD-95 palmitoylation. (A and B) DHHC2 but not DHHC3 mediates palmitoylation of PSD-95 upon activity blockade. Knockdown of DHHC2 but not DHHC3 inhibited Kyn-induced PSD-95–GFP recruitment at the synaptic membrane. miDHHC2-resistant DHHC2 (WT) but not PAT-inactive DHHC2 (CS) rescued Kyn-induced PSD-95 accumulation. (A) TIRFM intensity of representative five punctae from a neuron was plotted with time. (B) n = 3 each; ***, P < 0.001. Error bars indicate SD. Bar, 5 µm.
Dendritic DHHC2 translocates near postsynaptic sites upon activity blockade
We next investigated whether DHHC2 PAT activity, monitored by autopalmitoylation (Fukata et al., 2004), was regulated by synaptic activity. Whereas PSD-95 palmitoylation increased upon TTX or Kyn treatment, autopalmitoylation of DHHC2 and -3 did not change (Fig. 6 A), suggesting that DHHC2 activity may remain constant. We then investigated whether DHHC2 localization is regulated by synaptic activity. TIRFM imaging revealed that more DHHC2 was recruited near the membrane upon Kyn or TTX treatment (Fig. 6, B and C; and Video 4), where PSD-95 localized (Fig. S4 A). This translocation was activity sensitive as it was reversible upon washing out of Kyn (Fig. 6 D). Furthermore, we found that Kyn or TTX steadily induced colocalization of endogenous DHHC2 with PSD-95 over 48 h (Fig. 6, E and F), whereas DHHC3 remained at the Golgi apparatus (Fig. S4 B).
Figure 6.

Activity-sensitive synaptic translocation of DHHC2. (A) No change in DHHC autopalmitoylation (detected by the ABE method) was seen upon activity blockade (TTX or Kyn) of hippocampal neurons, suggesting that DHHC activity remains constant. (B and C) TIRFM imaging revealed that treatment with Kyn or TTX translocated DHHC2-GFP near the plasma membrane. n = 3; *, P < 0.05; **, P < 0.01 compared with control. Kymographs (pseudocolor) represent the changes in the intensity of DHHC2-GFP over time. (D) The translocation of DHHC2-GFP induced by Kyn treatment was reversible upon washing out Kyn. (E and F) Colocalization of endogenous DHHC2 with PSD-95 steadily increased over prolonged TTX or Kyn treatment. (F) n = 5–7 each; **, P < 0.01; ***, P < 0.001. (C and F) Error bars indicate SD. Bars: (B) 2 µm; (D) 5 µm; (E [left]) 3 µm; (E [right]) 1 µm.
Activity-sensitive PSD-95 palmitoylation by DHHC2 is necessary for homeostatic increase of AMPARs
Because PSD-95 anchors AMPARs at the postsynaptic sites through interaction with stargazin and related transmembrane AMPAR regulatory proteins (TARPs; Chen et al., 2000; Nicoll et al., 2006), we investigated changes in synaptic AMPARs upon activity blockade. We took advantage of the pHluorin-tagged GluR1 (pH-GluR1) subunit (Ashby et al., 2004; Yudowski et al., 2007) and TIRFM imaging to monitor specifically surface-expressed GluR1 (Video 5). pH-GluR1 punctate intensity was invariant over 12 h (Fig. 7, A and D; and Video 6). In contrast, TTX treatment gradually and continually increased pH-GluR1 intensity (Fig. 7, B and D; and Video 7). By post hoc immunostaining with PSD-95, we found that pH-GluR1 punctae by TIRFM completely overlapped PSD-95 clusters (Fig. 7 C). Furthermore, knockdown of DHHC2 or PSD-95 completely blocked this increase of pH-GluR1 intensity (Fig. 7, D and E). The effect of DHHC2 knockdown was rescued by miDHHC2-resistant WT DHHC2 (WT) but not by PAT-inactive DHHC2 (CS) (Fig. 7 E and Video 8). The effect of PSD-95 knockdown was rescued by short hairpin RNA–resistant WT PSD-95 (WT) but not by palmitoylation-deficient PSD-95 (CS) (Fig. 7 E, Fig. S4 D, and Video 9). We also found that knockdown of DHHC2 or PSD-95 similarly inhibited Kyn-induced increase of pH-GluR1 (Fig. 7 D). pH-GluR2, endogenous GluR1, GluR2, and stargazin-like TARPs but neither NR2A NMDA receptor nor VGLUT1 showed an increase similar to that of pH-GluR1 (Fig. S5). Similar inhibitory effects of DHHC2 or PSD-95 knockdown were observed for endogenous GluR1, GluR2, and TARPs (Fig. S5 G). These results indicate that activity-sensitive PSD-95 palmitoylation by DHHC2 mediates the homeostatic increase of AMPARs.
Figure 7.

Essential role of DHHC2-mediated PSD-95 palmitoylation in AMPAR homeostasis. (A and B) Upon 2 µM TTX treatment, TIRFM intensity of pH-GluR1 punctae gradually and continually increased over a 12-h observation. Fluorescence intensity was displayed in pseudocolor and was plotted with time. Kymographs represent the changes in the intensity of pH-GluR1. White lines indicate the regions used for the kymographs. Insets are magnified in the middle panels. (C) Post hoc immunostaining with PSD-95 showed that all GluR1 punctae by TIRFM overlapped synaptic PSD-95 clusters (arrows). (D) Quantification of fluorescent intensities of pH-GluR1 by TIRFM at 12 h after TTX or Kyn treatment. Knockdown of DHHC2 or PSD-95 completely inhibited the homeostatic increase of surface GluR1. n = 3 each; ***, P < 0.001 compared with nontreated control. miLacZ is a control miRNA targeting LacZ. (E) The inhibitory effect of DHHC2 knockdown was rescued by miDHHC2-resistant DHHC2 (WT) but not by PAT-inactive DHHC2 (CS). Furthermore, the inhibitory effect of PSD-95 knockdown was rescued by short hairpin RNA–resistant PSD-95 (WT) but not by palmitoylation-deficient PSD-95 (CS). n = 3 for each; ***, P < 0.001. (D and E) Error bars indicate SD. Bars: (A and B [top] and C) 10 µm; (A and B [insets]) 0.5 µm; (A and B [bottom] and E) 5 µm.
Discussion
By contrasting two representative PSD-95 palmitoylating enzymes, this study is the first to define differential regulation of DHHC-type palmitoylating enzymes. DHHC3 stably localizes at the Golgi apparatus and constitutively palmitoylates numerous substrates, including Gα, GluR2, and PSD-95. In contrast, dendritically localized DHHC2 senses changes in synaptic activity and rapidly translocates near postsynaptic membranes. Synaptically translocated DHHC2 induces rapid, specific, and stoichiometric palmitoylation and synaptic accumulation of PSD-95 and thereby AMPAR recruitment at postsynaptic sites. Thus, activity-sensitive DHHC2 translocation marks sites for AMPAR accumulation through compartmentalized PSD-95 palmitoylation. Complementing these findings, previous works showed that the Drosophila melanogaster DHHC17/HIP14 homologue localizes primarily to presynaptic terminals and acts on presynaptic Cys string protein and SNAP-25 (Ohyama et al., 2007; Stowers and Isacoff, 2007). The DHHC family members show distinctive subcellular distributions and different intracellular dynamics upon physiological stimuli. These distinctive properties provide mechanisms for specific control of protein palmitoylation by the large family of DHHC proteins.
Recent fluorescence recovery after photobleaching and photoconversion analyses revealed that several palmitoylated proteins such as Harvey Ras/neuroblastoma Ras (Rocks et al., 2005), Gαo (Chisari et al., 2007), and Gαq (Tsutsumi et al., 2009) rapidly shuttle between the plasma membrane and the Golgi apparatus. This constitutive shuttling comprises four steps: (1) palmitoylation by the Golgi-resident DHHC proteins, (2) trafficking to the plasma membrane, (3) depalmitoylation by a putative PPT and rapid cytosolic diffusion, and (4) transient trapping at the Golgi for repalmitoylation (Fig. 8 A; Rocks et al., 2006; Tsutsumi et al., 2009). In neurons, where the Golgi apparatus is segregated from the axon and dendrites, we suggest that Golgi-localized DHHC3 mediates the constitutive palmitoylation of PSD-95 at the cell body. In dendrites, PSD-95 depalmitoylated at the postsynaptic membrane diffuses from dendritic spine to shaft, is repalmitoylated on DHHC2-positive vesicles, and is redirected to postsynaptic membranes (Fig. 8 B). When synaptic activity is blocked, DHHC2 vesicles translocate from dendritic shafts to sites near the postsynaptic membrane. This allows DHHC2 to repalmitoylate PSD-95 in the spine (Fig. 8 C). Thus, mobile DHHC2 induces a local increase of PSD-95 palmitoylation, which leads to AMPAR recruitment. We propose that extracellular signals translocate specific DHHC PATs and create a new route for substrate shuttling between loci of palmitoylation and depalmitoylation, leading to efficient and precise substrate targeting. Such a compartmentalized regulatory mechanism of DHHC PATs may contribute to spatiotemporal regulation of signaling molecules in polarized neurons, epithelial cells, and migrating cells.
Figure 8.

Model for compartmentalized synaptic palmitoylation of PSD-95 by mobile DHHC2. (A) Certain palmitoylated proteins such as the Gα subunit shuttle between the plasma membrane and the Golgi in nonpolarized cells. Golgi-resident DHHC3 and a putative PPT at the plasma membrane can mediate this constitutive shuttling. In neurons, DHHC3 localizes at the Golgi apparatus and mediates constitutive palmitoylation of various substrates, including Gα and PSD-95. (B and C) PSD-95 shuttling in dendrites. (B) A dynamic equilibrium exists between palmitoylated postsynaptic PSD-95 and nonpalmitoylated cytosolic PSD-95. Depalmitoylated PSD-95 diffuses into the spine and dendritic shaft. DHHC2 PAT mainly localizes in dendritic shafts on vesicles and mediates dendritic PSD-95 palmitoylation. PSD, postsynaptic density. (C) When synaptic activity is blocked, DHHC2 vesicles move into spines. This translocated DHHC2 palmitoylates spinous PSD-95; the increased postsynaptic PSD-95 thereby contributes to AMPAR homeostasis.
This study monitors intracellular dynamics of palmitoylated proteins by taking advantage of time-lapse TIRFM, which visualizes membrane-associated proteins with exquisite sensitivity. This approach allowed us to follow dynamic changes in membrane-associated PSD-95–GFP over time in individual neurons. However, one may ask whether endogenous PSD-95 shows similar dynamics and whether TIRFM visualizes a limited set of synaptic contacts. The TIRFM limitation was supplemented with epifluorescent and confocal microscopic analyses on endogenous PSD-95. Also, our biohemical approaches, including metabolic labeling and the ABE method, showed that blocking synaptic activity quantitatively increases endogenously palmitoylated PSD-95, supporting the specificity of TIRFM imaging as a method for monitoring palmitoylated proteins in living cells.
A previous study reported that glutamate receptor activation accelerates depalmitoylation of PSD-95, dissociates PSD-95 from postsynaptic sites, and causes AMPAR endocytosis (El-Husseini et al., 2002). This sequence suggested that a PPT serves as the regulatory trigger. In contrast, our work demonstrates that activity blockade–induced PSD-95 palmitoylation up-regulates synaptic AMPARs. Thus, the palmitoylation/depalmitoylation cycle of PSD-95 bidirectionally contributes to AMPAR homeostasis (O’Brien et al., 1998; Turrigiano et al., 1998; Stellwagen and Malenka, 2006). By analogy, β-adrenergic receptor activation markedly accelerates depalmitoylation of Gαs, shifts Gαs to the cytoplasm, and down-regulates β-adrenergic receptor–mediated signaling (Wedegaertner and Bourne, 1994). Collectively, these studies suggest that palmitate cycling may generally mediate homeostasis of receptor-mediated signaling.
Recently, it was shown that both PSD-95 and PSD-93 play important roles in AMPAR trafficking (Elias et al., 2006). In this study, we found that knockdown of PSD-95 alone completely blocks the TTX- or Kyn-induced recruitment of AMPARs to the synapse. One may wonder why knockdown of PSD-95 alone completely blocks the TTX- or Kyn-induced AMPAR recruitment. We quantified expression levels of PSD-95 and PSD-93 in our cultured 18-d in vitro (DIV) hippocampal neurons by quantitative Western blotting. We found that PSD-95 expresses about eight times as much as PSD-93 (Fig. S3 B). Furthermore, we found that PSD-93–β, one of the major PSD-93 isoforms (Firestein et al., 2000; Parker et al., 2004), is specifically palmitoylated by the DHHC3 and -7 subfamily but not by the DHHC2 and -15 subfamily (Fig. S3 C), indicating that PSD-93–β palmitoylation is differently regulated from PSD-95 palmitoylation. Furthermore, it was reported that palmitoylation of PSD-93–α and PSD-93–β is not necessary for their postsynaptic targeting (Firestein et al., 2000). Collectively, we conclude that PSD-95 plays a major role in DHHC2-mediated AMPAR recruitment upon activity blockade.
Global proteomic studies indicate that palmitoylation represents a common posttranslational modification (Roth et al., 2006; Kang et al., 2008). Importantly, many palmitoylated proteins are key signaling molecules that subserve physiological processes. Furthermore, mutations of DHHC family members have been detected in cancers (Oyama et al., 2000; Mansilla et al., 2007; Yamamoto et al., 2007) and neurological disorders (Mukai et al., 2004, 2008; Mansouri et al., 2005; Yanai et al., 2006; Raymond et al., 2007). Elucidation of molecular mechanisms for palmitoylation lays a foundation to understand its role in physiological and pathological conditions. Because DHHC enzymes show substrate specificity, DHHC PATs represent exciting therapeutic targets. Our experiments of differential partitioning and regulation on DHHC PATs should serve as a prototype for understanding how dynamic protein palmitoylation is regulated in divergent signaling environments.
Materials and methods
Materials
The following antibodies were used: rabbit polyclonal antibodies to DHHC3/GODZ (Abcam), Gαq (Santa Cruz Biotechnology, Inc.), GluR1 (EMD; Millipore), GRIP1 (Millipore), PSD-93 (Millipore), and stargazin/TARP (Millipore); a guinea pig polyclonal antibody to VGLUT1 (Millipore); and mouse monoclonal antibodies to β-catenin (BD), GluR2 (Millipore), HA (Covance), NMDAR1 (Millipore), PSD-95 (Thermo Fisher Scientific), and synaptophysin (Sigma-Aldrich). Anti–PSD-93 antibody was raised against (aa 336–379) and detected all isoforms of PSD-93. Rabbit polyclonal antibodies to GFP, PSD-95, and moesin were raised against GFP (aa 1–239), GST–PSD-95 (aa 1–434), and GST-moesin (aa 307–577), respectively, and affinity purified. A mouse monoclonal antibody to DHHC2 was raised by a baculovirus display method, which is useful for the production of antibodies against membrane proteins (Masuda et al., 2003; Saitoh et al., 2007). The following reagents were used: Kyn and APV (Tocris); TTX (Nacalai Tesque, Inc.); CNQX and CHX (Sigma-Aldrich); and 2-bromohexadecanoic acid (2-BP; Fluka).
For knockdown experiments in HEK293 cells, siRNAs from QIAGEN were used: scramble (Allstars negative control), siDHHC2, and siDHHC3. siRNA and plasmid-based miRNA for DHHCs were validated by two methods: (1) reduced expression of exogenously expressed DHHC proteins in HEK293 cells (Western blotting) and (2) down-regulation of endogenous mRNAs in HEK293 cells (quantitative real-time PCR).
Cloning and plasmid constructions
The rat cDNAs of synaptophysin (GenBank/EMBL/DDBJ accession no. NM_012664), DHHC2 (GenBank/EMBL/DDBJ accession no. AF228917), DHHC3 (GenBank/EMBL/DDBJ accession no. NM_001039014), DHHC7 (GenBank/EMBL/DDBJ accession no. NM_133394), and DHHC15 (GenBank/EMBL/DDBJ accession no. AY886531) were cloned from rat brain total RNA by RT-PCR. Synaptophysin was subcloned into pCAGGS-GFP, and DHHC2, -3, -7, and -15 were subcloned into pEF-Bos-HA and pcDNA3.1. The mutant rat DHHC2(C156S) was generated by using site-directed mutagenesis. pGW1-rat PSD-95–GFP, pGW1-rat PSD-93–β-GFP and pEF-Bos-HA-mouse DHHC constructs were described previously (El-Husseini et al., 2002; Fukata et al., 2004; Parker et al., 2004). pGW1–PSD-95–HA was constructed by replacing a GFP fragment with a synthetic DNA fragment encoding HA. pEGFP-C1-Rac1-CLLL was described previously (Nakagawa et al., 2001). To obtain the antigen for antibody production, DHHC2 was subcloned into the pcDNA-His-Flag vector. His-Flag–tagged DHHC2 was then subcloned into pAcYM1 for baculovirus production. DHHC2 was also subcloned into pGW1-GFP. To obtain Thy1/pH-GluR1, we first inserted pHluorin between residues 21 and 22 of rat GluR1 (Tomita et al., 2004) and subcloned pH-GluR1 into a Thy1 expression cassette. pCAGGS–pH-GluR2 was made by inserting pHluorin between residues 23 and 24 of mouse GluR2 (Fukata et al., 2005). cDNA of NR2A-GFP (Luo et al., 2002) was subcloned into the pCAGGS vector.
In cultured hippocampal neurons, DHHC2 and -3 were knocked down by the miR-RNAi system (Invitrogen). We used BLOCK-iT RNAi Designer (Invitrogen) to select the targeting sequences, and the following targeting sequences were used (targeting both rat and human sequences): miDHHC2, 5′-GGTGAACAATTGTGTTGGATT-3′ (alternative sequence, 5′-TGTGCATAGTGTCCATGGAAA-3′; both sequences yielded similar results); miDHHC3, 5′-TGAGACGGGAATAGAACAATT-3′; and miLacZ, 5′-GACTACACAAATCAGCGATTT-3′ (as a negative control). After subcloning these oligonucleotides into pcDNA6.2-EmGFP-miR (Invitrogen), EmGFP was replaced with mCherry, and the pre-miRNA expression cassette of pcDNA6.2-mCherry-miR (or pcDNA6.2-EmGFP-miR) was transferred to the pCAGGS vector with a β-actin promoter. PSD-95 was knocked down as described previously (Elias et al., 2006), replacing GFP of pLLox3.7 (American Type Culture Collection) with mCherry. DHHC2 (on pEF-Bos-HA-rat DHHC2) and PSD-95 (on pGW1-rat PSD-95–HA) rescue constructs that have two and four different nucleotides in the target sequences were generated by using site-directed mutagenesis (DHHC2, 5′-GGTGAACAACTGCGTTGGATT-3′; PSD-95, 5′-TCACAATAATAGCCCAGTATA-3′; changed nucleotides are underlined). All PCR products were analyzed by DNA sequencing.
pcDNAI-Gαq-GFP was provided by C.A. Berlot (Weis Center for Research, Danville, PA; Hughes et al., 2001). The cDNAs of pHluorin and mCherry were provided by J.E. Rothman (Columbia University, New York, NY; Miesenbock et al., 1998) and R.Y. Tsien (University of California, San Diego, La Jolla, CA; Shaner et al., 2005), respectively. cDNAs of rat GluR1, NR2A-GFP, and pEGFP-C1-Rac1-CLLL were gifts from R. Huganir (Johns Hopkins University, Baltimore, MD), S. Vicini (Georgetown University School of Medicine, Washington, DC), and K. Kaibuchi (Nagoya University, Showa-Ku, Nagoya, Japan), respectively. Thy1 expression cassette was provided by D. Monard (Friedrich Miescher Institute, Basel, Switzerland; Luthi et al., 1997).
Time-lapse imaging with TIRFM
Hippocampal neuron cultures were prepared from rat embryonic day 18–19 embryos. All animal experiments described herein were reviewed and approved by the ethical committee in our institutes and were performed according to the institutional guidelines concerning the care and handling of experimental animals. Neurons were seeded at a density of 2.5 × 105 cells per 3.5-cm glass-based dish (Iwaki) in neurobasal medium (Invitrogen) supplemented with B-27 supplement (Invitrogen) and 2 mM Glutamax (Invitrogen). 8–10-DIV neurons were transfected by Lipofectamine 2000 (Invitrogen) and observed (18–21 DIV) at 37°C in a microincubator (MI-IBC-IF; Olympus) with an inverted microscope (IX81; Olympus) equipped with a Plan-Apochromat 100× NA 1.45 oil TIRFM objective lens, an ImageEM charge-coupled device (CCD) camera (C9100-13; Hamamatsu Photonics) and Meta Imaging software version 7.1 (MDS Analytical Technologies). A 488-nm laser was used as a light source. Time-lapse images were taken every 10 min with a laser-based zero drift autofocusing system (IX81-ZDC; Olympus), which adjusts the focal plane to the initial focal plane just before each imaging frame. The video files (QuickTime Movie) were produced with ImageReady 2.0 (Adobe Systems, Inc.). To quantitate changes in PSD-95–GFP, DHHC2-GFP, or pH-GluR1 intensity by TIRFM, we randomly chose fields, and the punctae (>1.25 µm in diameter) were quantitated in every frame. Fluorescent intensities from TIRF images were analyzed using MetaMorph software version 7.1 (MDS Analytical Technologies). The ratios of intensities at 0–120 min (for PSD-95 and DHHC2) or 0–12 h (for GluR1) in 50–100 randomly selected punctae (three to eight independent experiments) are shown. Kymographs were produced using Meta Imaging software version 7.1.
Immunofluorescence analysis of hippocampal neuron culture
Cultured hippocampal neurons (5 × 104 cells) were seeded onto 12-mm coverslips in 24-well dishes. 18–28-DIV neurons were fixed with 4% paraformaldehyde/120 mM sucrose/100 mM Hepes, pH 7.4, at room temperature for 10 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with PBS containing 10 mg/ml BSA for 10 min on ice. For staining of DHHC2 and VGLUT1, neurons were fixed with methanol for 10 min at −30°C. For mouse anti-DHHC2 antibody, Alexa Fluor 488–conjugated anti–mouse IgG1 subtype specific (Invitrogen) was used as a secondary antibody. For surface GluR1 and GluR2 staining, GluR1 and GluR2 receptors were “live” labeled with an antibody to an extracellular epitope of GluR1 (EMD) or GluR2 (Millipore) by incubating neurons in conditioned medium for 15 min at 37°C. Neurons were then fixed with 2% paraformaldehyde for 20 min and blocked as described above. Surface GluR1 and GluR2 were visualized with Alexa Fluor 488–conjugated secondary antibody. Fluorescent images were taken with a confocal laser-scanning microscopy system (LSM5 Exciter; Carl Zeiss, Inc.) equipped with a Plan-Apochromat 63× NA 1.40 oil immersion objective lens. For knockdown experiments, 8–12-DIV neurons were transfected with pCAGGS-mCherry-miR vectors by Lipofectamine 2000. At 8–10 d after transfection, neurons were stained with anti-DHHC2, DHHC3, PSD-95, GluR1, GluR2, and stargazin/TARP antibodies. To quantitate the intensity of PSD-95 clusters and surface GluR1, surface GluR2, or TARP clusters (costained with PSD-95), we randomly chose 10–15 fields from two independent neuronal cultures (on treated and age-matched sister control cultures) and analyzed the three largest caliber proximal dendrites (20 µm long; at least 400 clusters). We measured the mean intensities of individual clusters (>1 µm diameter) along these dendritic segments. Microscope control and all image analyses were performed with ZEN software (Carl Zeiss, Inc.). Brightness and contrast adjustments were applied to the whole image using Photoshop CS3 (Adobe Systems, Inc.). For some experiments, immunolabeled samples were observed by TIRFM and epifluorescent microscopy.
ABE method
The ABE method was performed as previously described (Roth et al., 2006; Kang et al., 2008) and modified for cultured neurons. After treating 18–28-DIV hippocampal neurons (5 × 105 cells/6-well dish) with the indicated antagonists or inhibitors, neurons were washed with PBS containing 10 mM N-ethyl-maleimide (NEM) twice and solubilized with 0.1 ml of lysis buffer (LB; 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, and 50 mM NaCl) containing 2% SDS and 10 mM NEM. After 15 min of extraction, LB with 2% Triton X-100 and 10 mM NEM was added to a final volume of 1 ml and incubated for 1 h at 4°C. After centrifugation at 20,000 g for 10 min, the supernatants were precipitated by the chloroform-methanol (CM) method (Wessel and Flugge, 1984). Precipitated protein was solubilized in 0.2 ml SB (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, and 4% SDS) containing 10 mM NEM at 37°C for 10 min. The protein was diluted into 0.8 ml LB with 0.2% Triton X-100 and 1 mM NEM and incubated overnight at 4°C. NEM was removed by three sequential CM precipitations. Precipitated protein was solubilized in 0.2 ml of buffer SB, and then 0.8 ml HB (1 M hydroxylamine, pH 7.5, 150 mM NaCl, 0.2% Triton X-100, and 1 mM biotin-HPDP) or buffer TB (1 M Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% Triton X-100, and 1 mM biotin-HPDP) was added. The mixture was incubated for 1 h at room temperature and subjected to CM precipitation. The precipitated protein was dissolved in 0.2 ml SB, diluted into 0.8 ml LB containing 150 mM NaCl, 0.2% Triton X-100, and 200 µM biotin-HPDP, and incubated for 1 h at room temperature. Free biotin-HPDP was removed by CM precipitation. The precipitated protein was solubilized in 100 µl of buffer UB (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, and 2% SDS) and diluted in 900 µl LB with 0.2% Triton X-100. After brief centrifugation, the supernatant was incubated with 30 µl neutravidin-agarose (Thermo Fisher Scientific) for 1 h at 4°C. After washing the beads with LB containing 0.1% SDS and 0.2% Triton X-100, bound proteins were suspended in SDS-PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, and 0.001% bromophenol blue) with 10 mM DTT (without βME; −βME) and boiled at 90°C for 2 min. To see the palmitoylation-dependent mobility shift of PSD-95, the cell lysate was treated similarly (−βME). To reduce the sample completely, the lysate was separately treated with SDS-PAGE sample buffer with 2% βME at 100°C for 5 min (+βME). Samples were subjected to SDS-PAGE and Western blotting with the indicated antibodies.
In vivo palmitate labeling
Hippocampal neurons were infected with Semliki forest virus to express GFP or GFP-tagged DN DHHC2/15 (Fukata et al., 2004). At 24 h after infection, cells were labeled for 2 h in neurobasal media containing 3 mCi/ml [3H]palmitic acid (PerkinElmer) either in the presence or absence of 10 mM Kyn. Labeled cells were washed with PBS and resuspended in 0.15 ml LB A (20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 100 mM NaCl, and 1% SDS). After 5 min of extraction, 1% Triton X-100 was added to a final volume of 1.5 ml. After centrifugation at 20,000 g for 10 min, the supernatants were incubated with rabbit anti–PSD-95 antibody for 1 h and then incubated for 1 h with 30 µl protein A–Sepharose (GE Healthcare) at 4°C. Immunoprecipitates were washed three times with buffer containing 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 100 mM NaCl, and 1% Triton X-100. Immunoprecipitated PSD-95 was suspended in SDS sample buffer. For fluorography, protein samples were separated by SDS-PAGE. Gels were treated with Amplify (GE Healthcare) for 30 min, dried under vacuum, and exposed to Biomax MS (Kodak) at −80°C for 2 wk. After autoradiography, the bands were scanned and analyzed with National Institutes of Health software.
Transfected HEK293 cells were preincubated for 30 min in serum-free DME with 5 mg/ml fatty acid–free BSA (Sigma-Aldrich). Cells were then labeled with 0.25 mCi/ml [3H]palmitic acid for 4 h in the preincubation medium. Cells were washed with PBS and scraped with SDS-PAGE sample buffer with 10 mM DTT. The cell lysate was resolved by SDS-PAGE, followed by fluorography (36-h exposure) and Western blotting.
In situ hybridization
In situ hybridization on 7-µm paraffin-embedded 3-wk-old rat brain sections (Genostaff) was performed by using digoxigenin-labeled RNA probes. cDNAs of mouse DHHC2 (nt 1–1,098 from initiating ATG), rat DHHC3 (nt 1–900), rat DHHC7 (nt 1–927), rat DHHC15 (nt 1–1,014), and rat PSD-95 (nt 1,212–1,444) were used for probe templates. An antidigoxigenin antibody linked to alkaline phosphatase (Dako) and NBT/BCIP (nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate; Dako) substrate was used to detect hybridization signals. All sections were developed for 1 h. Images were taken with a dissection microscope (SZ61; Olympus) equipped with a digital camera (DP20; Olympus) and with an upright microscope (BX51; Olympus) equipped with a UPlan SApo 20× NA 0.75 objective lens and a CCD camera (DP72; Olympus).
Quantitative Western blotting
Bands on blotted membranes were visualized with a cooled CCD camera (Light-Capture II; ATTO), and the optimal specific bands were analyzed with the CS Analyzer 3.0 software (ATTO). For calibration, immunopurified PSD-95–GFP and PSD-93–β-GFP from transfected HEK293 cells were quantitated by Coomassie brilliant staining using BSA.
Subcellular fractionation
The method was basically followed as described previously (Carlin et al., 1980). In brief, five rat adult brains were homogenized in buffer containing 320 mM sucrose and 10 mM Hepes-NaOH, pH 7.4 (containing 0.2 mM PMSF). Homogenate was centrifuged for 10 min at 1,000 g to remove crude nuclear fraction (P1). The supernatant (S1) was centrifuged at 9,000 g for 15 min to produce a pellet (P2) and supernatant (S2). The S2 was centrifuged at 100,000 g for 1 h to produce a pellet (P3; microsomal fraction) and supernatant (S3). The P2 fraction was resuspended in the homogenization buffer. Discontinuous sucrose gradients containing 3 ml of the resuspended P2 material and 3 ml each of 0.8, 1.0, and 1.2 M sucrose solutions in 10 mM Hepes-NaOH, pH 7.4, were run for 2 h at 58,000 g (SW41 rotor; Beckman Coulter). The band between 1.0 and 1.2 M sucrose was obtained as a synaptosome fraction. This synaptosome fraction was extracted with ice-cold 0.5% Triton X-100 in 0.16 M sucrose and 6 mM Tris-HCl, pH 8.1, and then centrifuged at 32,800 g for 20 min to divide into soluble and insoluble fractions (Ins1; PSD-1). The pellet was resuspended in 0.5% Triton X-100, 0.16 M sucrose, and 6 mM Tris-HCl, pH 8.1, and centrifuged at 200,000 g for 1 h to produce a pellet (Ins2; PSD-2). 50 µg of proteins of each fraction was analyzed by Western blotting.
Statistical analysis
The results are expressed as mean ± SD. Statistical comparisons between groups were performed by the Student’s t test.
Online supplemental material
Fig. S1 shows that synaptic PSD-95 accumulation upon activity blockade does not require protein synthesis. Fig. S2 shows the specific detection of PSD-95 palmitoylation by biochemical approaches. Fig. S3 shows that DHHC2 and -3 and PSD-95 dominantly express as compared with other family proteins in the hippocampus. Fig. S4 shows that DHHC2 translocates near the postsynaptic sites upon activity blockade. Fig. S5 shows that DHHC2 and PSD-95 are necessary for homeostatic increase of GluR2 as well as GluR1. Video 1 shows PSD-95–GFP dynamics in neurons by time-lapse TIRFM imaging and shows the increased accumulation of PSD-95–GFP upon Kyn treatment. Video 2 shows that CHX treatment does not inhibit Kyn-induced PSD-95–GFP increase at the synapse. Video 3 shows that activity-sensitive trafficking of PSD-95 is regulated by DHHC2. Video 4 shows activity-sensitive trafficking of DHHC2. Video 5 shows the specificity of pH-GluR1 imaging. Video 6 shows AMPAR (pH-GluR1) dynamics by time-lapse TIRFM imaging. Video 7 shows that pH-GluR1 intensity gradually and continually increases over 12 h upon TTX treatment. Video 8 shows the requirement of palmitoylating activity of DHHC2 for homeostatic increase of GluR1. Video 9 shows the requirement of PSD-95 palmitoylation for the homeostatic increase of GluR1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200903101/DC1.
Acknowledgments
We thank K. Imoto (National Institute for Physiological Sciences, Okazaki, Aichi, Japan) and M. Nishijima (National Institute of Health Sciences, Setagaya-ku,Tokyo, Japan and Japan Science and Technology Agency, Chiyoda-ku, Tokyo, Japan) for suggestions and encouragement, K. Kaibuchi for sharing reagents, F. Perez (Institut Curie, Paris, France), A.S. Kato (Eli Lilly and Company, Indianapolis, IN), and T. Watanabe (Nagoya University, Chikusa-ku, Nagoya, Japan) for valuable suggestions, and N. Takahashi for technical support.
J. Noritake and R. Tsutsumi are supported by the Japan Society for the Promotion of Science. Y. Fukata is supported by grants from the Human Frontier Science Program (HFSP; CDA0015-07) and Ministry of Education, Culture, Sports, Science and Technology (MEXT; 21680029). M. Fukata is also supported by grants from the HFSP (RGY0059-06) and MEXT (20670005, 20022043, and 20054022).
Footnotes
Abbreviations used in this paper: 2-BP, 2-bromopalmitate; ABE, acyl-biotin exchange; AMPAR, AMPA receptor; βME, β-mercaptoethanol; CCD, charge-coupled device; CHX, cycloheximide; CM, chloroform-methanol; DIV, day in vitro; DN, dominant-negative; Kyn, kynurenic acid; LB, lysis buffer; miRNA, microRNA; NEM, N-ethyl-maleimide; NMDA, N-methyl-d-aspartate; PAT, palmitoyl acyltransferase; PPT, palmitoyl protein thioesterase; TARP, transmembrane AMPAR regulatory protein; TIRFM, total internal reflection fluorescence microscopy; TTX, tetrodotoxin; WT, wild type.
References
- Ashby M.C., De La Rue S.A., Ralph G.S., Uney J., Collingridge G.L., Henley J.M. 2004. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs.J. Neurosci. 24:5172–5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels D.J., Mitchell D.A., Dong X., Deschenes R.J. 1999. Erf2, a novel gene product that affects the localization and palmitoylation of Ras2 in Saccharomyces cerevisiae.Mol. Cell. Biol. 19:6775–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin R.K., Grab D.J., Cohen R.S., Siekevitz P. 1980. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities.J. Cell Biol. 86:831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Chetkovich D.M., Petralia R.S., Sweeney N.T., Kawasaki Y., Wenthold R.J., Bredt D.S., Nicoll R.A. 2000. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms.Nature. 408:936–943 [DOI] [PubMed] [Google Scholar]
- Chisari M., Saini D.K., Kalyanaraman V., Gautam N. 2007. Shuttling of G protein subunits between the plasma membrane and intracellular membranes.J. Biol. Chem. 282:24092–24098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini Ael-D., Bredt D.S. 2002. Protein palmitoylation: a regulator of neuronal development and function.Nat. Rev. Neurosci. 3:791–802 [DOI] [PubMed] [Google Scholar]
- El-Husseini A.E., Schnell E., Chetkovich D.M., Nicoll R.A., Bredt D.S. 2000. PSD-95 involvement in maturation of excitatory synapses.Science. 290:1364–1368 [PubMed] [Google Scholar]
- El-Husseini Ael-D., Schnell E., Dakoji S., Sweeney N., Zhou Q., Prange O., Gauthier-Campbell C., Aguilera-Moreno A., Nicoll R.A., Bredt D.S. 2002. Synaptic strength regulated by palmitate cycling on PSD-95.Cell. 108:849–863 [DOI] [PubMed] [Google Scholar]
- Elias G.M., Funke L., Stein V., Grant S.G., Bredt D.S., Nicoll R.A. 2006. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins.Neuron. 52:307–320 [DOI] [PubMed] [Google Scholar]
- Fang C., Deng L., Keller C.A., Fukata M., Fukata Y., Chen G., Luscher B. 2006. GODZ-mediated palmitoylation of GABA(A) receptors is required for normal assembly and function of GABAergic inhibitory synapses.J. Neurosci. 26:12758–12768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernando C., Fukata M., Bernatchez P.N., Fukata Y., Lin M.I., Bredt D.S., Sessa W.C. 2006. Identification of Golgi-localized acyl transferases that palmitoylate and regulate endothelial nitric oxide synthase.J. Cell Biol. 174:369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein B.L., Craven S.E., Bredt D.S. 2000. Postsynaptic targeting of MAGUKs mediated by distinct N-terminal domains.Neuroreport. 11:3479–3484 [DOI] [PubMed] [Google Scholar]
- Fukata M., Fukata Y., Adesnik H., Nicoll R.A., Bredt D.S. 2004. Identification of PSD-95 palmitoylating enzymes.Neuron. 44:987–996 [DOI] [PubMed] [Google Scholar]
- Fukata Y., Tzingounis A.V., Trinidad J.C., Fukata M., Burlingame A.L., Nicoll R.A., Bredt D.S. 2005. Molecular constituents of neuronal AMPA receptors.J. Cell Biol. 169:399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y., Iwanaga T., Fukata M. 2006. Systematic screening for palmitoyl transferase activity of the DHHC protein family in mammalian cells.Methods. 40:177–182 [DOI] [PubMed] [Google Scholar]
- Funke L., Dakoji S., Bredt D.S. 2005. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions.Annu. Rev. Biochem. 74:219–245 [DOI] [PubMed] [Google Scholar]
- Hayashi T., Rumbaugh G., Huganir R.L. 2005. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites.Neuron. 47:709–723 [DOI] [PubMed] [Google Scholar]
- Hemsley P.A., Grierson C.S. 2008. Multiple roles for protein palmitoylation in plants.Trends Plant Sci. 13:295–302 [DOI] [PubMed] [Google Scholar]
- Hemsley P.A., Kemp A.C., Grierson C.S. 2005. The TIP GROWTH DEFECTIVE1 S-acyl transferase regulates plant cell growth in Arabidopsis.Plant Cell. 17:2554–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T.E., Zhang H., Logothetis D.E., Berlot C.H. 2001. Visualization of a functional Galpha q-green fluorescent protein fusion in living cells. Association with the plasma membrane is disrupted by mutational activation and by elimination of palmitoylation sites, but not be activation mediated by receptors or AlF4.J. Biol. Chem. 276:4227–4235 [DOI] [PubMed] [Google Scholar]
- Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A.O., Thompson J.X., Roth A.F., Drisdel R.C., Mastro R., et al. 2008. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation.Nature. 456:904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C.A., Yuan X., Panzanelli P., Martin M.L., Alldred M., Sassoe-Pognetto M., Luscher B. 2004. The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ.J. Neurosci. 24:5881–5891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M.B. 2000. Signal-processing machines at the postsynaptic density.Science. 290:750–754 [DOI] [PubMed] [Google Scholar]
- Kim E., Sheng M. 2004. PDZ domain proteins of synapses.Nat. Rev. Neurosci. 5:771–781 [DOI] [PubMed] [Google Scholar]
- Linder M.E., Deschenes R.J. 2004. Model organisms lead the way to protein palmitoyltransferases.J. Cell Sci. 117:521–526 [DOI] [PubMed] [Google Scholar]
- Linder M.E., Deschenes R.J. 2007. Palmitoylation: policing protein stability and traffic.Nat. Rev. Mol. Cell Biol. 8:74–84 [DOI] [PubMed] [Google Scholar]
- Lobo S., Greentree W.K., Linder M.E., Deschenes R.J. 2002. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae.J. Biol. Chem. 277:41268–41273 [DOI] [PubMed] [Google Scholar]
- Luo J.H., Fu Z.Y., Losi G., Kim B.G., Prybylowski K., Vissel B., Vicini S. 2002. Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture.Neuropharmacology. 42:306–318 [DOI] [PubMed] [Google Scholar]
- Luthi A., Van der Putten H., Botteri F.M., Mansuy I.M., Meins M., Frey U., Sansig G., Portet C., Schmutz M., Schroder M., et al. 1997. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation.J. Neurosci. 17:4688–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla F., Birkenkamp-Demtroder K., Kruhoffer M., Sorensen F.B., Andersen C.L., Laiho P., Aaltonen L.A., Verspaget H.W., Orntoft T.F. 2007. Differential expression of DHHC9 in microsatellite stable and instable human colorectal cancer subgroups.Br. J. Cancer. 96:1896–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri M.R., Marklund L., Gustavsson P., Davey E., Carlsson B., Larsson C., White I., Gustavson K.H., Dahl N. 2005. Loss of ZDHHC15 expression in a woman with a balanced translocation t(X;15)(q13.3;cen) and severe mental retardation.Eur. J. Hum. Genet. 13:970–977 [DOI] [PubMed] [Google Scholar]
- Masuda K., Itoh H., Sakihama T., Akiyama C., Takahashi K., Fukuda R., Yokomizo T., Shimizu T., Kodama T., Hamakubo T. 2003. A combinatorial G protein-coupled receptor reconstitution system on budded baculovirus. Evidence for Galpha and Galphao coupling to a human leukotriene B4 receptor.J. Biol. Chem. 278:24552–24562 [DOI] [PubMed] [Google Scholar]
- Miesenbock G., De Angelis D.A., Rothman J.E. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins.Nature. 394:192–195 [DOI] [PubMed] [Google Scholar]
- Migaud M., Charlesworth P., Dempster M., Webster L.C., Watabe A.M., Makhinson M., He Y., Ramsay M.F., Morris R.G., Morrison J.H., et al. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein.Nature. 396:433–439 [DOI] [PubMed] [Google Scholar]
- Mukai J., Liu H., Burt R.A., Swor D.E., Lai W.S., Karayiorgou M., Gogos J.A. 2004. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia.Nat. Genet. 36:725–731 [DOI] [PubMed] [Google Scholar]
- Mukai J., Dhilla A., Drew L.J., Stark K.L., Cao L., MacDermott A.B., Karayiorgou M., Gogos J.A. 2008. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion.Nat. Neurosci. 11:1302–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M., Fukata M., Yamaga M., Itoh N., Kaibuchi K. 2001. Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites.J. Cell Sci. 114:1829–1838 [DOI] [PubMed] [Google Scholar]
- Nicoll R.A., Tomita S., Bredt D.S. 2006. Auxiliary subunits assist AMPA-type glutamate receptors.Science. 311:1253–1256 [DOI] [PubMed] [Google Scholar]
- O’Brien R.J., Kamboj S., Ehlers M.D., Rosen K.R., Fischbach G.D., Huganir R.L. 1998. Activity-dependent modulation of synaptic AMPA receptor accumulation.Neuron. 21:1067–1078 [DOI] [PubMed] [Google Scholar]
- Ohyama T., Verstreken P., Ly C.V., Rosenmund T., Rajan A., Tien A.C., Haueter C., Schulze K.L., Bellen H.J. 2007. Huntingtin-interacting protein 14, a palmitoyl transferase required for exocytosis and targeting of CSP to synaptic vesicles.J. Cell Biol. 179:1481–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama T., Miyoshi Y., Koyama K., Nakagawa H., Yamori T., Ito T., Matsuda H., Arakawa H., Nakamura Y. 2000. Isolation of a novel gene on 8p21.3-22 whose expression is reduced significantly in human colorectal cancers with liver metastasis.Genes Chromosomes Cancer. 29:9–15 [DOI] [PubMed] [Google Scholar]
- Parker M.J., Zhao S., Bredt D.S., Sanes J.R., Feng G. 2004. PSD93 regulates synaptic stability at neuronal cholinergic synapses.J. Neurosci. 24:378–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponimaskin E., Dityateva G., Ruonala M.O., Fukata M., Fukata Y., Kobe F., Wouters F.S., Delling M., Bredt D.S., Schachner M., Dityatev A. 2008. Fibroblast growth factor-regulated palmitoylation of the neural cell adhesion molecule determines neuronal morphogenesis.J. Neurosci. 28:8897–8907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond F.L., Tarpey P.S., Edkins S., Tofts C., O’Meara S., Teague J., Butler A., Stevens C., Barthorpe S., Buck G., et al. 2007. Mutations in ZDHHC9, which encodes a palmitoyltransferase of NRAS and HRAS, cause X-linked mental retardation associated with a Marfanoid habitus.Am. J. Hum. Genet. 80:982–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M.D. 2006. Palmitoylation of ligands, receptors, and intracellular signaling molecules.Sci. STKE. doi:10.1126/stke.3592006re14 [DOI] [PubMed] [Google Scholar]
- Rocks O., Peyker A., Kahms M., Verveer P.J., Koerner C., Lumbierres M., Kuhlmann J., Waldmann H., Wittinghofer A., Bastiaens P.I. 2005. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms.Science. 307:1746–1752 [DOI] [PubMed] [Google Scholar]
- Rocks O., Peyker A., Bastiaens P.I. 2006. Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors.Curr. Opin. Cell Biol. 18:351–357 [DOI] [PubMed] [Google Scholar]
- Roth A.F., Feng Y., Chen L., Davis N.G. 2002. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase.J. Cell Biol. 159:23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A.F., Wan J., Bailey A.O., Sun B., Kuchar J.A., Green W.N., Phinney B.S., Yates J.R., III, Davis N.G. 2006. Global analysis of protein palmitoylation in yeast.Cell. 125:1003–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh R., Ohtomo T., Yamada Y., Kamada N., Nezu J., Kimura N., Funahashi S., Furugaki K., Yoshino T., Kawase Y., et al. 2007. Viral envelope protein gp64 transgenic mouse facilitates the generation of monoclonal antibodies against exogenous membrane proteins displayed on baculovirus.J. Immunol. Methods. 322:104–117 [DOI] [PubMed] [Google Scholar]
- Shaner N.C., Steinbach P.A., Tsien R.Y. 2005. A guide to choosing fluorescent proteins.Nat. Methods. 2:905–909 [DOI] [PubMed] [Google Scholar]
- Stellwagen D., Malenka R.C. 2006. Synaptic scaling mediated by glial TNF-alpha.Nature. 440:1054–1059 [DOI] [PubMed] [Google Scholar]
- Stowers R.S., Isacoff E.Y. 2007. Drosophila huntingtin-interacting protein 14 is a presynaptic protein required for photoreceptor synaptic transmission and expression of the palmitoylated proteins synaptosome-associated protein 25 and cysteine string protein.J. Neurosci. 27:12874–12883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S., Fukata M., Nicoll R.A., Bredt D.S. 2004. Dynamic interaction of stargazin-like TARPs with cycling AMPA receptors at synapses.Science. 303:1508–1511 [DOI] [PubMed] [Google Scholar]
- Topinka J.R., Bredt D.S. 1998. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel, Kv1.4.Neuron. 20:125–134 [DOI] [PubMed] [Google Scholar]
- Tsutsumi R., Fukata Y., Noritake J., Iwanaga T., Perez F., Fukata M. 2009. Identification of G protein alpha subunit-palmitoylating enzyme.Mol. Cell. Biol. 29:435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G.G., Leslie K.R., Desai N.S., Rutherford L.C., Nelson S.B. 1998. Activity-dependent scaling of quantal amplitude in neocortical neurons.Nature. 391:892–896 [DOI] [PubMed] [Google Scholar]
- Wedegaertner P.B., Bourne H.R. 1994. Activation and depalmitoylation of Gs alpha.Cell. 77:1063–1070 [DOI] [PubMed] [Google Scholar]
- Wessel D., Flugge U.I. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids.Anal. Biochem. 138:141–143 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Chochi Y., Matsuyama H., Eguchi S., Kawauchi S., Furuya T., Oga A., Kang J.J., Naito K., Sasaki K. 2007. Gain of 5p15.33 is associated with progression of bladder cancer.Oncology. 72:132–138 [DOI] [PubMed] [Google Scholar]
- Yanai A., Huang K., Kang R., Singaraja R.R., Arstikaitis P., Gan L., Orban P.C., Mullard A., Cowan C.M., Raymond L.A., et al. 2006. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function.Nat. Neurosci. 9:824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski G.A., Puthenveedu M.A., Leonoudakis D., Panicker S., Thorn K.S., Beattie E.C., von Zastrow M. 2007. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors.J. Neurosci. 27:11112–11121 [DOI] [PMC free article] [PubMed] [Google Scholar]