

Abstract
In critically ill patients various conditions may lead to the activation of poly(ADP-ribose) polymerase (PARP). By promoting cellular energetic dysfunction, and by enhancing pro-inflammatory gene expression, PARP activation significantly contributes to the pathogenesis of shock. PARP activation is usually triggered by DNA strand breakage, which is typically the result of the overproduction of various reactive oxidant species. One of the pathophysiological conditions associated with PARP activation is hyperglycemia, where the reactive species are produced from the mitochondria and other cellular sources. In the present study we tested whether endotoxin-induced PARP activation and pro-inflammatory mediator production can be modified by insulin therapy. Rats subjected to bacterial lipopolysaccharide (LPS) with or without insulin co-treatment were studied. LPS-induced PARP activation in circulating lymphocytes was measured by flow cytometry, tumor necrosis factor alpha (TNF-α) production was measured by ELISA. The direct effect of insulin on the PARP activity of mononuclear leukocytes and human umbilical vein endothelial cells (HUVEC) in elevated glucose conditions was tested in vitro. LPS-induced significant hyperglycemic response, activated PARP in circulating lymphocytes and induced TNF-α production. Insulin treatment prevented LPS-induced hyperglycemic response, blocked PARP activation and blunted LPS-induced TNF-α response. Insulin treatment caused a slight reduction in the PARP activity of mononuclear cells and HUVECs in vitro. We demonstrate that insulin treatment blocks LPS-induced PARP activation in vivo. We propose that this effect is mainly indirect, and occur due to the prevention of stress induced hyperglycemia, with a direct cellular effect of insulin playing a potential supplemental role. The current findings may have significant implications in the context of the emerging concept of tight glycemic control and insulin treatment for critically ill patients.
Keywords: sepsis, diabetes, glycemic control, PARP, oxidative stress, inflammation, TNF-α
Introduction
Oxygen- and nitrogen-derived reactive oxidants and free radicals play a significant role in the pathogenesis of shock. These species can induce DNA single-strand breakage, which subsequently triggers the activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) (Jagtap and Szabo, 2005). PARP uses NAD+ as substrate and its overactivation may result in rapid depletion of intracellular NAD+ and ATP, leading to cell dysfunction and necrotic cell death. PARP is also involved in the upregulation of multiple pro-inflammatory cytokines and chemokines, including tumor necrosis factor alpha (TNF-α) (Jagtap and Szabo, 2005).
Pharmacological inhibition of PARP has been shown to provide benefit in rodent and large animal models of shock, in terms of improvement in hemodynamic parameters, vascular function, intestinal function, pulmonary function as well as survival rate (Jagtap et al., 2002; Goldfarb et al., 2002; Soriano et al., 2002). PARP activation has also been demonstrated in tissue samples from patients dying from septic shock, and the degree of PARP activation in the hearts of these patients showed a significant correlation with the degree of cardiac dysfunction (Soriano et al. 2006).
Many conditions of critically ill patients are associated with hyperglycemia. Insulin therapy, in order to maintain tight glycemic control has been demonstrated to provide significant benefit: in a prospective, randomized, controlled study involving adults admitted to surgical intensive care units and receiving mechanical ventilation, intensive insulin therapy substantially reduced mortality and morbidity. Intensive insulin treatment reduced the number of deaths from multiple organ failure with sepsis. Markers of inflammation were found to be less frequently abnormal in the intensive insulin treatment group than in the conventional treatment group (Ellger et al., 2006; Langley and Adams, 2007). Although a number of outstanding issues remain to be addressed with this concept, and the degree of benefit that normalization of circulating glucose offers may be different in different groups of patients (Preiser and Devos, 2007; Vanhorebeek et al., 2007; Nazer et al., 2007), intensive glucose control emerges as a potential therapeutic tool for critically ill patients.
The aim of the current study was to test whether there is a link between hyperglycemia and PARP activation in endotoxin shock. We have tested, therefore, in a rat model of endotoxemia whether insulin therapy affects PARP activation and TNF-α production. In addition, the potential direct effect of insulin on the PARP activation of mononuclear cells and human umbilical vein endothelial cells (HUVEC) was also tested in vitro.
Materials and methods
All investigations conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1985), and were approved by the local ethics committee.
Animal model
Age-matched male Wistar rats (n=16, weighing 350–400g) were treated with a single dose of 15mg/kg Escherichia coli endotoxin lipopolysaccharide (LPS) (Sigma/Aldrich), followed by treatment with vehicle (n=8) or 5 units of rapid insulin subcutaneously (Novorapid, Novo Nordisk) (n=8). The control group (n=8) remained untreated. Blood glucose levels of the animals were periodically monitored (Accu-Check). Sample collection started immediately before LPS administration and was repeated every ten minutes in the first hour, every 30 minutes in the second hour and at 3 hours. At 3 hours, animals were anesthetized with N2O-Narcothan mixture (Sigma/Aldrich). Peripheral blood leukocytes were isolated for the measurement of PARP activation, and serum was collected for TNF-α measurement.
PARP activation in circulating cells was measured by a flow cytometric method (Mabley et al., 2005) based on the immunohistochemical detection of the product of the enzyme, poly(ADP-ribose) (PAR). Circulating mononuclear leukocytes were isolated from whole blood using Histopaque-1083 according to the users’ manual (Sigma/Aldrich). After the fixation and permeabilization of the cells with Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (Becton-Dickinson), monoclonal mouse anti-PAR antibody was used as primary antibody to stain intracellular PAR (Tulip Biolabs, West Point, PA). All procedures were performed in Cytoperm solution after the fixation of the cells. We used purified mouse IgG3κ isotype control (anti-KLH) antibody (Beckton Dickinson). As secondary antibody FITC-conjugated anti-mouse immunoglobulin specific polyclonal antibody was used (multiple adsorption) (Becton Dickinson). Flow cytometric measurements were performed on single cell suspension of rat leukocytes using FACSCalibur (Becton Dickinson, San Jose, CA). Region 1 (R1) was defined to contain cells having typical Forward Scatter and Side Scatter properties of lymphocytes. Isotype control stained cells served as negative control for each sample. Fluorescence data were collected using logarithmic amplification until we reached 10,000 counts of R1 cells. On the PAR histograms the gate was R1.
Serum TNF-α levels were measured using sandwich ELISA method (R&D Systems).
In vitro experiments
Whole blood collected from male Wistar rats (300–350g) was diluted 1:10 in RPMI medium containing 30mM glucose with or without insulin (10nM, Sigma/Aldrich) and incubated for 24hrs in 37°C (5% CO2). Mononuclear cells were isolated using Histopaque-1083.
Human umbilical endothelial cells (HUVEC, Cambrex, East Rutherford, NJ) were cultured in Endothelial Basal Medium containing 2% FBS, recombinant human EGF 0.1%, bovine brain extract 0.4%, gentamycin 0.1%, hydrocortisone 0.1% and 30mM glucose with or without insulin (10 nM) for 24 hrs.
HUVECs and mononuclear cells were lysated in hot lysis buffer (1% TRIS, 1% SDS). The lysates were centrifuged and the supernatant were used for western blot analysis. 10μg protein was loaded in each well of 4–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA). After electrophoresis proteins were transferred to nitrocellulose membrane (Invitrogen). Unspecific labeling was prevented by incubating the membranes in 10% milk (1hr, room temperature). Anti-PAR monoclonal mouse antibody was used as primary antibody (1:8000, 3hrs, room temperature, Calbiochem), secondary antibody was horseradish peroxidase linked anti-mouse IgG antibody (1:5000, 1hr, room temperature, Cell Signaling, Danvers, MA). The blots were then developed using chemiluminescence method (Amersham, Arlington Heights, IL).
For statistical analysis, analysis of variance was used. For the comparison of columns Tukey’s post hoc test was applied.
Results
LPS treatment caused significant hyperglycemia for the first 120min. The highest blood glucose level was measured 60min after LPS injection. In the second hour the blood glucose levels of the animals gradually decreased and returned to the normal even hypoglycemic range by 180min after LPS treatment (Fig. 1). Insulin treatment prevented the LPS-induced hyperglycemia (Fig. 1).
Fig. 1.
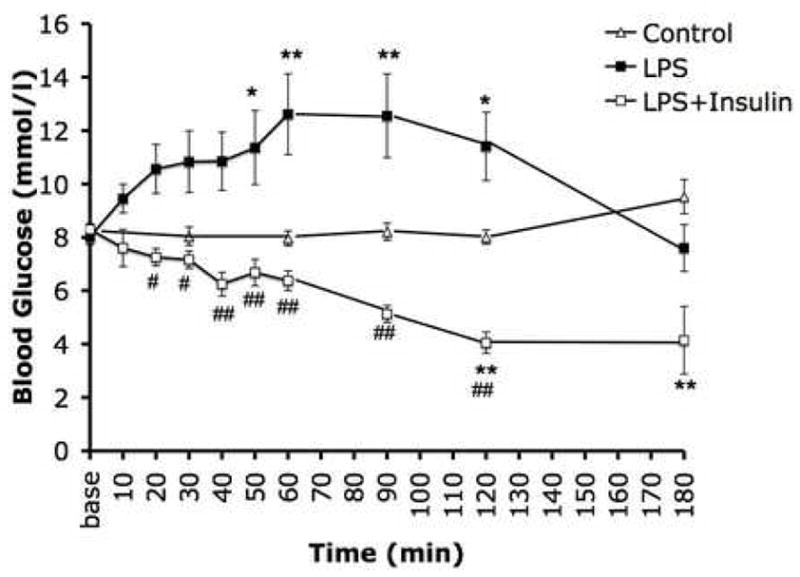
Blood glucose profiles of rats injected with E. coli LPS (15 mg/kg i.v.) with or without insulin treatment (5 U s.c.). Endotoxin induced a significant degree of hyperglycemia, which was normalized by insulin treatment. In the control group, a slight decline of glucose levels occurred over time, perhaps due to a spontaneous restitution of a slight degree of hyperglycemia associated with the surgical procedure itself. Values are mean ± SEM *: p≤0.05 vs. Control, **: p≤0.01 vs. Control, #: p≤0.05 Insulin+LPS vs. LPS alone, ##: p≤0.01 Insulin+LPS vs. LPS alone. N=8 animal per experimental groups.
The PAR content of circulating lymphocytes – representing their PARP activity – was significantly increased in response to LPS. This activation of PARP was prevented by the administration of insulin (Fig. 2a, 2b).
Fig. 2.
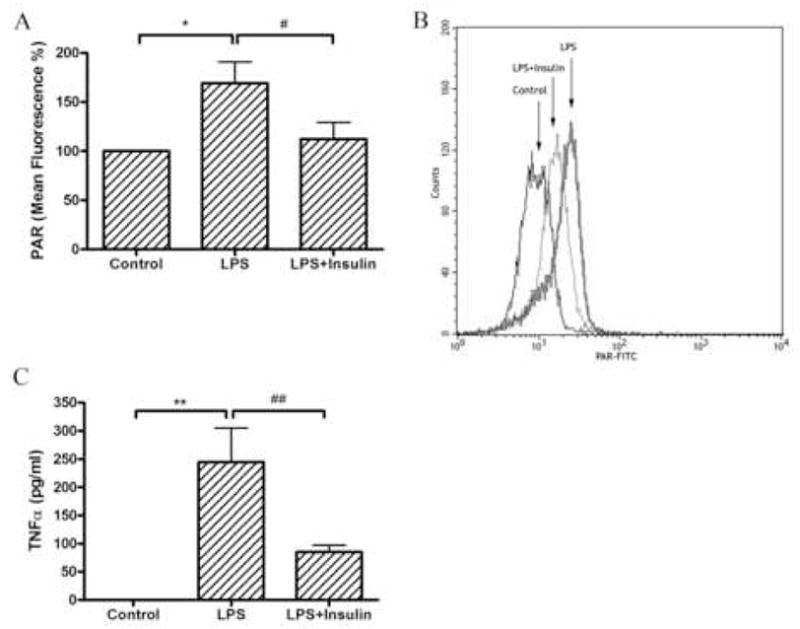
Panel A.Differences in PAR content of circulating lymphocytes in response to lipopolysaccharide and insulin treatment. Mean fluorescence intensity of R1 cells (lymphocytes) stained with anti-PAR antibody. Endotoxin caused a significant increase of the PAR content of these cells, which was attenuated by insulin treatment. Values are mean ± SEM *: p≤0.05 vs. Control, #: p≤0.05 Insulin+LPS vs. LPS alone. Panel B. Representative flow cytometric measurement. PAR-FITC shows the amount of fluorescence labeling in the cells. Panel C. Serum TNF-α levels at 180 min after LPS injection with or without insulin treatment. Endotoxin caused a marked increase in serum TNF-α levels, which was abolished by insulin treatment. Values are mean ± SEM. **: p≤0.01 vs. Control, ##: p≤0.01 Insulin+LPS vs. LPS alone. N=4–6 determinations per experimental group.
Serum TNF-α levels also showed a significant increase, as measured at 3h after LPS injection, which was prevented by insulin treatment (Fig. 2c). It is noteworthy that while the inhibition of PARP activation by insulin in the circulating cells was a complete response, the inhibition of TNF-α production was a partial one. These findings may suggest that the promotion of TNF-α release into the serum during endotoxemia is triggered by multiple factors; PARP activation being one of these stimuli.
In vitro experiments using Western blot analysis showed that insulin slightly, but significantly decreases poly(ADP-ribosylation) of proteins in HUVECs and mononuclear cells cultured in medium containing 30mM glucose, indicating that insulin has a slight, but significant direct effect on PARP activation, independent from its role in altering extracellular glucose concentration in vivo (Fig. 3).
Fig. 3.
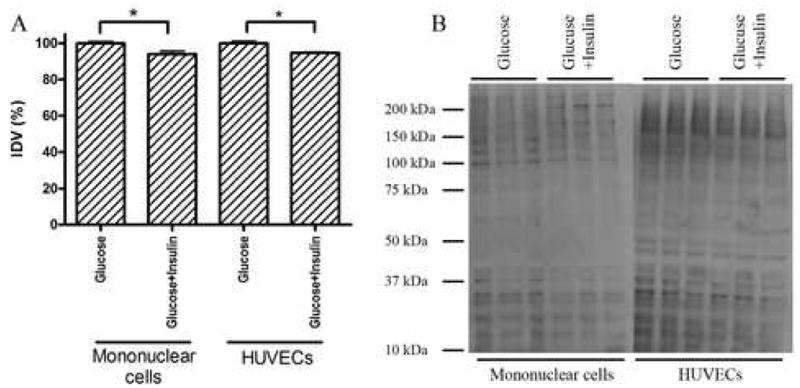
Panel A.Densitometric analysis of western blots. Insulin treatment slightly decreased the degree of protein poly(ADP-ribosylation) in both cell types. N=3 determinations per experimental group. Panel B. Representative Western blot image. In case of mononuclear cells two PAR positive bands that can be seen in the untreated group around 60 and 75 kDa disappears due to insulin treatment.
Discussion
It is generally assumed that reactive oxygen and nitrogen species produced during a variety of pathophysiological conditions including stroke, myocardial infarction, circulatory shock, endotoxin shock induce DNA strand breakage, which subsequently triggers the activation of PARP in various organs as well as in circulating leukocytes (Jagtap and Szabo, 2005; Pacher et al., 2007). Indeed, antioxidant therapy suppresses, while depletion of the endogenous antioxidant pool enhances oxidant-induced cell damage and PARP activation in these conditions (Cuzzocrea et al., 1998; Pieper et al., 2005; Horvath and Szabo, 2007). However, the potential role of hyperglycemia in triggering oxidative and nitrosative stress and PARP activation in endotoxin shock has not yet been investigated.
The importance of hyperglycemia in the context of critically ill patients has been highlighted by experimental and clinical studies demonstrating that tight glycemic control with insulin provides significant protective effects in patients in medical and surgical intensive care units (Ellger et al., 2006; Langley and Adams, 2007; Soop et al., 2007; Preiser and Devos, 2007). There is a separate field of investigation, in connection with the pathomechanisms of diabetic complications that is involved in studying the mechanisms of free radical production and cellular alterations in conjunction with elevated extracellular glucose concentrations. This line of investigation demonstrated that elevated extracellular glucose levels induces the overproduction of mitochondrial oxidants and free radicals, which, in turn, results in the activation of PARP, and the modulation of various downstream pathways of inflammation and cell response (Soriano et al., 2001; Pacher et al., 2002; Du et al., 2003). By linking the findings of these studies with the results of the experiments described in the current report, we propose that a possible mechanism whereby endotoxin shock induces PARP activation involves hyperglycemia, subsequent leak of oxidants and free radicals from the mitochondria, followed by DNA strand breaks. The exact source of the reactive species during hyperglycemia is a subject of on-going investigations; there is evidence, in various experimental models, for a role for mitochondrial reactive oxidant species, as well as for NAD(P)H oxidase, myeloperoxidase and aldose reductase in the production of reactive oxygen and nitrogen species in the vascular endothelium (Du et al., 2003; Inoguchi et al., 2003; Zhang et al., 2004; Obrosova et al., 2005; Nishikawa and Araki, 2007; Quagliaro et al., 2007). Whatever the source of reactive species is, it appears that this oxidative stress is sufficient to induce the activation of PARP in circulating leukocytes. Further work is required to determine whether PARP activation in circulating leukocytes serves as a ‘damage signal’ that precedes the death of these cells, or possibly PARP activation might act as an enhancer of leukocyte-derived inflammatory responses, thereby perpetuating the systemic inflammatory response.
The finding that insulin therapy reduces TNF-α production (which has been previously reported by other groups as well (Jeschke el al., 2004) can also be integrated with the PARP concept, as PARP is known to upregulate a variety of pro-inflammatory genes and PARP inhibitors have previously been shown to suppress the endotoxin-induced production of TNF-α in multiple experimental systems (Goldfarb et al., 2002).
Insulin exerts a variety of cellular and biological effects additional to its effect on glucose control. In HUVECs insulin stimulates adenosine and L-arginine transport and nitric oxide (NO) synthesis (Munoz et al., 2006). Likewise, recent data indicate that PARP can be regulated by alternative pathways that do not involve free radical generation or DNA damage (Szabo et al., 2006). Our in vitro experiments showed that insulin has a slight, but noticeable direct inhibitory effect on PARP activity in HUVECs and also in mononuclear leukocytes.
In the context of critical care illnesses, the glucose-mediated therapeutic effects of insulin appear to be predominant over glycemic control-independent actions (Ellger et al., 2006; Van den Berghe et al., 2006). Our observation that insulin decreases the degree of protein PARylation in vitro only by approximately 5% may also support this hypothesis and is consistent with the hypothesis that insulin reduces PARP activation in the current model of endotoxemia by normalizing plasma glucose levels, and hence reducing the amount of reactive oxygen and nitrogen species capable of PARP activation via DNA injury.
Conclusion
In summary, the current report demonstrates that insulin therapy in a rat model of endotoxemia blocks PARP activation and prevents inflammatory mediator production. Thus, the beneficial effects of insulin therapy in critical illness may be related, at least in part, to an inhibition of the pathophysiological consequences of the excessive overactivation of PARP.
Acknowledgments
This work was supported by the National Institutes of Health (NIH R01 GM060915 and HL71254) the OTKA (Hungarian Scientific Research Fund, Budapest, Hungary).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL, Szabo C. Effect of L-buthionine-(S,R)-sulphoximine, an inhibitor of gamma-glutamylcysteine synthetase on peroxynitrite- and endotoxic shock-induced vascular failure. British Journal of Pharmacology. 1998;123(3):525–37. doi: 10.1038/sj.bjp.0701612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of Clinical Investigation. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellger B, Debaveye Y, Vanhorebeek I, Langouche L, Giulietti A, Van Etten E, Herijgers P, Mathieu C, Van den Berghe G. Survival benefits of intensive insulin therapy in critical illness: Impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes. 2006;55:1096–1105. doi: 10.2337/diabetes.55.04.06.db05-1434. [DOI] [PubMed] [Google Scholar]
- Goldfarb RD, Marton A, Szabo E, Virag L, Salzman AL, Glock D, Akhter I, McCarthy R, Parrillo JE, Szabo C. Protective effect of a novel, potent inhibitor of poly(adenosine 5′-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis. Critical Care Medicine. 2002;30:974–980. doi: 10.1097/00003246-200205000-00004. [DOI] [PubMed] [Google Scholar]
- Horvath EM, Szabo C. Poly(ADP-ribose) polymerase as a drug target for cardiovascular disease and cancer: an update. Drug News & Perspectives. 2007;20(3):171–81. doi: 10.1358/dnp.2007.20.3.1092098. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, Sato N, Sekiguchi N, Kobayashi K, Sumimoto H, Utsumi H, Nawata H. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. Journal of American Society of Nephrology. 2003;14(8 Suppl 3):S227–32. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Soriano FG, Virag L, Liaudet L, Mabley J, Szabo E, Hasko G, Marton A, Lorigados CB, Gallyas F, Jr, Sumegi B, Hoyt DG, Baloglu E, VanDuzer J, Salzman AL, Southan GJ, Szabo C. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: Potent cytoprotective and antishock agents. Critical Care Medicine. 2002;30:1071–1082. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nature Reviews Drug Discovery. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Jeschke MG, Klein D, Bolder U, Einspanier R. Insulin attenuates the systemic inflammatory response in endotoxemic rats. Endocrinology. 2004;145:4084–4093. doi: 10.1210/en.2004-0592. [DOI] [PubMed] [Google Scholar]
- Langley J, Adams G. Insulin-based regimens decrease mortality rates in critically ill patients: A systematic review. Diabetes/Metabolism Research and Reviews. 2007;23(3):184–92. doi: 10.1002/dmrr.696. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M, Szabo C. Gender differences in the endotoxin-induced inflammatory and vascular responses: Potential role of poly(ADP-ribose) polymerase activation. The Journal of Pharmacology and Experimental Therapeutics. 2005;315:812–820. doi: 10.1124/jpet.105.090480. [DOI] [PubMed] [Google Scholar]
- Munoz G, San Martin R, Farias M, Cea L, Vecchiola A, Casanello P, Sobrevia L. Insulin restores glucose inhibition of adenosine transport by increasing the expression and activity of the equilibrative nucleoside transporter 2 in human umbilical vein endothelium. Journal of Cellular Physiology. 2006;209(3):826–35. doi: 10.1002/jcp.20769. [DOI] [PubMed] [Google Scholar]
- Nazer LH, Chow SL, Moghissi ES. Insulin infusion protocols for critically ill patients: a highlight of differences and similarities. Endocrine Practice. 2007;13(2):137–46. doi: 10.4158/EP.13.2.137. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxidants & Redox Signalling. 2007;9(3):343–53. doi: 10.1089/ars.2006.1458. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54(1):234–42. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–21. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper GM, Nilakantan V, Chen M, Zhou J, Khanna AK, Henderson JD, Jr, Johnson CP, Roza AM, Szabo C. Protective mechanisms of a metalloporphyrinic peroxynitrite decomposition catalyst, WW85, in rat cardiac transplants. The Journal of Pharmacolology and Experimental Therapy. 2005;314(1):53–60. doi: 10.1124/jpet.105.083493. [DOI] [PubMed] [Google Scholar]
- Preiser JC, Devos P. Clinical experience with tight glucose control by intensive insulin therapy. Critical Care Medicine. 2007;35(9):S503–7. doi: 10.1097/01.CCM.0000278046.24345.C7. [DOI] [PubMed] [Google Scholar]
- Quagliaro L, Piconi L, Assaloni R, Da Ros R, Szabo C, Ceriello A. Primary role of superoxide anion generation in the cascade of events leading to endothelial dysfunction and damage in high glucose treated HUVEC. Nutrition, Metabolism, and Cardiovascular Diseases. 2007;17(4):257–67. doi: 10.1016/j.numecd.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Soop M, Nygren J, Thorell A, Ljungqvist O. Stress-induced insulin resistance: recent developments. Current Opinion in Clinical Nutrition and Metabolic Care. 2007;10(2):181–6. doi: 10.1097/MCO.0b013e32801481df. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nature Medicine. 2001;7:108–13. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Liaudet L, Szabo E, Virag L, Mabley JG, Pacher P, Szabo C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17(4):286–92. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Nogueira AC, Caldini EG, Lins MH, Teixeira AC, Cappi SB, Lotufo PA, Bernik MM, Zsengeller Z, Chen M, Szabo C. Potential role of poly(adenosine 5′-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Critical Care Medicine. 2006;34:1073–1079. doi: 10.1097/01.CCM.0000206470.47721.8D. [DOI] [PubMed] [Google Scholar]
- Szabo C, Pacher P, Swanson RA. Novel modulators of poly(ADP-ribose) polymerase. Trends in Pharmacological Sciences. 2006;27(12):626–30. doi: 10.1016/j.tips.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berghe G, Wilmer A, Milants I, Wouters PJ, Bouckaert B, Bruninckx F, Bouillon R, Schetz M. Intensive insulin therapy in mixed medical/surgical intensive care units: Benefit versus harm. Diabetes. 2006;55:3151–3159. doi: 10.2337/db06-0855. [DOI] [PubMed] [Google Scholar]
- Vanhorebeek I, Langouche L, Van den Berghe G. Tight blood glucose control: what is the evidence? Critical Care Medicine. 2007;35(9):S496–502. doi: 10.1097/01.CCM.0000278051.48643.91. [DOI] [PubMed] [Google Scholar]
- Zhang C, Yang J, Jennings LK. Leukocyte-derived myeloperoxidase amplifies high-glucose--induced endothelial dysfunction through interaction with high-glucose--stimulated, vascular non-leukocyte-derived reactive oxygen species. Diabetes. 2004;53(11):2950–9. doi: 10.2337/diabetes.53.11.2950. [DOI] [PubMed] [Google Scholar]