

Abstract
OBJECTIVE
This study was aimed at ascertaining the efficacy of antisense oligonucleotide-formulated microspheres to prevent type 1 diabetes and to reverse new-onset disease.
RESEARCH DESIGN AND METHODS
Microspheres carrying antisense oligonucleotides to CD40, CD80, and CD86 were delivered into NOD mice. Glycemia was monitored to determine disease prevention and reversal. In recipients that remained and/or became diabetes free, spleen and lymph node T-cells were enriched to determine the prevalence of Foxp3+ putative regulatory T-cells (Treg cells). Splenocytes from diabetes-free microsphere-treated recipients were adoptively cotransferred with splenocytes from diabetic NOD mice into NOD-scid recipients. Live animal in vivo imaging measured the microsphere accumulation pattern. To rule out nonspecific systemic immunosuppression, splenocytes from successfully treated recipients were pulsed with β-cell antigen or ovalbumin or cocultured with allogeneic splenocytes.
RESULTS
The microspheres prevented type 1 diabetes and, most importantly, exhibited a capacity to reverse clinical hyperglycemia, suggesting reversal of new-onset disease. The microspheres augmented Foxp3+ Treg cells and induced hyporesponsiveness to NOD-derived pancreatic β-cell antigen, without compromising global immune responses to alloantigens and nominal antigens. T-cells from successfully treated mice suppressed adoptive transfer of disease by diabetogenic splenocytes into secondary immunodeficient recipients. Finally, microspheres accumulated within the pancreas and the spleen after either intraperitoneal or subcutaneous injection. Dendritic cells from spleen of the microsphere-treated mice exhibit decreased cell surface CD40, CD80, and CD86.
CONCLUSIONS
This novel microsphere formulation represents the first diabetes-suppressive and reversing nucleic acid vaccine that confers an immunoregulatory phenotype to endogenous dendritic cells.
Type 1 diabetes is a disorder of glucose homeostasis caused by a chronic autoimmune inflammation of the pancreatic islets of Langerhans (1). The ultimate outcome is the loss of insulin-producing cells to numbers below a threshold that is critically required to maintain physiological glucoregulation. Before this threshold, however, escalating inflammation around (peri-insulitis) and in the islets of Langerhans (insulitis) first renders the insulin-producing β-cells insensitive to glucose and incapable of appropriate insulin production mainly due to the actions of cytokines like interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin (IL)-1β (2,3).
On clinical confirmation, a large number of type 1 diabetic patients still exhibit evidence of residual β-cell mass that, for a limited time, is functionally responsive to glucose and produces insulin (the so-called “honeymoon period”) (4). In fact, patients with a residual β-cell mass manifest better glycemic control and improved prognosis for diabetic complications including retinopathy and nephropathy. These observations have compelled investigation into agents that can be used at the time of clinical diagnosis to preserve residual β-cell mass primarily by intervening with the ongoing autoimmunity. The use of pharmacological systemic immunosuppressive drugs met with initial success in controlling autoimmunity, however, on withdrawal, the autoimmunity recurred, indicating that systemic agents would need to be administered long-term with their associated adverse effects (5,6). More recently, clinical reversal of hyperglycemia has been achieved by anti-CD3 antibody administration, although some questions linger regarding mechanism of action in the transient immunodepletion and associated cytokine-related side effects (7,8). Finally, despite the initial observations in adults, administration of a peptide derived from HSP60 into new-onset diabetic children failed to exhibit any benefit compared with control subjects (9,10). A need therefore remains for a diabetes-suppressive immunotherapeutic agent that does not engender nonspecific systemic immunosuppression.
It is generally accepted that the initial wave of infiltrating immune cells in type 1 diabetes immunopathogenesis consists mainly of antigen-presenting cells homing into the islets in response to an as-yet-unidentified microenvironmental anomaly (11). Although not completely resolved mechanistically and temporally, this anomaly, in a chronic process, compels migratory antigen-presenting cells, and most prominently dendritic cells, to acquire β-cell-resident antigens derived from apoptotic and/or necrotic β-cells. The migratory dendritic cells then undergo an intrinsic “maturation” program that renders them capable of activating T-cells (including autoreactive, β-cell-specific T-cells) as they accumulate inside the draining pancreatic lymph nodes (12-14).
Dendritic cells, however, also have the capacity to activate and maintain immunoregulatory, “suppressive” cell networks. Apparently, they are regulatory when in a state of functional “immaturity” (15-17). Functional immaturity can be conferred to dendritic cells partly by downregulating costimulatory pathways using systemic and molecule-specific approaches (18). Numerous studies have confirmed that exogenous administration of functionally immature dendritic cells can facilitate allograft survival and can also prevent autoimmune disease and its recurrence (18). We have shown that administration of dendritic cells from NOD mice with low-level expression of CD40, CD80, and CD86 (induced by ex vivo treatment with antisense oligonucleotides targeting the 5′ ends of the respective primary transcripts) into syngeneic recipients can considerably delay and prevent the onset of disease (19,20). This approach is now in a phase I clinical trial in which autologous dendritic cells generated in vitro from leukapheresis products are being administered to established type 1 adult patients to determine safety (M.T. and N.G., personal communication; FDA IND BB-12858). Despite the promise of this study, we have encountered cumbersome logistical requirements to generate these dendritic cell embodiments. We are concurrently pursuing an alternative method to stabilize dendritic cell immaturity directly in vivo.
Many studies confirm that microparticle carriers can direct dendritic cells to the administration site, and once phagocytosed, the contents can shape the dendritic cell functional phenotype (21,22). Yoshida and Babensee (22) showed that biodegradable poly-(lactic-co-glycolic acid) (PLGA) microspheres actually induce dendritic cell maturation by upregulating the CD40, CD80, and CD86 costimulatory molecules. Our studies required a nucleic acid delivery system that would be phagocytosed by dendritic cells without upregulating these costimulatory molecules. We therefore chose to incorporate antisense oligonucleotides directed against CD40, CD80, and CD86 into PROMAXX microsphere delivery system (Baxter Healthcare). The inert PROMAXX microsphere technology has been shown to be safe and effective in human trials (23). More importantly, when administered in vivo, this technology is neutral with respect to dendritic cell maturation state, compared with the known immunostimulatory properties of PLGA-based formulations (22). This neutrality on dendritic cell maturation is a critical criterion in adapting microsphere chemistry for immunosuppressive objectives in which dendritic cells are involved as mediators. Herein, we report a PROMAXX-microsphere-based vaccine in which the antisense oligonucleotides were shown to render dendritic cells diabetes suppressive (19,20) and to prevent and even reverse new-onset autoimmune diabetes.
RESEARCH DESIGN AND METHODS
PROMAXX antisense oligonucleotide microsphere formulation and characterization
Three phosphorothioated (*) antisense oligonucleotides targeted to the CD40, CD80, and CD86 primary transcripts were synthesized by the DNA synthesis facility at University of Pittsburgh. The antisense oligonucleotide sequences are CD40-antisense, 5′ C*A*C* A*G*C* C*G*A* G*G*C* A*A*A G*A*C* A*C*C* A*T*G* C*A*G* G*G*C* A-3′ CD80-antisense, 5′ -G*G*G* A*A*A G*C*C* A*G*G* A*A*T* C*T*A* G*A*G* C*C*A* A*T*G* G*A-3′ and CD86-antisense, 5′-T*G*G* G*T*G* C*T*T* C*C*G* T*A*A* G*T*T* C*T*G* G*A*A* C*A*C* G*T*C-3′. Scrambled antisense oligonucleotides were also formulated into microspheres and used as nonsense controls in several experiments.
An aqueous solution of the oligonucleotide mixture was prepared by combining aliquots of three oligonucleotide solutions to form a 10 mg/ml solution. Ten milligrams per milliliter poly-L-lysine-HBr in diH2O (poly-L-lysine-HBr with an average molecular weight of 50,000 Da; Bachem, King of Prussia, PA) was prepared. Poly-L-lysine-HBr was added to the oligonucleotide solution at a volumetric ratio of 1:1. The mixture was vortexed gently. A 25% polymer solution was prepared containing 12.5% polyvinyl pyrrolidone (PVP; with an average molecular weight of 40,000 Da; Spectrum Chemicals, Gardena, CA) and 12.5% polyethylene glycol (PEG; with an average molecular weight of 3,350 Da; Spectrum Chemicals) in 0.1 mol/l sodium acetate (Spectrum Chemicals) at pH 5.5. The polymer solution was added in a 2:1 volumetric ratio as follows: 750 μl antisense oligonucleotides, 750 μl poly-L-lysine-HBr, 3.0 ml PEG/PVP, to a final total volume of 4.5 ml.
The 4.5-ml preparation was incubated for 30 min at 70°C and then cooled to 23°C. The solution became turbid on cooling, and a precipitate was formed. The suspension was then centrifuged, and the excess PEG/PVP was aspirated. The resulting pellet was washed by resuspending the pellet in deionized water, followed by centrifugation and removal of the supernatant. The washing process was repeated three times. The aqueous suspension was frozen and lyophilized to form a dry powder of microspheres comprising oligonucleotide and poly-L-lysine. Particle size was determined using dynamic light scattering (LB-550 Nanoparticle Size Analyzer; Horiba, Irvine, CA). Microsphere morphology was examined by scanning electron microscopy (S-4800; Hitachi, Pleasanton, CA).
The weight percent load of the antisense oligonucleotide components in the microsphere was determined using gradient reverse-phase high-performance liquid chromatography (HPLC) with UV detection at 260 nm (Waters, Milford, MA). The microspheres were deformulated using competitive displacement of the DNA oligonucleotides from the poly-L-lysine using an excess of poly-L-aspartic acid sodium salt (molecular weight 5,000 -15,000 Da; Sigma) in Tris EDTA buffer (pH 7.8) at 55°C for 24 h. The reverse-phase HPLC was performed on a Waters XTerra MS C18 column (4.6 × 50 mm). Mobile phase A was 8.6 mmol/l tetraethylammonium and 100 mmol/l HFIP, pH 8.2. Mobile phase B was methanol. The oligonucleotides were eluted with a 30-min gradient of 15% B to 18% B at a flow rate of 0.5 ml/min. The column temperature was maintained at 60°C.
Approximately 1.1 mg microspheres was suspended into 1.1 ml 1× PBS (pH 7.4) to measure in vitro release. The microspheres were centrifuged at several time points, and the release medium was aspirated and measured at UV 260 nm. Fresh release medium was added to the microspheres until the next time point. The release studies were conducted at 22 and 37°C.
Experimental animals
Female NOD/LtJ, NOD-scid, C57BL/6, and Balb/c mice were purchased from The Jackson Laboratories (Bar Harbor, ME) and used between the ages of 5-22 weeks. Animals were maintained in a specific pathogen-free environment in the Animal Facility of the Rangos Research Center and used in full compliance with experimentation protocols approved by the Animal Research Care Committee of the Children's Hospital of Pittsburgh.
Reagents, biochemicals, and cell culture
All biochemical and cell culture reagents were purchased from Invitrogen (Carlsbad, CA). Antibodies were purchased from BD Biosciences (San Diego, CA) either as directly conjugated fluorescent embodiments or as affinity-purified preparations in concert with fluorescently labeled isotype-matched secondary products. The specific clones used were CD4 clone, RM4-5; and CD25 clone, 7D4. To ascertain the prevalence of Foxp3+ cells, we used the kit commercially available from eBioscience (San Diego, CA), which includes the FJK-16S Foxp3 clone. Spleen or lymph node-derived cells were enriched into T-cells using column methodology (R&D Systems, Indianapolis, IN). The NIT-1 insulinoma cell line (CRL-2055; American Type Culture Collection) was maintained in Ham's F-12K medium with 2 mmol/l L-glutamine and 10% heat-inactivated fetal bovine serum until 70% confluence. The cells were then gently removed by collagenase digestion and made into lysates by repeated freeze-thaw cycles. The lysate was dispensed into aliquots in sterile PBS.
Antisense microsphere administration
Microspheres formulated with the mixture of the CD40, CD80, and CD86 antisense oligonucleotides (AS-MSPs) or with scrambled control sequences (SCR-MSP) were dispensed into aliquots of 50-μg oligonucleotide formulations in sterile PBS. One hundred microliters AS-MSP, SCR-MSP, or PBS was injected subcutaneously, at a site anatomically proximal to the pancreatic lymph nodes, into 5- to 8-week-old NOD female mice in the initial prevention study. To determine their efficacy in new-onset diabetic NOD mice, we first treated diabetic mice (determined by two consecutive blood glucose measurements of >300 mg/dl) once daily with 2 units 1:1 Humulin R:Humulin N mix in PBS until nonfasting blood glucose stabilized to below 300 mg/dl. Insulin treatment was immediately stopped, and the microsphere formulations were injected subcutaneously, at a site anatomically proximal to the pancreatic lymph nodes, three times a week until the mice were killed for further study.
Adoptive transfer of immune cells into NOD-scid recipients
To determine whether the microsphere administration induced regulatory immune cell populations, we isolated spleen from diabetes-free NOD mice administered the AS-MSP and made single cells. Parallel single-cell preparations were enriched into T-cells. Splenocytes and enriched T-cells were cotransferred in equal numbers (1 × 107) into 7- to 10-week-old female NOD-scid recipients by intravenous injection. Diabetes was monitored on consecutive days, twice weekly.
Fluorescence-activated cell sorter analysis
Fluorescence-activated cell sorter (FACS) analyses were performed on spleen or lymph node cells of successfully treated NOD mice periodically. These were mice that were treated at <10 weeks of age and remained diabetes free or new-onset diabetic mice that were “reversed.” Specifically, cell surface phenotype of the T-cells was assessed by FACS analysis in a FACSVantage SE instrument using FACSDiva and CellQuest modules (BD Biosciences). In addition to the antibodies described above, relevant isotype-specific, fluorescently conjugated antibodies were used throughout as controls for nonspecific cell surface binding. Cells were incubated with mixtures of specific antibodies, and in parallel, with the isotype controls at titers between 1:100 and 1:500 as per the manufacturer's suggestions. They were also labeled with propidium iodide, 7-AAD, and/or the annexin V-staining reagent (Invitrogen-Molecular Probes) and then used directly for FACS analysis. Percentage of positive cells or mean fluorescence intensity was measured for cells gated on forward and side scatter properties representing T-lymphocytes. Dead cells and cell clumps were excluded from the analyses, which were performed using the CellQuest software package.
T-cell proliferation assays in culture
To ascertain proliferation of T-cells from AS-MSP-treated NOD mice to alloantigens or nominal antigens, spleens were isolated from randomly selected mice and enriched into T-cells. These were then cocultured with equal numbers of irradiated splenocytes (1 × 105) from allogeneic mice or syngeneic mice in the presence/absence of 1 μg intact ovalbumin. Proliferation was measured 5 days later using the CyQuant fluorometric reagent (Invitrogen-Molecular Probes) as directed by the manufacturer. The coculture supernatants were retained to measure cytokine levels by Luminex-based fluorescence methods (Beadlyte; Upstate Biotechnology). To measure proliferation in response to the NIT-1 cell line-derived lysate, 1 × 105 splenocytes from treated NOD mice were cocultured with an equal number of syngeneic and age-matched irradiated splenocytes with or without the addition of 5 μg NIT-1 cell lysate. After 5 days, the supernatant was removed for cytokine analysis, and proliferation was measured.
Histology
The degree of insulitis in the pancreata of randomly selected successfully treated NOD mice was ascertained in serial sections of formalinfixed tissue by hematoxylin-eosin treatment. Additional sections were probed for insulin content using a commercially available method (Vector BioLabs) with an insulin antibody (DakoCytomation, Carpinteria, CA).
In vivo live-animal imaging
NOD female mice (8 weeks of age) were anesthetized with isoflurane delivered by the XGI-8 Gas Anesthesia System (Xenogen). Initial isoflurane concentration was set to 2.5% and was reduced to 1.5% once the animal was anesthetized. Mice were imaged before injection on the IVIS Lumina (Xenogen) system using a dsRed filter set. Fluorescent exposure times of 0.8 s were used for in vivo imaging and of 0.1 s for ex vivo imaging. Once imaged for background fluorescence, mice received a 300-μl intraperitoneal injection that contained either a mixture of 150 μl fluorescent microspheres (FluoSpheres 580 nm/605 nm; Molecular Probes-Invitrogen) and 100 μl PBS (control) or a mixture of 150 μl fluorescent microspheres (FluoSpheres 580 nm/605 nm) and 0.5 μg/μl AS-MSP microspheres. Mice were then imaged just before euthanasia (and organ excision) at 3, 24, and 48 h after injection. The spleen and pancreas were then removed and imaged immediately afterward. In a subsequent study, a PROMAXX formulation of a Cy3-labeled oligonucleotide siRNA targeting CD86 was injected subcutaneously at an anatomically distal and proximal site to the pancreatic lymph nodes (scruff and flank, respectively) and the pancreas and spleen were excised 3 h after the injection. Average radiance of organs was calculated using Living Image version 3.0 (Xenogen-Caliper Life Sciences), and mean values were graphed.
Determination of costimulatory molecule levels on dendritic cells loaded with AS-MSP in spleen in vivo
Female NOD mice (8-12 weeks of age) were treated with fluorescent microspheres 0.2 μm in diameter with the excitation emission profiles of 505 nm/515 nm (FluoSpheres 505 nm/515 nm; Molecular Probes-Invitrogen). The mice were injected subcutaneously with a mix of either 100 μl fluorescent microspheres and 100 μl PBS (control) or 100 μl fluorescent microspheres and 100 μl 0.5 μg/μl antisense oligonucleotide mixture (CD40, CD80, and CD86) in a total volume of 300 μl. Spleens where excised at 1, 2, 3, and 6 days after injection. Single splenocytes were treated with Mouse BD Fc block (BD Pharmingen) for 5 min to reduce nonspecific antibody binding. Cells were then treated with anti-mouse CD11c APC and CD40 PE, CD80 PE, or CD86 PE for 30 min (BD Pharmingen), after which the cells were washed and fixed in a 2% paraformaldehyde solution. Cells were then gated by FACS into CD11c and fluorescent microsphere double-positive cells, and within this gate, CD40, CD80, and CD86 levels were further ascertained. Control isotypes were used throughout this experiment.
Statistical analysis
Student's t test, ANOVA, and Kaplan-Meier log-rank analysis where described were facilitated using the Prism version 4 software by GraphPad (San Diego, CA).
RESULTS
Characterization of antisense oligonucleotide-formulated PROMAXX microspheres (AS-MSPs)
Scanning electron micrographs of the PROMAXX microspheres exhibited a relatively smooth surface (Fig. 1A). The particle size of the microspheres determined by light scattering was 0.5-4 μm in size with an average particle size of ~2.5 μm. Thus, the calculated surface area of a single microsphere is ~19.6 μm2. The loading of oligonucleotides determined by reverse-phase HPLC in the microspheres was ~70% weight by weight. Based on these measurements, the calculated number of copies of antisense oligonucleotide per microsphere was ~1.05 × 108.
FIG. 1.

A: Scanning electron micrograph of the AS-MSP. The micrograph exhibits an essentially smooth surface with particle diameters in the 1-to 4-μm size range. Size bar is shown at bottom of the micrograph.B: The cumulative percent release of antisense oligonucleotides from the microspheres. The cumulative percent release was observed to be directly proportional to the square root of time. At 22°C, ~0.8% of the oligonucleotide was released, and at 37°C,1.1% of the incorporated oligonucleotide was released. The release kinetics appears to conform to matrix diffusion release mechanism.C: AS-MSP administration into NOD mice at 5-8 weeks of age delays diabetes onset. Two groups of NOD female mice (5-8 weeks old) were given a single subcutaneous injection of microsphere-formulated antisense oligonucleotides at a site anatomically proximal to the pancreatic lymph nodes. The formulation was injected in the amount of what was considered to contain 50 mg of a 1:1:1 mixture of each antisense oligonucleotide (anti-CD40, anti-CD80, and anti-CD86) or scrambled sequences (SCR-MSP) or PBS vehicle (control). Tail vein blood glucose was measured weekly. Diabetes was confirmed after two consecutive readings of>280 -300 mg/dl. The graph shows cumulative survival of two independently treated cohorts.P<0.0001, Kaplan-Meier analysis.D: Frequent AS-MSP administration into NOD mice at 5-8 weeks of age prevents diabetes onset. NOD female mice (5-8 weeks old) were given eight consecutive single subcutaneous injections, at a site anatomically proximal to the pancreatic lymph nodes (once weekly), of microsphere-formulated antisense oligonucleotides. The formulation was injected in the amount of what was considered to contain 50 μg of a 1:1:1 mixture of each antisense oligonucleotide (anti-CD40, anti-CD80, and anti-CD86; AS-MSP) or scrambled sequences (SCR-MSP) or PBS vehicle (control). Tail vein blood glucose was measured twice weekly. Diabetes was confirmed after two consecutive readings of >280 -300 mg/dl.P < 0.0001, Kaplan-Meier analysis.
The in vitro release kinetics of the antisense oligonucleotides from the microspheres is shown in Fig. 1B at 22 and 37°C. The data show that after a small initial burst effect, the cumulative percent release is proportional to the square root of time. However, after 120 h, <1.1% of the incorporated antisense oligonucleotides have been released at 37°C and <0.8% release was observed at 22°C. This suggests that despite the 70% weight percent loading of oligonucleotide in the microspheres, most of the drug release occurs only after the microsphere has been up-taken by cells in vivo.
Administration of diabetes-suppressive antisense oligonucleotide-formulated PROMAXX microspheres into pre-diabetic NOD female mice delays/prevents diabetes and reverses it in new-onset diabetic animals
Our previous studies demonstrated that NOD-derived dendritic cells treated ex vivo with a mixture of phosphorothioated antisense oligonucleotides targeting the 5′ end of the CD40, CD80, and CD86 primary transcripts prevented diabetes in syngeneic recipients (19,20). To determine whether the microsphere-formulated anti-sense mixture was as efficacious, we administered AS-MSP targeting the 5′ end of the CD40, CD80, and CD86 primary transcripts into NOD female mice between the ages of 5 and 8 weeks old. As controls, we concurrently treated parallel groups with PROMAXX formulation containing SCR-MSP and PBS vehicle. One single injection of AS-MSP, at a site anatomically proximal to the pancreatic lymph nodes, significantly delayed onset of diabetes (Fig. 1C), and eight consecutive injections (as illustrated in Fig. 1D) were very efficacious in preventing the disease altogether. All NOD mice treated with control formulations and untreated NOD mice developed diabetes by 22 weeks of age. We are now determining whether fewer consecutive injections can be as efficacious.
We then proceeded to determine whether the AS-MSP could reverse new-onset hyperglycemia, which could suggest reversal of autoimmunity and mechanisms promoting preservation of residual β-cell mass in NOD mice. NOD mice between 12 and 16 weeks of age developed diabetes confirmed by two consecutive blood glucose readings of >300 mg/dl. These mice were treated with daily insulin injections intraperitoneally until glycemia was stabilized to <300 mg/dl. Decreased blood glucose was generally observed over a 10- to 18-day period of insulin administration. The insulin was then immediately discontinued and AS-MSP, SCR-MSP, or PBS was injected subcutaneously, at a site anatomically proximal to the pancreatic lymph nodes, twice weekly for no more than 25 days after the first microsphere administration (refer to diagram in Fig. 2A). Figure 2B and C demonstrates that AS-MSP administration can reverse new-onset hyperglycemia that is stably maintained even after cessation of the microsphere treatment. This outcome is reproducible, and in Fig. 2D, we show the outcome from three additional study groups. An additional 7 of 15 mice exhibited reversal of hyperglycemia and stable maintenance after cessation of the AS-MSP administration.
FIG. 2.

Multiple rounds of AS-MSP administration into new-onset diabetic NOD female mice with improved blood glucose levels; stable fasting euglycemia even after AS-MSP withdrawal. Diabetes onset was confirmed by two consecutive blood readings of >300 mg/dl. Insulin was administered daily until blood glucose fell below 300 mg/dl. Insulin was immediately stopped and the AS-MSP administrations (at a site anatomically proximal to the pancreatic lymph nodes) began as shown in the timeline (A). In some mice, AS-MSP administration was withdrawn as shown inA. The data show the mean nonfasting blood glucose (B) and the mean fasting blood glucose (C) ± SE. The graphs (D) show reversal of new-onset diabetes in separate groups of diabetic mice after multiple AS-MSP administrations as described in RESEARCH DESIGN AND METHODS.
To ascertain the degree to which pancreatic inflammation was affected in NOD recipients of the AS-MSP that exhibited long-term protection from diabetes (treated with AS-MSP at <8 weeks of age; cohort shown in Fig. 1D), we isolated pancreata from three randomly selected AS-MSP recipients, SCR-MSP controls, and diabetic NOD mice. Whereas SCR-MSP-treated mice and diabetic controls exhibited significant insulitis and indistinguishable islet mass, respectively, we observed normal islet architecture with an absence of insulitis in AS-MSP recipients (Fig. 3). AS-MSP treatment of NOD female mice augments the prevalence of Foxp3+CD25+ putative regulatory T-cells in vivo Our previous data suggested that the ex vivo antisense-treated dendritic cells were suppressive at least in part by augmenting the prevalence of CD4+ CD25+ putative regulatory T-cells (Treg cells) (19,20). We have now specifically addressed the question of whether AS-MSP directly augment Treg cells by enumerating the percentage of Foxp3+ CD25+ T-cells inside a CD4+ gate in the spleen and lymph nodes of pre-diabetic NOD females. AS-MSP alone, and not the control microspheres or PBS, augmented the prevalence of Foxp3+ CD25+ T-cells (Fig. 4).
FIG. 3.

Absence of insulitis and normal insulin content in pancreata of AS-MSP-treated NOD mice.Top: Hematoxylin-eosin staining of representative serial sections from diabetic, SCR-MSP diabetic, and diabetes-free AS-MSP recipients.Bottom: Insulin staining of representative serial sections from diabetic, SCR-MSP diabetic, and diabetes-free AS-MSP. Sections are representative of three randomly selected NOD mice in each group.
FIG. 4.

T-cells from AS-MSP-treated, diabetes-free NOD mice exhibit increased prevalence of Foxp3+ CD25+ putative Treg cells. T-cells were enriched from the spleen or the pooled lymph nodes of AS-MSP-treated diabetes-free mice selected at random from the AS-MSP diabetes-free cohort shown in Figure 1D. All mice treated with PBS or SCR-MSP developed diabetes as shown in Figure 1D. At the time of diabetes confirmation, T-cells were harvested from spleen and pooled lymph nodes. The cells were then stained intracellularly for Foxp3 and with CD25, and the percentage of double-positive cells in a lymphocyte population was determined by FACS analysis. An example of the gating is shown inA. The scatter gram inBshows the percentage of double-positive cells in individual mice at the time of euthanasia in spleen, and inC, the percentage of putative Treg cells in pooled lymph node is shown.P < 0.001 between AS-MSP-treated mice and the two controls in both graphs by Mann-WhitneyUtest.
Given that the AS-MSP treatment yielded augmented numbers of CD4+ CD25+ Foxp3+ putative Treg cells, it was logical to posit that a splenocyte population from AS-MSP-treated mice should be able to confer some degree of suppression to diabetes inducement in immunodeficient mice administered splenocytes from diabetic NOD mice. Toward this objective, a 1:1 mix of splenocytes from AS-MSP-treated diabetes-free NOD mice and new-onset diabetic NOD mice was injected into female NOD-scid mice between 6 and 10 weeks of age. As controls, a 1:1 mix of splenocytes from nondiabetic 10 week-old NOD mice was treated with either SCR-MSP (eight consecutive subcutaneous injections spaced every 3 days apart), and splenocytes from diabetic mice were injected into 6- to 10-week-old NOD-scid females. In addition, another control group of NOD-scid mice was injected with splenocytes from new-onset diabetic NOD mice. Each cell population consisted of 1 × 107 splenocytes freshly isolated. Blood glucose was monitored twice weekly, and two consecutive readings of nonfasting glucose >300 mg/dl was considered as the diabetic threshold. Figure 5 demonstrates that adoptive transfer of diabetes in NOD-scid recipients was almost completely abrogated in the presence of splenocytes derived from AS-MSP recipients that were diabetes free.
FIG. 5.
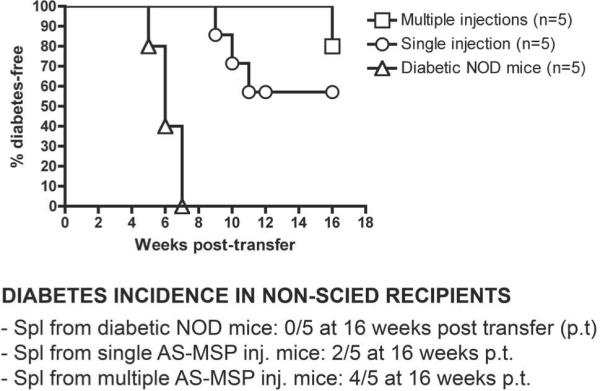
Cotransfer of splenocytes from AS-MSP-treated, diabetes-free NOD mice suppresses the adoptive transfer of diabetes into NOD-scid mice by splenocytes from diabetic NOD donors. Four of five NOD-scid recipients of splenocytes from AS-MSP-treated NOD mice and diabetogenic splenocytes remained diabetes-free at 16 weeks after cell transfer, whereas only two of five and zero of five were diabetes-free after cotransfer of diabetogenic splenocytes and splenocytes from SCR-MSP and PBS-treated NOD mice, respectively. In the graphinset, Spl refers to splenocytes and p.t. refers to the time the mice were killed for further analysis after transfer. Splenocytes from randomly selected diabetes-free AS-MSP-treated mice from the treatment groups shown in Figure 1C and D(single or multiple AS-MSP injections) were cotransferred into female NOD-scid mice of 10 weeks of age along with an equal number of splenocytes (1 × 107) from new-onset diabetic NOD female mice (15-18 weeks old). Diabetes was monitored once weekly in tail vein blood. Levels of >280 mg/dl at two consecutive readings were deemed to indicate diabetes. There were five NOD-scid recipients per cotransfer groups as shown below in the graph.P = 0.0003, between control and AS-MSP splenocyte recipients, Kaplan-Meier analysis.
In vivo-injected AS-MSPs accumulate within the pancreatic lymph nodes and spleen and confer decreased costimulatory molecule surface expression on splenic dendritic cells in vivo. To determine the route of migration of the AS-MSP, which could offer insight into the mechanism of immunoregulation and diabetes suppression, we used in vivo imaging technology to observe the temporal accumulation of a 1:1 mix of commercially available fluorescent microspheres and the AS-MSPs. The sizes of the commercially purchased fluorescent microspheres (FluoSpheres) were considerably smaller than the particle size of the AS-MSP. The AS-MSP were 2.5 μm on average and the FluoSpheres were 0.2 μm on average. Thus the FluoSpheres should not interfere with the uptake of the AS-MSP by dendritic cells. In Fig. 6, we show that as early as 3 h after intraperitoneal injection, the fluorescent microspheres accumulated at anatomical sites where the pancreas and the spleen reside (Fig. 6A). The intensity of the fluorescence did not change over a 72-h monitoring period (data not shown). To confirm the accumulation of the microspheres within the pancreas and spleen, we excised these two organs from the mice that were being monitored live, and in Fig. 6B, we confirm that the fluorescent microspheres accumulated as early as 3 h after injection at distinct foci in pancreas, which we interpret to be the pancreatic lymph nodes, and at distinct foci within the spleen. Although accumulation of fluorescent microspheres was observed in these two organs regardless of whether AS-MSP were included in the mix or not, the actual intensity of the fluorescence was different in organs excised at the times indicated in Fig. 6C and D. The fluorescence was evident in the spleen and pancreata of all mice at all time points studied (from 3 h to 6 days after injection) (Fig. 6; data not shown).
FIG. 6.


In vivo accumulation of AS-MSP. NOD mice received a subcutaneous injection containing a 1:1 mix of 0.2-μm-diameter fluorescent microspheres and sterile PBS or fluorescent microspheres with 50 μg AS-MSP. A: In vivo imaging of mice was performed 3 h after injection. The injection site is clearly visible as well as regions with microsphere accumulation (anatomically located in the area of the pancreas and the spleen). B: The spleen and pancreas were removed from animals at 3, 24, and 48 h after injection and imaged. The excised spleens are in the top two panels of each quadrant (spleen from mice receiving the fluorescent microspheres+PBS on left and spleen from mice receiving the fluorescent microspheres+AS-MSP on the right), and the pancreata are shown in the bottom panels. C and D: The mean radiance per area was quantified for the excised spleen and pancreas and graphically shown below the imaging figures. The graphs represent organs from one mouse and the differences in the magnitudes of radiance are representative of organs from three separate mice. E: Comparison of intraperitoneal versus subcutaneous administration of directly labeled oligonucleotide microspheres on accumulation inside the pancreas. NOD mice were injected with. a PROMAXX formulation of a Cy3-labeled siRNA targeting the CD86 gene via subcutaneous route at a site anatomically distal and proximal to the pancreatic lymph nodes. Three hours after injection, the pancreata and spleens were harvested and imaged as described inRESEARCH DESIGN AND METHODS. CN refers to organs from mice administered PBS vehicle alone, IP refers to animals receiving microsphere-formulated Cy3-conjugated oligonucleotide by intraperitoneal route, and SubQ refers to animals receiving microsphere-formulated Cy3-conjugated oligonucleotide by subcutaneous route. Subcutaneous delivery was made into the scruff of the animal (close to the neck; NECK) and into the flank anatomically proximal to the pancreatic lymph nodes (FLANK). F: AS-MSP administration does not increase costimulatory levels on spleen-derived dendritic cells in vivo. NOD mice were treated with a 1:1 mix of fluorescent microspheres and PBS (CN) or with fluorescent microspheres and 50 μg antisense oligonucleotide mixture (AS) by subcutaneous injection. Spleens were harvested, and single cells were stained with CD40, CD80, CD86, and CD11c antibodies. The cells were analyzed by flow cytometry at days 1, 2, 3, and 6 after injection. Cell populations that stained positive for CD11c and fluorescent microspheres were then gated to measure the presence and levels of CD40, CD80, or CD86. The graphs show the median of the costimulatory molecule levels of spleen cells from individual mice (horizontal bar) as the percentage of costimulatory molecule inside a CD11c+ fluorescent bead+ gate. P < 0.05 by Mann-WhitneyU test between control and AS-MSP mix-treated mice for CD86 on day 1; for CD40, CD80, and CD86 on day 2; for CD40 on day 3
As an additional confirmation that oligonucleotide-formulated microspheres accumulate inside the pancreas and spleen, we injected PROMAXX-formulated Cy3-labeled siRNA to CD86 identically as described above. We also compared the pancreatic accumulation of this Cy3-labeled formulation after intraperitoneal injection, subcutaneous injection at a site distal to the anatomic location of the pancreatic lymph nodes (at the scruff of the mouse), and subcutaneous injection at the site used in the prevention and reversal studies documented herein (flank of the mouse). Figure 6E confirms the pancreatic accumulation of PROMAXX-formulated Cy3-labeled oligonucleotide at 3 h after injection but only when the subcutaneous injection was performed anatomically proximal to the pancreatic lymph nodes.
To confirm that AS-MSP administration conferred a decrease in CD40, CD80, and CD86 in dendritic cells in vivo, we injected NOD mice intraperitoneally with a mixture of the FluoSpheres with PBS as control or with AS-MSP as described in RESEARCH DESIGN AND METHODS. At the times indicated at the top of each graph in Fig. 6F, we excised the spleen and measured CD40, CD80, and CD86 levels in CD11c+ fluorescence+ double-positive single cells. We observed a decreased level of all three costimulatory molecules on CD11c+ cells that had concentrated the mixture of fluorescent microspheres and AS-MSP as early as 1 day after injection with maintenance of these levels compared with CD11c+ cells from control mix-treated NOD mice over a 6-day monitoring period. The only exception was seen at day 3 for CD40.
AS-MSP treatment of NOD pre-diabetic female mice yields T-cells hyporesponsive to NIT-1 cell lysate in vitro without inducing nonspecific immunosuppression
A major concern for eventual translation of diabetes-suppressive therapies into human trials is the antigen specificity and therefore the cell specificity of the approach and whether the treatment confers global and nonspecific suppression. To address these issues, we killed randomly selected diabetes-free mice from the cohorts shown in Fig. 1D, and we then proceeded to ascertain the proliferation of splenic and lymph node T-cells to alloantigens, nominal antigens using intact ovalbumin and also using syngeneic β-cell-derived antigen in the form of cell lysate from the NOD-derived insulinoma cell line NIT-1 (24,25). Although insulin and GAD are viable candidate autoantigens with mechanistic and teleological involvement (26), the nature of the initiating autoantigen remains unclear. Nevertheless, it is reasonable to consider that it should be β-cell resident. Therefore, we used the NIT-1 cell line that derives from an NOD insulinoma as a source of β-cell antigen in cocultures of T-cells from diabetes-free NOD mice treated with the AS-MSP to determine the possibility of antigen-specific hyporesponsiveness. Supplementary Fig. 7, which is detailed in the online appendix (available at http://dx.doi.org/10.2337/db07-0507), shows that T-cell proliferation to nominal and alloantigen is maintained, whereas there is T-cell hypoproliferation in cocultures with NIT-1 cell lysate. Furthermore, ascertaining the cytokine profile in the coculture supernatants, we observed a significant decrease in TNF-α production by T-cells from AS-MSP-treated, diabetes-free NOD mice even in the presence of NIT-1 lysate (Supplementary Fig. 7F; from those diabetes-free NOD mice in Fig. 1D). Although IFN-γ production was slightly decreased in the cocultures of T-cells from the AS-MSP-treated mice, it was not statistically distinguishable from the cocultures with T-cells from PBS-treated mice in the presence of NIT-1 lysate. The assay, finally, could not detect the presence of IL-4, IL-10, or TGF-α in the supernatants.
DISCUSSION
Formulation of bioactive agents into microparticles offers a versatile means of delivering these molecules in vivo especially for the purpose of modulating the immune system (21,27). An important component of the modulatory properties of microspheres is the polymer backbone that often stimulates potent antigen-presenting cell activation, which is beneficial for tumor immunotherapy or antipathogen interventions (28,29). For the purposes of immunosuppression, however, the polymer and the component chemistries should be such that at the very minimum, they should be neutral on antigen-presenting cell state. PROMAXX nucleic acid microspheres offers this versatility (23) as a result of the minimal quantity of formulation excipients. In this regard, we now show its utility as a key component of a diabetes-suppressive vaccine.
The most noteworthy finding in these studies is the capacity of the AS-MSP to reverse new-onset hyperglycemia, which we believe is underlined by preservation of residual β-cell mass. Nevertheless, we cannot yet formally distinguish this from a potential regenerative process of β-cells (division of existing β-cells or differentiation of progenitor/stem cells from ductal epithelium [30 -32]). There is, however, considerable support from previous studies for a sufficiency in β-cell mass that is functionally responsive to glucose at the time of diagnosis of type 1 diabetes (32-36). This mass, if permitted to recover from autoimmune attack by modulating the aggressive and β-cell-specific immune cells, may provide the patient with normoglycemic metabolic control.
Although our data herein are not the first to demonstrate small molecule-based diabetes prevention in the NOD mouse, they are the first to show a well-defined microparticle system that can reverse hyperglycemia in new-onset disease whose mechanism of action is decipherable. In preliminary studies, we have observed that when the formulation was injected into NOD-scid immunodeficient mice reconstituted with splenocytes from NOD mice of various diabetes stages (young, 12-15 weeks old where autoimmunity is already established and where β-cell function is impaired, and from new-onset diabetics), the prevalence of CD4+ CD25+ putative Treg cells increased. We have now confirmed herein in a more direct experiment that CD4+ CD25+ Foxp3+ T-cells numbers increase in female NOD mice administered AS-MSP. These findings may explain the capacity of T-cells from AS-MSP-treated NOD mice free of diabetes to prevent the adoptive cotransfer of the disease to NOD-scid recipients. More importantly, T-cells from diabetes-free AS-MSP exhibited poor proliferation to NIT-1 lysate in vitro while proliferating vigorously in cocultures with allogeneic irradiated splenocytes or when pulsed with ovalbumin.
Many lines of evidence conclude that injected microsphere formulations are rapidly taken up by resident and/or migratory antigen-presenting cells, especially dendritic cells, and accumulate inside lymphoid organs anatomically proximal to the site of injection. Our studies in the past have also confirmed this uptake/trafficking (K.N., J.H., N.G., unpublished observations). Herein, we have provided some of these data, which confirm that subcutaneously injected microspheres accumulate as early as 3 h after injection within the pancreas, very possibly in the lymph nodes, and eventually in the spleen. Although we do not explicitly demonstrate it, we are very confident that the most likely method of microsphere accumulation inside the pancreas is via dendritic cells. Of wide interest is the mechanism by which antigen and therefore cell/tissue specificity is acquired by the microsphere-loaded dendritic cells. A series of elegant studies (12-14,17) point to a process in which migratory dendritic cells potentially acquire the microspheres that are injected physically proximal to a site of ongoing inflammation and, guided by pro-inflammatory signals deriving from the diabetic pancreas, acquire pancreatic β-cell antigens in the form of apoptotic cells at a site of inflammation. Then, these dendritic cells will exit the inflamed tissue and accumulate within the regional lymphoid organs where they can engage not only effector T-cells but Treg cells as well (12-14,17,33). It has been shown that dendritic cells that acquire apoptotic cells enter into a state of functional immaturity, which may result in tolerance to the acquired antigens (17,34). We hypothesize that a similar, if not identical, process is occurring immediately after AS-MSP administration in NOD mice. The inflammation inside the pancreas with associated β-cell apoptosis will drive AS-MSP-loaded dendritic cells to acquire β-cell antigen, and immediately thereafter, their accumulation inside the pancreatic lymph nodes will facilitate their interaction with Treg cells which may, in themselves or in concert with AS-MSP-stabilized dendritic cell or other endogenous dendritic cell subsets, induce β-cell-specific immune hyporesponsiveness or functional tolerance to β-cell-restricted antigens (35-37). We show that microsphereloaded dendritic cells in the spleen express decreased levels of CD40, CD80, and CD86 at their surface, and we anticipate that this is true for dendritic cells in the pancreatic lymph nodes. Although we do not currently have an explanation for the increase in CD86 levels in fluorescent dendritic cells from the spleen of mice treated with the AS-MSP mixture at day 6 after administration, this could reflect the eventual degradation of the antisense nucleic acid in the dendritic cells by day 6, the disappearance of the migratory dendritic cells and acquisition of the fluorescent particles by endogenous, secondary dendritic cells in a cross-priming mechanism, or both.
What has become apparent in these preliminary accumulation studies is the differential accumulation of microspheres inside the pancreas after subcutaneous injection depending on the proximity of the injection site to the pancreas. When administered subcutaneously at a site that flanks the site of the pancreatic lymph nodes, the microspheres accumulate within 3 h. In contrast, there is no detectable accumulation when the microspheres are administered subcutaneously at a site that does not drain to the pancreatic lymphatics. This observation is relevant for two reasons. First, it offers insight into which administration route may be more clinically useful. Second, it suggests that immunoregulatory interventions, at least those that involve dendritic cells as intermediates, are mechanistically active only when the pancreatic lymph nodes are involved (i.e., the nexus of autoimmune cells and regulatory cells that exhibit antigen specificity). We are, consequently, very interested in identifying the precise mechanism involved in costimulatory protein surface density changes at the dendritic cells in spleen and in pancreatic lymph nodes in response to the AS-MSP treatment. The scarcity of dendritic cells from the pancreatic lymph nodes at the times shown in Fig. 6 did not permit us to pursue an effective enrichment of these cells, but it is one of our objectives in future studies in which the precise mechanisms and temporal sequence of cell migration and interaction with other cells in the pancreatic lymph nodes and/or at the islets of Langerhans remain to be established.
Many investigators support immunotherapy approaches for autoimmunity where putative autoantigen supply provides the antigen, and hence the tissue, specificity (38-41). It is worth noting, however, that suppression of autoimmune disease in animal models need not require antigen supply (37). Mechanistically, such interventions may involve bystander tolerance, linked suppression, or similar phenomena (37,42,43). Although we did not supply autoantigen to the AS-MSP regimen, we did inject diabetic NOD mice with insulin to normalize the glycemia. In one possible mechanism, the exogenous insulin supply (as a well-characterized putative autoantigen), before AS-MSP administration may be acting similar to or in an identical manner as earlier insulin-based tolerance strategies (1,38-41). However, we think a more likely mechanism is that the AS-MSPs stabilized subsets of endogenous dendritic cells toward diabetes-suppressive states by downregulating cell surface CD40, CD80, and CD86. We are currently actively investigating the possible mechanism experimentally. It must be stressed, concurrently, that insulin administration alone cannot account for the AS-MSP effects in its physiological glucoregulatory capacity. No mouse in groups treated with insulin alone was capable of maintaining normoglycemia for more than 1 week after withdrawal of the insulin (Fig. 5). This argues against the possibility that in the AS-MSP-treated animals, new-onset diabetes reversal was due to insulin-induced β-cell rest phenomena (44,45). Taken together, these findings may be readily translatable clinically with an immediate aim of preserving residual β-cell mass in newly onset or preclinical human autoimmune diabetes.
Supplementary Material
ACKNOWLEDGMENTS
M.T. has received National Institutes of Health Grant DK-063499. N.G. has received National Institutes of Health Grant DK-063499, Juvenile Diabetes Research Foundation Grant 17-2007-1066, and a preclinical discovery research contract from Epic Therapeutics, a wholly owned subsidiary of Baxter Health Care.
William Fowle at the Electronic Materials Research Institute at Northeastern University (Boston, MA) conducted the scanning electron microscopy studies.
Glossary
- AS-MSP
antisense microsphere
- FACS
fluorescence-activated cell sorter
- HPLC
high-performance liquid chromatography
- IFN-γ
interferon-γ
- IL
interleukin
- PEG
polyethylene glycol
- PLGA
poly-(lactic-co-glycolic acid)
- PVP
polyvinyl pyrrolidone
- SCR-MSP
scrambled control sequences microsphere
- TNF-α
tumor necrosis factor-α
- Treg cell
regulatory T-cell
Footnotes
Additional information for this article can be found in an online appendix at http://dx.doi.org/10.2337/db07-0507.
REFERENCES
- 1.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 2.Thomas HE, Darwiche R, Corbett JA, Kay TW. Interleukin-1 plus gamma-interferon-induced pancreatic β-cell dysfunction is mediated by β-cell nitric oxide production. Diabetes. 2002;51:311–316. doi: 10.2337/diabetes.51.2.311. [DOI] [PubMed] [Google Scholar]
- 3.Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest. 1998;102:516–526. doi: 10.1172/JCI844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdul-Rasoul M, Habib H, Al-Khouly M. “The honeymoon phase ” in children with type 1 diabetes mellitus: frequency, duration, and influential factors. Pediatr Diabetes. 2006;7:101–107. doi: 10.1111/j.1399-543X.2006.00155.x. [DOI] [PubMed] [Google Scholar]
- 5.Bougneres PF, Landais P, Boisson C, Carel JC, Frament N, Boitard C, Chaussain JL, Bach JF. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes. 1990;39:1264–1272. doi: 10.2337/diab.39.10.1264. [DOI] [PubMed] [Google Scholar]
- 6.Lipton R, LaPorte RE, Becker DJ, Dorman JS, Orchard TJ, Atchison J, Drash AL. Cyclosporin therapy for prevention and cure of IDDM: epidemiological perspective of benefits and risks. Diabetes Care. 1990;13:776–784. doi: 10.2337/diacare.13.7.776. [DOI] [PubMed] [Google Scholar]
- 7.Chatenoud L. CD3-specific antibodies restore self-tolerance: mechanisms and clinical applications. Curr Opin Immunol. 2005;17:632–637. doi: 10.1016/j.coi.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elias D, Avron A, Tamir M, Raz I. DiaPep277 preserves endogenous insulin production by immunomodulation in type 1 diabetes. Ann N Y Acad Sci. 2006;1079:340–344. doi: 10.1196/annals.1375.052. [DOI] [PubMed] [Google Scholar]
- 10.Lazar L, Ofan R, Weintrob N, Avron A, Tamir M, Elias D, Phillip M, Josefsberg Z. Heat-shock protein peptide DiaPep277 treatment in children with newly diagnosed type 1 diabetes: a randomised, double-blind phase II study. Diabete Metab Res Rev. 2007;23:286–291. doi: 10.1002/dmrr.711. [DOI] [PubMed] [Google Scholar]
- 11.Babaya N, Nakayama M, Eisenbarth GS. The stages of type 1A diabetes. Ann N Y Acad Sci. 2005;1051:194–204. doi: 10.1196/annals.1361.061. [DOI] [PubMed] [Google Scholar]
- 12.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Carbone FR, Belz GT, Heath WR. Transfer of antigen between migrating and lymph node-resident DCs in peripheral T-cell tolerance and immunity. Trends Immunol. 2004;25:655–658. doi: 10.1016/j.it.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. 2002;196:1079–1090. doi: 10.1084/jem.20020991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 16.Steinman RM. The control of immunity and tolerance by dendritic cell. Pathol Biol (Paris) 2003;51:59–60. doi: 10.1016/s0369-8114(03)00096-8. [DOI] [PubMed] [Google Scholar]
- 17.Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, Iyoda T, Ravetch J, Dhodapkar M, Inaba K, Nussenzweig M. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. 2003;987:15–25. doi: 10.1111/j.1749-6632.2003.tb06029.x. [DOI] [PubMed] [Google Scholar]
- 18.McCurry KR, Colvin BL, Zahorchak AF, Thomson AW. Regulatory dendritic cell therapy in organ transplantation. Transpl Int. 2006;19:525–538. doi: 10.1111/j.1432-2277.2006.00306.x. [DOI] [PubMed] [Google Scholar]
- 19.Harnaha J, Machen J, Wright M, Lakomy R, Styche A, Trucco M, Makaroun S, Giannoukakis N. Interleukin-7 is a survival factor for CD4+ CD25+ T-cells and is expressed by diabetes-suppressive dendritic cells. Diabetes. 2006;55:158–170. [PubMed] [Google Scholar]
- 20.Machen J, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol. 2004;173:4331–4341. doi: 10.4049/jimmunol.173.7.4331. [DOI] [PubMed] [Google Scholar]
- 21.Waeckerle-Men Y, Allmen EU, Gander B, Scandella E, Schlosser E, Schmidtke G, Merkle HP, Groettrup M. Encapsulation of proteins and peptides into biodegradable poly(D,L-lactide-co-glycolide) microspheres prolongs and enhances antigen presentation by human dendritic cells. Vaccine. 2006;24:1847–1857. doi: 10.1016/j.vaccine.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida M, Babensee JE. Molecular aspects of microparticle phagocytosis by dendritic cells. J Biomater Sci Polym Ed. 2006;17:893–907. doi: 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- 23.Mandal TK. Inhaled insulin for diabetes mellitus. Am J Health Syst Pharm. 2005;62:1359–1364. doi: 10.2146/ajhp040249. [DOI] [PubMed] [Google Scholar]
- 24.Reid BD, Qin HY, Prange S, Lee-chan E, Yu Q, Elliott JF, Singh B. Modulation and detection of IDDM by membrane associated antigens from the islet beta cell line NIT-1. J Autoimmun. 1997;10:27–34. doi: 10.1006/jaut.1996.9999. [DOI] [PubMed] [Google Scholar]
- 25.Hamaguchi K, Gaskins HR, Leiter EH. NIT-1, a pancreatic β-cell line established from a transgenic NOD/Lt mouse. Diabetes. 1991;40:842–849. doi: 10.2337/diab.40.7.842. [DOI] [PubMed] [Google Scholar]
- 26.Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol. 2004;552:306–310. [PubMed] [Google Scholar]
- 27.Bramwell VW, Perrie Y. Particulate delivery systems for vaccines. Crit Rev Ther Drug Carrier Syst. 2005;22:151–214. doi: 10.1615/critrevtherdrugcarriersyst.v22.i2.20. [DOI] [PubMed] [Google Scholar]
- 28.Keegan ME, Saltzman WM. Surface-modified biodegradable microspheres for DNA vaccine delivery. Methods Mol Med. 2006;127:107–113. doi: 10.1385/1-59745-168-1:107. [DOI] [PubMed] [Google Scholar]
- 29.Davis SS. The use of soluble polymers and polymer microparticles to provide improved vaccine responses after parenteral and mucosal delivery. Vaccine. 2006;24(Suppl 2):S2-7–S2-10. doi: 10.1016/j.vaccine.2005.01.102. [DOI] [PubMed] [Google Scholar]
- 30.Zorina TD, Subbotin VM, Bertera S, Alexander AM, Haluszczak C, Gambrell B, Bottino R, Styche AJ, Trucco M. Recovery of the endogenous beta cell function in the NOD model of autoimmune diabetes. Stem Cells. 2003;21:377–388. doi: 10.1634/stemcells.21-4-377. [DOI] [PubMed] [Google Scholar]
- 31.Trucco M. Is facilitating pancreatic beta cell regeneration a valid option for clinical therapy? Cell Transplant. 2006;15(Suppl 1):S75–S84. doi: 10.3727/000000006783982322. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, D'Hoker J, Stange G, Bonne S, DeLeu N, Xiao X, VanDeCasteele M, Mellitzer G, Ling Z, Pipeleers D, Bowens L, Scharfmann R, Gradwohl G, Heimberg H. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Belz GT, Heath WR, Carbone FR. The role of dendritic cell subsets in selection between tolerance and immunity. Immunol Cell Biol. 2002;80:463–468. doi: 10.1046/j.1440-1711.2002.01116.x. [DOI] [PubMed] [Google Scholar]
- 34.Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev. 2006;212:314–329. doi: 10.1111/j.0105-2896.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 36.Yamazaki S, Patel M, Harper A, Bonito A, Fukuyama H, Pack M, Tarbell KV, Talmor M, Ravetch JV, Inaba K, Steinman RM. Effective expansion of alloantigen-specific Foxp3+ CD25+ CD4+ regulatory T cells by dendritic cells during the mixed leukocyte reaction. Proc Natl Acad Sci U S A. 2006;103:2758–2763. doi: 10.1073/pnas.0510606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol. 2006;18:93–102. doi: 10.1016/j.smim.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, vonHerrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alleva DG, Maki RA, Putnam AL, Robinson JM, Kipnes MS, Dandona P, Marks JB, Simmons DL, Greenbaum CJ, Jimenez RG, Conlon PJ, Gottlieb PA. Immunomodulation in type 1 diabetes by NBI-6024, an altered peptide ligand of the insulin B epitope. Scand J Immunol. 2006;63:59–69. doi: 10.1111/j.1365-3083.2005.01705.x. [DOI] [PubMed] [Google Scholar]
- 40.Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L, Jackson DC, Harrison LC. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest. 2003;111:1365–1371. doi: 10.1172/JCI17166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homann D, Dyrberg T, Petersen J, Oldstone MB, von Herrath MG. Insulin in oral immune “tolerance”: a one-amino acid change in the B chain makes the difference. J Immunol. 1999;163:1833–1838. [PubMed] [Google Scholar]
- 42.Derks R, Jankowska-Gan E, Burlingham WJ. Dendritic cell type determines the mode of linked suppression (TGF-beta vs. IDO) by adaptive T regulatory cells. Transplantation. 2006;82:183. [Google Scholar]
- 43.Frasca L, Carmichael P, Lechler R, Lombardi G. Anergic T cells effect linked suppression. Eur J Immunol. 1997;27:3191–3197. doi: 10.1002/eji.1830271216. [DOI] [PubMed] [Google Scholar]
- 44.Ortqvist E, Bjork E, Wallensteen M, Ludvigsson J, Aman J, Johansson C, Forsander G, Lindgren F, Berglund L, Bengtsson M, Berne C, Persson B, Karlsson FA. Temporary preservation of β-cell function by diazoxide treatment in childhood type 1 diabetes. Diabetes Care. 2004;27:2191–2197. doi: 10.2337/diacare.27.9.2191. [DOI] [PubMed] [Google Scholar]
- 45.Palmer JP. Beta cell rest and recovery: does it bring patients with latent autoimmune diabetes in adults to euglycemia? Ann N Y Acad Sci. 2002;958:89–98. doi: 10.1111/j.1749-6632.2002.tb02950.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.