

Abstract
L1 cell adhesion molecule (L1), a protein critical for appropriate development of the central nervous system, is a target for ethanol teratogenicity. Ethanol inhibits both L1 mediated cell adhesion as well as L1 mediated neurite outgrowth. L1 has been shown to increase cell survival in cerebellar granule cells while ethanol has been shown to increase cell death. We sought to determine if L1 protected cells from ethanol induced cell death. Cerebellar granule cells from postnatal day 6 rat pups were cultured on either poly L-lysine with or without an L1 substratum. Alcohol was added at 2 hours post plating and cell survival was measured at various times. L1 substratum significantly increased cell survival at 72 and 120 hours. Ethanol significantly reduced cell survival at 48 hours, with no effect at 72 or 120 hours, both in the presence and absence of L1. At 48 hours, L1 significantly increased cell survival in the presence of ethanol. We conclude that ethanol interferes with processes other than L1-L1 interactions in causing cell death, and that ethanol effects would be more severe in the absence of L1.
Keywords: cell death, ethanol, L1 cell adhesion molecule, cerebellar granule neurons, fetal alcohol syndrome, fetal alcohol spectrum disorder
INTRODUCTION
L1 neural cell adhesion molecule (L1), a highly conserved member of the Ig superfamily of cell adhesion molecules (Lindner et al. 1983), is critical to the development of the central nervous system. L1 is a transmembrane glycoprotein of approximately 200 kDa with six Ig-like domains, five fibronectin type III domains, a single-pass transmembrane region and a cytoplasmic domain (Wong et al. 1995). The genes for both human and mouse L1 are located on the X chromosome (Djabali et al. 1990). L1 is a single-copy gene of 28 exons, two of which are alternatively spliced (Jouet et al. 1994). One form containing both alternatively spliced exons, is located on surfaces of long axons and on growth cones during development, and continues to be expressed in the adult nervous system on unmyelinated axons. The functions of L1 include cell adhesion, neurite outgrowth, axon fascicle formation and neural migration (Stallcup and Beasley 1985; Landmesser et al. 1988). In addition to these functions, L1 decreases cell death both in vitro and in vivo. Surviving cerebellar granule neurons increase by 60% when grown in serum free media in the presence of L1 (Chen et al. 1999). L1 knockout mice have 30% fewer pyramidal and granule neurons throughout the pyramidal and granular layers of the hippocampus (Demyanenko et al. 1999).
There is considerable overlap of the neuropathological abnormalities observed in fetal alcohol syndrome (FAS) with those of patients with L1 mutations (Charness et al. 1994; Ramanathan et al. 1996; Bearer 2001), implicating L1 in the pathogenesis of FAS. Heavy drinking during pregnancy is the cause of FAS, one of the leading known forms of mental retardation (Abel and Sokol 1991). FAS is marked by distinctive craniofacial abnormalities, growth retardation, and central nervous system damage (Jones et al. 1973). Drinking during pregnancy can also result in a spectrum of effects known as fetal alcohol spectrum disorder (FASD), which range from severe cognitive and behavioral impairment without the classic facial dysmorphology to relatively subtle neurobehavioral deficits (Stratton et al. 1996). It is estimated that 1% of all newborns are affected by prenatal ethanol exposure (Sampson et al. 1997; Bearer et al. 1999). Recently, an increase in cell death of the neurons of the central nervous system is proposed as one underlying mechanism of alcohol developmental toxicity (Olney 2002; Bonthius et al. 2003; Bonthius et al. 2006). Increased cell death in the developing cerebellum following ethanol exposure is observed by many authors, both in vivo (Ikonomidou et al. 2000) (Heaton et al. 2003; Xu et al. 2003), and in vitro (Oberdoerster and Rabin 1999; Saito et al. 1999; de la Monte et al. 2001; Bonthius et al. 2003).
Several lines of evidence implicate ethanol’s toxic effects on the central nervous system to inhibition of functions of L1. L1 binds to another molecule of L1 on an opposed surface in homophilic binding (Lemmon et al. 1989) and enables growth cones to extend rapidly along a bundle of pre-existing axons. Ethanol has been shown to inhibit both of these L1 functions; L1 homophilic binding (Charness et al. 1994; Ramanathan et al. 1996) and L1-mediated neurite outgrowth (Bearer et al. 1999; Watanabe et al. 2004).
We hypothesize that the increase in cell death of the developing central nervous system induced by ethanol is mediated in part by ethanol inhibition of L1 enhancement of cell survival. We undertook the present investigation to determine if ethanol induced cell death would be affected by the presence of L1.
METHODS
Preparation of cerebellar granule cells
Rat cerebellar granule cells are obtained from postnatal day 6 Sprague-Dawley rat pups (Zivic-Miller). Cerebellums are dissected in Ca++, Mg++ free phosphate buffered saline and incubated in 1% trypsin-EDTA for 15 minutes on ice, then triturated with fire-polished Pasteur pipettes in the presence of 0.05% DNase. The cells are allowed to settle for 5 minutes, the supernatant is removed and centrifuged at 200g for 5 min. The cell pellet is resuspended in serum free defined media consisting of (K5 recipe), and counted. Viability is assessed with trypan blue. Cells are seeded onto previously prepared 8 well Lab-Tek II slides (Nalge Nunc International) at 1.5×105viable cells/well. These cells have been extensively characterized as > 90% cerebellar granule cells (Hockberger et al. 1987; Beattie and Siegel 1993).
Preparation of L1-Fc
L1-Fc is prepared as previously described (Bearer et al. 1999). Polymerase chain reaction are used to amplify a fragment of clone 17 which contains the extracellular domain of L1 with primers from 2901 to 2918 and 3336 to 3319 to create a whole extracellular domain of human L1 cDNA. The latter primer also has a 3′ splice donor site and EcoRI restriction site. The amplified fragment is digested with BsiwI and EcoRI and ligated into a BsiwI/EcoRI digested pBluescript vector containing the full length L1 cDNA. The vector is sequenced across the entire amplified region and insertion sites. The truncated L1 cDNA containing the whole extracellular domain of L1 is excised from the vector with Hind III and EcoRI and ligated into the pIG vector (Ingenius) which contains the Fc region of human immunoglobin isotype 1. The completed vector is electroporated into Escherichia coli MC1061 cells. Plasmid DNA is purified by alkaline lysis and checked by agarose gel electrophoresis.
The plasmid is stably expressed in NIH 3T3 cells as compared to HEK293 cells as previously described (Haspel et al. 2000). Stably transfected 3T3 cells are propagated using a new technique, the artificial mouse by Cellmax and serum free media. L1-Fc is purified from the harvested media by modification of the method of Sakurai (Sakurai et al. 1998). Briefly, proteins are ammonium sulfate precipitated, then resuspended in 50% DE52 slurry in 17 mM NaH2PO4, pH 6.3 (Buffer A), and dialyzed overnight at 4°C. 20 ml of Buffer A is added to the dialysate, and rocked at 4°C overnight. The DE52 slurry is centrifuged, and washed three times with Buffer A. L1-Fc is eluted with Buffer A containing 0.4 M KCl and the protein concentration of the eluant is determined. Protein A-conjugated sepharose beads at 0.1 ml/mg protein are added, the pH adjusted with 0.1 ml 1 M Hepes, pH 8.0 per ml of eluant and Na Azide added to give a final concentration of 0.01%. The slurry is rocked overnight and stored at 4°C. To determine purity and L1-Fc content, an aliquot of the L1-Fc-bound sepharose beads is washed, and L1-Fc is eluted with (50 mM glycine, pH 2.80) into test tubes containing 1 M Tris, pH 8.0 to neutralize the pH. Protein concentration is determined with the BCA protein assay kit (Pierce). The purity and immunoreactivity of remaining eluted L1-Fc is determined by polyacrylamide electrophoresis and either silver stained for protein, or Western blot using anti-Fc antibodies for immunoreactivity. To show that this new technique using stably transfected NIH-3T3 cells and the artificial mouse yield pure L1-Fc, a representative silver stain/Western blot is shown in Figure 1. For each experiment, the appropriate amount of L1-Fc containing protein A-sepharose slurry is eluted for L1-Fc as described. Following elution, an aliquot of L1-Fc is taken for protein determination, and 1% BSA is added to each fraction.
Figure 1.
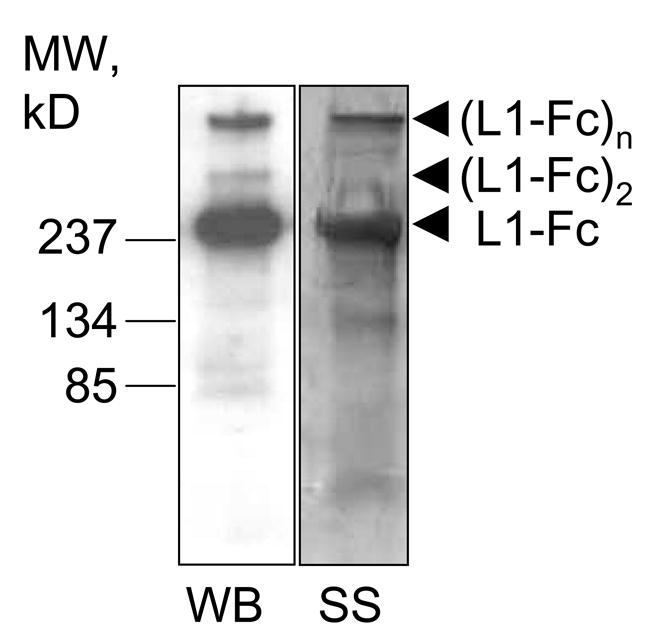
Western blot (WB) using anti-Fc as primary antibody and silver stain (SS) of L1-Fc. L1-Fc under reduced conditions shown here is predominantly a monomer (L1-Fc) with some dimer ((L1-Fc)2) and multimers ((L1-Fc)n) remaining.
Preparation of control slides and slides coated with L1-Fc
8-well Lab-Tek II slides (Nalge Nunc International) are precoated with 250 μl of 0.01% poly L-lysine (Sigma)(PL). After 10 min, the solution is aspirated and the wells washed twice with phosphate buffered saline (PBS). For plates coated with L1-Fc, protein A is added first to bind to the Fc domain, thus orienting the L1 domain upward into the media. Protein A was added to each well as a 1 mg/ml solution for 10 minutes, then aspirated, and the well rinsed twice with PBS. L1-Fc is added to the appropriate wells. After 10 minutes, the solution is aspirated, and the wells are again rinsed with PBS. The substrates are then blocked with 1 ml of DMEM containing 10% fetal calf serum (DMEM complete) for 30 min in a 37°C 10% CO2 incubator. The substrate plates are washed twice with DMEM complete and are incubated at 37°C in 10% CO2 while cerebellar granule cells are being prepared.
Measurement of neurite length
Neurite outgrowth is measured as previously described(Bearer et al. 1999). Briefly, cerebellar granule cells are grown on prepared LabTek slides for 18 h, then washed with ice cold PBS and fixed with 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Neurons are examined using a Zeiss microscope equipped with an Image-1 analysis system. Neurite length is measured as the distance between the center of the cell soma and the tip of its longest neurite. The neurite has to meet the following requirements: it must emerge from an isolated cell (not a clump of cells), it must not contact another cell or neurite, and it must be longer than the diameter of the soma.
Measurement of cell death
At 24 hours after plating the cells, the media is removed and replaced with fresh media with or without 25 mM ethanol. For ethanol treated cells, the slides are placed in a separate incubator with a water bath containing 25 mM ethanol. To ensure continual exposure to 25 mM ethanol, the water bath and the media are replaced daily. Previous work in our lab shows less than 10% loss of ethanol under these conditions (Bearer et al. 1999). The following are the experimental conditions : poly L-lysine alone (Control), poly L-lysine with 25 mM ethanol (EtOH), L1-Fc alone (L1-Fc), and L1-Fc with 20 mM EtOH (L1-Fc + EtOH).
At various time points post addition of ethanol, the slides are removed from the incubator and washed twice with PBS. Live and dead cells are determined by Molecular Probes’ LIVE/DEAD Viability/Cytotoxicity Kit (L-3224) following manufacturer’s directions. The number of live cells is determined by measuring intracellular esterase activity with calcein AM (hydrolyzed by intracellular esterases to produce a green-fluorescent product). Dead cells are counted by staining with ethidium bromide homodimer (red stain, Dead). Results are expressed as % survival (live/(live + dead) at time t divided by live/(live+dead) at time 24 h (the time of addition of ethanol)).
RESULTS
Neurite outgrowth is dependent on the amount of L1-Fc on the culture plate
Neurite length of cerebellar granule cells at 18 hours is dependent on the amount of L1-Fc expressed by NIH-3T3 cells on the plate(Figure 2). Maximal neurite length is obtained at 2 μg L1-Fc added per well. Therefore, we use 2 μg L1-Fc per well for the rest of the experiments as the minimal concentration needed for maximal physiologic effect.
Figure 2.

Cerebellar granule neurons grown on increasing amounts of L1-Fc. Cerebellar granule cells are prepared from postnatal day 6 rat pups, and plated onto multiwell slides precoated with poly L-lysine, protein A and indicated amounts of L1-Fc. Cultures are grown in serum-free media containing 5 mM KCl in 10% CO2 at 37°C. At 18 hours, cells are fixed and neurite length was measured using computer assisted microscopy (mean +/− SE).
CGN in low potassium media die by 6 days in vitro
Cell survival is measured using the Live/Dead stain. To measure the percent survival, living and dead cells are counted, and the percent survival is calculated as described in Methods. Cultures are counted at 24, 48, 72, 120, and 168 hours and cell survival is calculated as the percent of cells alive at time X compared to 24 hours. There are no surviving cells in any of the culture conditions at 168 hours as previously published (Chen et al. 1999). Therefore data is shown for 48, 72 and 120 hours.
L1-Fc increases cell survival at 72 and 120 hours and reduces the rate of cell death at 48 hours
The addition of L1-Fc to the cultures results in an increase in cell survival at 72 and 120 hours (Figure 3). At 72 hours, 41% of cells survives in the absence of L1-Fc (Control) whereas 65% survives in the presence of L1-Fc (L1-Fc), an increase of 61%. At 120 hours, 34% of cells survives in the absence of L1-Fc (Control), whereas 54% survives in the presence of L1-Fc (L1-Fc), an increase of 54%. This increase in survival using human L1-Fc from NIH-3T3 cells is comparable to that previously reported with mouse L1-Fc from CHOK1 cells (Chen et al. 1999) and indicates that the experimental conditions are adequate to measure the survival effect of L1-Fc. While the effect of L1-Fc on cell survival cannot be measured at 48 hours because both control and L1-Fc cultures have 100% survival, the rate of cell death between 48 h and 72 h is reduced by 40% (48%/d in control versus 28%/d in L1-Fc) in the presence of L1-Fc.
Figure 3.

Cerebellar granule cells plated on either poly L-lysine alone (Control), or poly L-lysine, protein A and 2 μg/well of L1-Fc (L1-Fc) are grown in serum-free, low potassium media. At 24 hours post-plating, the media is changed to fresh media. Cells are fixed and stained for living cells at 24, 48, 72 and 120 hrs post-plating. % survival is calculated as the percent of cells alive compared to cells alive at 24 h as described (mean +/− SE).
Ethanol reduces cell survival at 48 hours
The concentration of 25 mM ethanol is chosen for these studies as this concentration gives maximal inhibition of L1-mediated neurite outgrowth (Bearer et al. 1999). As can be seen in Figure 4, 25 mM ethanol causes a significant reduction in cell survival at 48 hours of cells plated on PL alone (Control) (88% survival Control vs 55% survival EtOH; a 37.5% reduction in survival). However, at 72 and 120 hours, ethanol has no effect on cell survival. Thus, CGN in culture become ethanol insensitive with time, either as an adaptive or a maturational process (Bonthius et al. 2003).
Figure 4.

Cerebellar granule cells plated on poly L-lysine alone are grown in serum-free, low potassium media. At 24 hours post-plating, the media is changed to either fresh media (Control) or fresh media containing 25 mM ethanol (Ethanol). Cells are fixed and stained for living cells at 24, 48, 72 and 120 hrs in culture. % survival is calculated as the percent of cells alive compared to cells alive at 24 h as described (mean +/− SE).
Ethanol decreases cell survival in the presence of L1-Fc
We next investigate if ethanol inhibits the survival of cerebellar granule cells in the presence of L1-Fc. Addition of ethanol to cultures containing L1-Fc significantly reduces cell survival, from 94% to 75% at 48 hours, but has no effect at 72 h or 120 h (Figure 5, L1-Fc vs. L1-Fc + EtOH). Thus, it appears that ethanol increases cell death even in the presence of L1-Fc.
Figure 5.

Cerebellar granule cells plated on poly L-lysine, protein A and 2 mcg/well of L1-Fc are grown in serum-free, low potassium media. At 24 hours post-plating, the media is changed to either fresh media (L1-Fc) or fresh media containing 25 mM ethanol (L1-Fc + EtOH). Cells are fixed and stained for living and dead cells at 24, 48, 72 and 120 hrs following ethanol addition. % survival is calculated as the percent of cells alive compared to cells alive at 24 h as described (mean +/− SE).
Ethanol induced cell death is less in the presence of L1-Fc
To determine whether CGN are more vulnerable to ethanol in the presence or absence of L1-Fc, we compare the % survival between cells grown in 25 mM ethanol with or without L1-Fc at 48 hours. Ethanol significantly reduces cell survival both of cells plated on poly-lysine alone (Control) or on L1-Fc (L1-Fc)(Figure 6). This result indicates that ethanol increases cell death in both undifferentiated (control) and differentiated (L1-Fc) cells. In addition, survival of cells exposed to ethanol is greater in the presence of L1-Fc showing that L1-Fc exerts a neuroprotective effect even in the presence of ethanol. This neuroprotective effect may be enhanced differentiation or some other pro-survival process.
Figure 6.

Cerebellar granule cells plated on either poly L-lysine alone (Control), or poly L-lysine, protein A and 2 μg/well of L1-Fc (L1-Fc) were grown in serum-free, low potassium media. At 24 hours post-plating, the media is changed to either fresh media or fresh media containing 25 mM ethanol (+ EtOH). Cells are fixed and stained for living and dead cells at 24 and 48 hrs following the change of media. % survival is calculated as the percent of cells alive compared to cells alive at 24 h as described.
DISCUSSION
This study yielded two important new findings. First, L1 cell adhesion molecule enhances cell survival in the presence of ethanol. Second, ethanol reduces cell survival even in the presence of L1-Fc. In addition, the results confirm previous findings that L1 increases CGN survival in vitro (Chen et al. 1999) and CGN become resistant to the effects of ethanol, either as a result of prolonged exposure and adaptation (Prendergast et al. 2000) or due to distinct developmental stages of the cells (Pantazis et al. 1993; Cartwright and Smith 1995).
There are several plausible mechanistic explanations for these results. One hypothesis is that L1 enhances the maturation process of the CGN. This enhanced maturation increases baseline cell survival and makes them less vulnerable to ethanol at 48 hours than CGN grown on PL alone. We and others have shown that L1 enhances neurite outgrowth which may be a physical correlate of increased maturation and decreased ethanol resistance (Bearer et al. 1999; Schmid et al. 2000).
A second possible explanation is that ethanol inhibits the ability of L1 to promote the maturation of the CGN, and hence increases their susceptibility to ethanol.
L1 is implicated in ethanol induced cell death in vivo. Octanol which antagonizes ethanol inhibition of L1 mediated cell adhesion (Wilkemeyer et al. 2000), also reduces cell death in whole embryo cultures exposed to ethanol (Chen et al. 2001). A neuroprotective peptide, NAPVSIPQ (NAP) both antagonizes ethanol inhibition of L1-mediated cell adhesion (Wilkemeyer et al. 2003), and prevents fetal demise and growth restriction in an animal model of FAS (Spong et al. 2001).
While the finding that L1 increases cell survival in the presence of ethanol is important, a more relevant finding to in vivo models is the impact of ethanol on L1-enhanced cell survival. In this report, low dose ethanol decreases cell survival significantly in the presence of L1. However, the effect of ethanol on cell survival is significantly greater in the absence of L1. Thus, this finding would predict, that if L1 is a central target for ethanol teratogenesis, ethanol will induce greater cell death in the L1 knockout mouse (Fransen et al. 1998) than wildtype. This model also predicts that L1-L1 interaction is not the only target for ethanol.
While this investigation does not characterize the cell death as apoptotic or necrotic, L1-Fc has been shown to decrease apoptosis, increase bcl-2 and reduce c-jun in CGN (Chen et al. 1999). L1 clustering on the cell surface has been shown to stimulate several pro-survival signaling cascades including P13-kinase, ERK1/2 and CREB (Schmid et al. 2000; Spencer et al. 2003; Tang et al. 2006). Inhibition of ERK1/2 phosphorylation with the MEK inhibitors PD98059 and U0126 inhibits L1-mediated neurite outgrowth. We have recently shown that ethanol inhibits L1-mediated phosphorylation of ERK1/2 (Tang et al. 2006) and L1-mediated neurite outgrowth (Bearer et al. 1999). Thus, one possible mechanism of ethanol’s inhibition of neurite outgrowth and increased cell death is by its effect on the ERK1/2 signaling pathway. Future experiments will further delineate the role of this pathway in ethanol induced cell death in the presence of L1.
Acknowledgments
This research was supported by NIAAA grant AA11839. We thank Kevin Buck for his technical assistance with these experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel E, Sokol R. A revised conservative estimate of the incidence of FAS and its economic impact. Alcoholism: Clinical and Experimental Research. 1991;15:514–524. doi: 10.1111/j.1530-0277.1991.tb00553.x. [DOI] [PubMed] [Google Scholar]
- Bearer CF. Mechanisms of brain injury: L1 cell adhesion molecule as a target for ethanol-induced prenatal brain injury. Seminars in Pediatric Neurology. 2001;8:100–107. doi: 10.1053/spen.2001.25227. [DOI] [PubMed] [Google Scholar]
- Bearer CF, Swick AR, O’Riordan MA, Cheng G. Ethanol inhibits L1-mediated neurite outgrowth in postnatal rat cerebellar granule cells. Journal of Biological Chemistry. 1999;274:13264–13270. doi: 10.1074/jbc.274.19.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie C, Siegel R. Developmental cues modulate GABAA receptor subunit mRNA expression in cultured cerebellar granule neurons. Journal of Neuroscience. 1993;13:1784–1792. doi: 10.1523/JNEUROSCI.13-04-01784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, Karacay B, Dai D, Pantazis NJ. FGF-2, NGF and IGF-1, but not BDNF, utilize a nitric oxide pathway to signal neurotrophic and neuroprotective effects against alcohol toxicity in cerebellar granule cell cultures. Developmental Brain Research. 2003;140:15–28. doi: 10.1016/s0165-3806(02)00549-7. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, McKim RA, Koele L, Harb H, Kehrberg AH, Mahoney J, Karacay B, Pantazis NJ. Severe alcohol-induced neuronal deficits in the hippocampus and neocortex of neonatal mice genetically deficient for neuronal nitric oxide synthase (nNOS) Journal of Comparative Neurology. 2006;499:290–305. doi: 10.1002/cne.21095. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Smith SM. Stage-dependent effects of ethanol on cranial neural crest cell development: partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol Clin Exp Res. 1995;19:1454–1462. doi: 10.1111/j.1530-0277.1995.tb01007.x. [DOI] [PubMed] [Google Scholar]
- Charness M, Safran R, Perides G. Ethanol inhibits neural cell-cell adhesion. Journal of Biological Chemistry. 1994;269:9304–9309. [PubMed] [Google Scholar]
- Chen S, Mantei N, Dong L, Schachner M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. Journal of Neurobiology. 1999;38:428–439. doi: 10.1002/(sici)1097-4695(19990215)38:3<428::aid-neu10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Chen SY, Wilkemeyer MF, Sulik KK, Charness ME. Octanol antagonism of ethanol teratogenesis. FASEB Journal. 2001;15:1649–1651. doi: 10.1096/fj.00-0862fje. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Neely TR, Cannon J, Wands JR. 1950–60 Nov;58(12–13) doi: 10.1007/PL00000829. [DOI] [PMC free article] [PubMed] [Google Scholar]; CMLS. Ethanol impairs insulin-stimulated mitochondrial function in cerebellar granule neurons. Cell Mol Life Sci. 2001;58:1950–1960. doi: 10.1007/PL00000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. Journal of Neuroscience. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djabali M, Mattei M, Nguyen C, Roux D, Demengeot J, Denizot F, Moos M, Schachner M, Goridis C, Jordan B. The gene encoding L1, a neural adhesion molecule of the immunoglobulin family, is located on the X chromosome in mice and man. Genomics. 1990;7:587–593. doi: 10.1016/0888-7543(90)90203-7. [DOI] [PubMed] [Google Scholar]
- Fransen E, D’Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SKE, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Human Molecular Genetics. 1998;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- Haspel J, Friedlander DR, Ivgy-May N, Chickramane S, Roonprapunt C, Chen S, Schachner M, Grumet M. Critical and optimal Ig domains for promotion of neurite outgrowth by L1/Ng-CAM. Journal of Neurobiology. 2000;42:287–302. [PubMed] [Google Scholar]
- Heaton MB, Moore DB, Paiva M, Madorsky I, Mayer J, Shaw G. The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol. Alcohol Clin Exp Res. 2003;27:657–669. doi: 10.1097/01.ALC.0000060527.55252.71. [DOI] [PubMed] [Google Scholar]
- Hockberger PE, Tseng HT, Connor JA. Immunocytochemical and electrophysiological differentiation of rat cerebellar granule cells in explant cultures. Journal of Neuroscience. 1987;7:1370–1383. doi: 10.1523/JNEUROSCI.07-05-01370.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Jones K, Smith D, Ulleland C, Streissguth P. Pattern of malformation in offspring of chronic alcoholic women. Lancet. 1973;1:1267–1271. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson J, Metzenberg A, Ionasescu V, Temple K, Kenwrick S. X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nature Genetics. 1994;7:402–407. doi: 10.1038/ng0794-402. [DOI] [PubMed] [Google Scholar]
- Landmesser L, Dahm L, Schultz K, Rutishauser U. Distinct roles for adhesion molecules during innervation of embryonic chick muscle. Developmental Biology. 1988;130:645–670. doi: 10.1016/0012-1606(88)90358-2. [DOI] [PubMed] [Google Scholar]
- Lemmon V, Farr K, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–1603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- Lindner J, Rathjen F, Schachner M. L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature. 1983;305:427–430. doi: 10.1038/305427a0. [DOI] [PubMed] [Google Scholar]
- Oberdoerster J, Rabin RA. Enhanced caspase activity during ethanol-induced apoptosis in rat cerebellar granule cells. European Journal of Pharmacology. 1999;385:273–282. doi: 10.1016/s0014-2999(99)00714-1. [DOI] [PubMed] [Google Scholar]
- Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23:659–668. doi: 10.1016/S0161-813X(01)00092-4. [DOI] [PubMed] [Google Scholar]
- Pantazis N, Dohrman D, Goodlett C, Cook R, West J. Vulnerability of cerebellar granule cells to alcohol-induced cell death diminishes with time in culture. Alcohol Clin Exp Res. 1993;17:1014–1021. doi: 10.1111/j.1530-0277.1993.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Harris BR, Blanchard JAn, Mayer S, Gibson DA, Littleton JM. In vitro effects of ethanol withdrawal and spermidine on viability of hippocampus from male and female rat. Alcohol Clin Exp Res. 2000;24:1855–1861. [PubMed] [Google Scholar]
- Ramanathan R, Wilkemeyer M, Mittal B, Perides G, Charness M. Alcohol inhibits cell-cell adhesion mediated by human L1. Journal of Cell Biology. 1996;133:381–390. doi: 10.1083/jcb.133.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Saito M, Berg MJ, Guidotti A, Marks N. Gangliosides attenuate ethanol-induced apoptosis in rat cerebellar granule neurons. Neurochemistry Research. 1999;24:1107–1115. doi: 10.1023/a:1020704218574. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Roonprapunt C, Grumet M. Purification of Ig-fusion proteins from medium containing Ig. BioTechniques. 1998;25:382–385. doi: 10.2144/98253bm09. [DOI] [PubMed] [Google Scholar]
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JMJ. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. Journal of Neuroscience. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JP, Rice-Evans C, Williams RJ. Modulation of pro-survival Akt/PKB and ERK1/2 signalling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. Journal of Biological Chemistry. 2003 June 24; doi: 10.1074/jbc.M305063200. epub. [DOI] [PubMed] [Google Scholar]
- Spong CY, Abebe DT, Gozes I, Brenneman DE, Hill JM. Prevention of fetal demise and growth restrition in a mouse model of fetal alcohol syndrome. Journal of Pharmacology and Experimental Therapeutics. 2001;297:774–779. [PubMed] [Google Scholar]
- Stallcup W, Beasley L. Involvement of the nerve growth factor-inducible large external glycoprotein (NILE) in neurite fasciculation in primary cultures of rat brain. Proceedings of the National Academy of Science U S A. 1985;82:1276–1280. doi: 10.1073/pnas.82.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton K, Howe C, Battaglia F. Summary. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. Vol. 57. National Academy Press; Washington, D.C.: 1996. [Google Scholar]
- Tang NHM, O’Riordan MA, Farkas C, Buck K, Lemmon V, Bearer CF. Ethanol inhibits L1 cell adhesion molecule activation of mitogen activated protein kinases. Journal of Neurochemistry. 2006;96:1480–1490. doi: 10.1111/j.1471-4159.2006.03649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Ymazaki M, Miyazaki H, Arikawa C, Itoh K, Sasaki T, Maehama T, Frohman MA, Kanaho Y. Phospholipase D2 functions as a downstream signaling molecule of MAP kinase pathway in L1-stimulated neurite outgrowth of cerebellar granule neurons. Journal of Neurochemistry. 2004;89:142–151. doi: 10.1111/j.1471-4159.2004.02308.x. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer MF, Sebastian AB, Smith SA, Charness ME. Antagonists of alcohol inhibition of cell adhesion. Proceedings of the National Academy of Science USA. 2000;97:3690–3695. doi: 10.1073/pnas.050545697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkemeyer MF, Chen SY, Menkari CE, Brenneman DE, Sulik KK, Charness ME. Differential effects of ethanol antagonism and neuroprotection in peptide fragment NAPVSIPQ prevention of ethanol-induced developmental toxicity. Proceedings of the National Academy of Sciences U S A. 2003;100:8543–8548. doi: 10.1073/pnas.1331636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cheng G, Payne H, Lemmon V. The cytoplasmic domain of the cell adhesion molecule L1 is not required for homophilic adhesion. Neuroscience Letters. 1995;200:155–158. doi: 10.1016/0304-3940(95)12100-i. [DOI] [PubMed] [Google Scholar]
- Xu J, Eun YJ, Chang H, Tison G, Jun CG, Wands JR, De La Monte SM. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: Role of PTEN phosphatase. Journal of Biological Chemistry. 2003;278:26929. doi: 10.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]