

Abstract
Synaptic vesicles (SVs) are composed of ∼10 types of transmembrane proteins that must be recycled after exocytosis of neurotransmitter. The mechanisms for resorting these proteins into synaptic vesicles once incorporated into the plasma membrane after exocytosis are poorly understood. The adaptor complex AP-2 is the major clathrin-associated adaptor for cargo recognition at the plasma membrane. Here, we have investigated its role in synaptic vesicle endocytosis. shRNA-mediated knockdown of the AP-2 complex results in an ∼96% reduction of this protein complex in primary neurons. We used simultaneous expression of shRNA and pHluorin-tagged vesicle components to show that the absence of AP-2 significantly slows but does not prevent the endocytosis of four of the major synaptic vesicle transmembrane proteins. We show that in the absence of AP-2, the AP-1 adaptor complex appears to functionally substitute for AP-2 but results in complex internalization kinetics that are now sensitive to the guanine-nucleotide exchange factor for ADP-ribosylation factor GTPase (ARF–GEF) inhibitor brefeldin-A (BFA). Simultaneous removal of both AP-2 and AP-1 prevents this compensatory substitution and results in slowed but functional endocytosis. These results demonstrate that in the absence of AP-2, SV proteins still become endocytosed, and synaptic vesicle recycling remains operational.
Introduction
Typical synapses of the CNS contain ∼100 synaptic vesicles (SVs) that participate in exocytosis and recycling to sustain synaptic transmission. SV retrieval pathways have long been debated (Wu et al., 2007), but a number of recent studies implicate both clathrin (Gad et al., 2000; Augustine et al., 2006; Granseth et al., 2006) and dynamin (Yamashita et al., 2005; Ferguson et al., 2007) for the majority of SV endocytosis. Cargo proteins destined for internalization via clathrin-coated pits are concentrated into these pits via the dual interaction of these proteins with an adaptor complex (AP) and the AP with coat-components of the endocytic bud. Four APs have been identified (AP-1, AP-2, AP-3, and AP-4) and function along different membrane trafficking routes within cells (Bonifacino and Traub, 2003). Of these complexes, only AP-2 functions during endocytosis at the plasma membrane. AP-2, a heterotetrameric complex composed of subunits α, β2, μ2, and σ2 is thought to act in cargo selection by direct interaction with selection motifs encoded in the cytoplasmic tails of cargo molecules. This complex is essential for survival as genetic ablation of AP-2 leads to embryonic lethality in fly (Gonzalez-Gaitan and Jackle, 1997), worm (Shim and Lee, 2000), and mouse (Mitsunari et al., 2005). Thus, AP-2 is a leading candidate to coordinate the sorting of SV proteins during recycling.
At the synapse, it has been identified as an interaction partner with synaptotagmin (Zhang et al., 1994; Haucke and De Camilli, 1999; Haucke et al., 2000), a critical SV cargo protein. Synaptotagmin, however, is but one of ∼10 different types of SV proteins (Takamori et al., 2006) that span the bilayer of the vesicle and therefore, in principle, need to be sorted back into synaptic vesicles after exocytosis. In addition to synaptotagmin, a number of SV proteins are considered essential cargo in the reformation of the vesicle. These include VAMP-2, the vesicle-associated SNARE (soluble N-ethylmaleimide-sensitive factor attached protein receptor) protein required for exocytosis, a vesicular transporter of neurotransmitter required for filling the vesicle with neurotransmitter (vGlut in the case of glutamate), and a proton pump (the V-type ATPase) required to generate the proton-motive force necessary to drive neurotransmitter uptake. Additionally, other abundant cargo proteins of lesser-known function such as synaptophysin, synaptogyrin, SV2, and the chloride channel CLC-3, presumably, also need to be resorted before endocytosis.
We tested the hypothesis that AP-2 is crucial for SV endocytosis using shRNA-mediated removal of AP-2 in individual dissociated primary neurons that simultaneously expressed pHluorin-tagged variants of one of four different SV cargos. Our experiments show that in the absence of AP-2, endocytosis and vesicle recycling still persist but with severely impaired kinetics. Our results also demonstrate that, in the absence of AP-2, partial compensation is driven by AP-1 rendering endocytosis sensitive to the guanine-nucleotide exchange factor for ADP-ribosylation factor GTPase (ARF–GEF) inhibitor brefeldin-A. In the absence of both AP-1 and AP-2, endocytosis still persists at a reduced rate. Our results demonstrate that AP-2 plays a critical role in efficient endocytosis for a large fraction of SV proteins but that other mechanisms allow for endocytosis to proceed in its absence.
Materials and Methods
Cell culture and optical setup.
Hippocampal CA3–CA1 regions were dissected from 1- to 3-d-old Sprague Dawley rats, dissociated, and plated onto poly-ornithine-coated glass for 14–21 d as described previously (Ryan, 1999). In the majority of experiments, constructs were transfected 8 d after plating, and imaging was performed 14–20 d after plating (6–12 d after transfection), and the coverslips were mounted in a rapid-switching, laminar-flow perfusion and stimulation chamber (volume ∼75 μl) on the stage of a custom-built laser-illuminated epifluorescence microscope. In some cases, neurons were transfected 12 d after plating and imaged 3 d later. Live-cell images were acquired with an Andor iXon+ (model #DU-897E-BV) back-illuminated electron-multiplying charge-coupled device camera in epifluorescence. An Ar+ ion laser or a solid-state diode pumped 488 nm laser for pHluorin or 532 nm laser for FM 4-64 that was shuttered using acousto-optic modulation in all nondata acquiring. Fluorescence excitation and collection was done through a 40× 1.3 NA Fluar Zeiss objective using 515–560 nm emission and 510 nm dichroic filters for pHluorin or 573–648 nm emission and 565 nm dichroic filters for FM 4-64. Action potentials were evoked by passing 1 ms current pulses, yielding fields of ∼10 V/cm via platinum–iridium electrodes. Cells were continuously perfused (0.5–1.0 ml/min; ∼25°C) in a saline solution containing (in mm) 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, buffered to pH 7.4, 30 glucose, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Sigma), and 50 μm d,l-2-amino-5-phosphonovaleric acid (AP5; Sigma). Unless otherwise noted, all chemicals were obtained from Sigma. NH4Cl applications were done with 50 mm NH4Cl in substitution of 50 mm of NaCl, buffered to pH 7.4. Advasep was obtained from CyDex and used at 1 mm where indicated. Bafilomycin (Calbiochem) was used at 1 μm where indicated. Brefeldin-A was obtained from Sigma and used at 10 μg/ml where indicated.
shRNA and shRNA-resistant μ2 plasmids.
Synthetic oligonucleotides (60-mers) containing the rat μ2 subunit cDNA target sequences (GTGGATGCCTTTCGCGTCA) of AP-2 or rat γ subunit cDNA target sequences (GCGCCTGTACAAGGCAATT) of AP-1 for cloning in pSUPER vector were synthesized (Invitrogen), annealed, and ligated into pSUPER using BglII and HindIII enzyme sites according to manufacturer's instructions. The HA-μ2 construct resistant to shRNA μ2 was generated using Quick Change Site-Directed mutagenesis (Stratagene). The sequence for shRNA μ2 corresponding to nucleotide positions, 85–103, was mutated to GTGGACGCTTTCCGGGTCA, leaving the amino acid sequence unchanged.
Immunofluorescence and quantification.
After live-cell imaging, neurons were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100, blocked with 5% BSA, and subsequently incubated with appropriated primary antibodies [anti-green fluorescent protein (GFP) (Invitrogen), anti-α adaptin (ABR), anti-clathrin (ABR), anti-γ adaptin (BD Biosciences), or anti-HA tag (Covance) antibodies]. Alexa 488- or Alexa 546-conjugated secondary antibodies were applied in primary antibody-incubated samples with different color combinations as needed. Immunofluorescence images of fixed cells were acquired using custom-built laser-scanning microscope (LSM) with custom-written control software for LSM. Images were acquired with Z-sectioning mode with 12 slices and 0.4 μm spacing and merged using maximum intensity. Expression levels of AP-2, clathrin heavy chain, and AP-1 were measured at cell bodies to avoid possible spatial overlap with other cells. For RNA-based knockdown of AP-2 (AP-2KD), we measured the fluorescence in the probe antibody channel in GFP-positive cells corrected for background in surrounding regions and a small amount of bleed through the filter set. This was compared with the fluorescence intensity in nontransfected (GFP-negative) cell bodies. Expression level (percentage) = (FGFP+ neuron − F back)/(F GFP−neuron − F back)*100.
Image and data analysis.
Images were analyzed in ImageJ (http://rsb.info.nih.gov/ij/) using a custom-written plugin (http://rsb.info.nih.gov/ij/plugins/time-series.html). All functionally visible varicosities were selected for analysis by testing their responsiveness to test stimuli of 25 action potential trains at 10 Hz. Fluorescence time course traces were analyzed using Origin Pro (version 7.5). Exponential fitting was done using a double exponential function. Traces whose fit values for each time constant were equal (within the error of the fit) were assigned to be single component traces. All others were assigned as double exponentials.
Results
shRNA-mediated knockdown of μ2 results in ∼96% loss of AP-2 complex in primary neurons
Our experiments make use of relatively low transfection efficiency of primary neurons to express pHluorin-tagged synaptic vesicle proteins that in turn express at only tracer levels of these reporter constructs (Fernández-Alfonso et al., 2006; Balaji and Ryan, 2007). To combine this with AP-2KD, we made use of simultaneous transfection of an shRNA plasmid encoding a hairpin sequence targeted to the μ2 subunit of AP-2 and a plasmid encoding a pHluorin-tagged synaptic vesicle protein. Most experiments were performed between 6 and 12 d after transfection to allow the targeted protein to significantly decrease in expression. We measured the expression level of the AP-2 complex in individual neurons by performing retrospective quantitative immunofluorescence on the transfected cells using an antibody against the nontargeted α subunit of AP-2 (Fig. 1A) and normalized the intensity to the staining in nontransfected cells in the same dish. These experiments showed that AP-2 levels are reduced by ∼96% (Fig. 1C), whereas the expression level of one of AP-2's major interacting partners, clathrin heavy chain (Fig. 1B,C), was unaffected.
Figure 1.
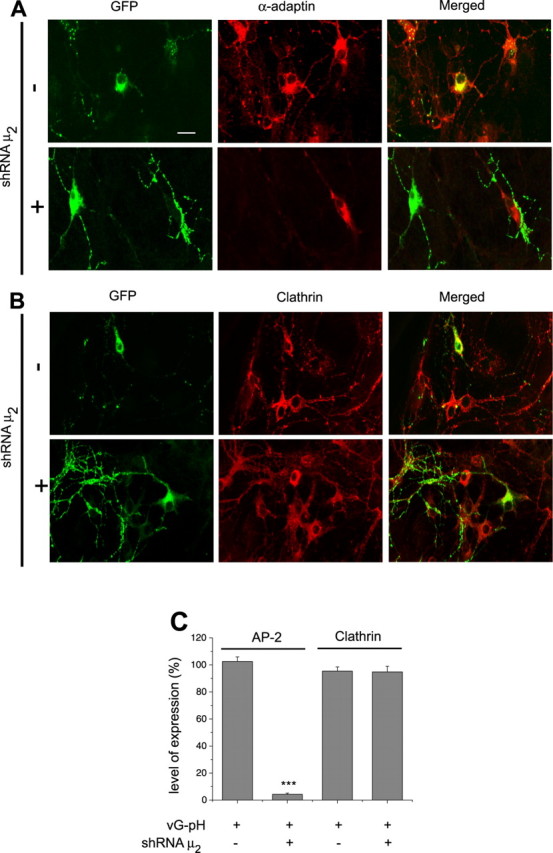
shRNA-targeting μ2 efficiently depletes AP-2 in hippocampal neurons. A, B, Primary hippocampal neurons transfected with vG–pH with/without shRNA-targeting μ2 were stained with both anti-GFP (green) and anti-α adaptin (red) (A) or anti-clathrin heavy chain (red; B) antibodies. Scale bar, 20 μm. C, In cells transfected with shRNA-targeting μ2, AP-2, but not clathrin, was severely depleted: [AP-2]AP-2KD = 4.3 ± 0.9% (n = 28) compared with controls (n = 20), whereas [AP-2] = 102.5 ± 3.4% in cells transfected with vG–pH alone compared with nontransfected cells. [Clathrin]AP-2KD = 94.8 ± 4.1% (n = 14) and [Clathrin] = 95.4 ± 3.1% (n = 17) in cells transfected with vG–pH alone compared with nontransfected cells. ***p < 0.01.
Surface fraction of vGlut–pHluorin increases significantly in the absence of AP-2
In neurons transfected with vGlut–pHluorin (vG–pH) alone, the majority of this reporter protein resides within an acidic, internal vesicle pool at nerve terminals with a surface expression of only 3.38 ± 0.6% (n = 10) (Fig. 2A, left panel) (Balaji and Ryan, 2007) as determined by measurements of the magnitude of the relative fluorescence change during alkalization with NH4Cl (Sankaranarayanan et al., 2000). After AP-2 knockdown, however, the resting vG–pH fluorescence is greater along both the axon and the nerve terminals (Fig. 2A, right). Measurements of fluorescence changes using NH4Cl reveal that the surface fraction has increased in the absence of AP-2 to13.0 ± 3.3% (n = 8). Similar results were obtained with pHluorin-tagged synaptophysin, synaptotagmin, and VAMP-2 (data not shown). This steady-state increase in surface fluorescence is consistent with the idea that endocytosis is impaired in the absence of AP-2, leaving synaptic vesicle proteins stranded on the axon, as has previously been observed with VAMP-2 for a number of endocytic protein mutants (Dittman and Kaplan, 2006).
Figure 2.

Depletion of AP-2 leads to increased surface fraction of vGlut–pHluorin and decreased recycling vesicle pool size. A, Surface expression of vG–pH was determined by measuring the fold change in fluorescence for boutons in normal saline for control and knockdown cells (AP-2KD) in response to application of NH4Cl. Scale bar, 10 μm. In control cells, the vG–pH surface expression at synaptic boutons was 3.38 ± 0.6% (n = 10), whereas in AP-2KD, the surface fraction was 13.0 ± 3.3% (n = 8). B, Schematic diagram of FM 4-64 staining and destaining experiment for measurement of total recycling pool. Cells were stimulated in the presence of FM 4-64 to turn over the entire recycling pool at 20 Hz for 100 s and further incubated for an additional 5 min after end of the stimulation. After washing for 3 min with Advasep buffer and additional 10 min with Ca2+-free Tyrodes, neurons were stimulated twice (10 Hz for 150 s and 10 Hz 60 s) in normal Tyrodes and the total dye released was measured to determine the size of the recycling pool. The recycling pool was 30.0 ± 3.6% (n = 10) in the AP-2KD compared with controls (n = 10).
Depletion of recycling vesicle pools in the absence of AP-2
We determined the relative size of the synaptic vesicle recycling pool at nerve terminals in AP-2KD neurons compared with their wild-type counterparts using fluorescent styryl dye loading. The protocol for this experiment is shown in Figure 2B. FM 4-64 was bath applied to the dish of neurons and stimulated to turn over the entire recycling vesicle pool (2000 action potentials; 20 Hz). The dye was left on for an additional 5 min to allow for completion of endocytosis. After dye washout (10 min), the neurons were again stimulated maximally, and the amount of total dye destaining was compared in transfected (AP-2KD, vG–pH) and nontransfected neurons. These experiments indicate that in the absence of AP-2 the recycling vesicle pool is decreased to 30 ± 3.6% (n = 10) of that in control cells. Experiments measuring the kinetics of vesicle pool turnover using bafilomycin indicated that although the total recycling vesicle pool size is severely depleted, the kinetics of exocytosis of that pool was unaffected (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
After stimulus endocytosis of synaptotagmin, synaptophysin, vGlut, and VAMP-2 all significantly slowed in the absence of AP-2
pHluorin-tagged synaptic vesicle proteins have proven to be a useful probe for examining the kinetics of endocytosis at nerve terminals (Sankaranarayanan and Ryan, 2000; Granseth et al., 2006; Balaji and Ryan, 2007). Action potential firing-driven exocytosis leads to the exposure of the vesicle lumen to the more alkaline external environment leading to dequenching of the pHluorin fluorescence. In control hippocampal nerve terminals, providing the amount of exocytosis-driven accumulation of SV protein, does not saturate the endocytic machinery (stimuli of <100 action potentials) (Balaji et al., 2008), endocytosis is well described by a single stochastic process with a time constant of ∼15 s (Fig. 3A) (Balaji and Ryan, 2007). The fluorescence decay is dominated by this process as the reacidification step requires only 3–4 s (Atluri and Ryan, 2006; Granseth et al., 2006). In the absence of AP-2, however, the fluorescence decay for vG–pH is significantly slowed showing a t1/e = 58.8 ± 6.9 s (n = 18). The kinetics of the decay is no longer well fit by a single exponential (see below), and for simplicity, we have simply chosen to measure the time for the decay to reach 1/e of the initial value. To determine if AP-2 is functionally important for other cargoes, we took advantage of the ability to label three other important SV proteins such that pHluorin is targeted to the vesicle lumen to determine if the absence of AP-2 would change their endocytosis kinetics. In the absence of AP-2 endocytosis kinetics of synaptophysin (Fig. 3B), synaptotagmin I (Fig. 3C) and VAMP-2 (Fig. 3D) were also severely impaired with fluorescence decay times t1/e = 40.8 ± 3.3 s (n = 16), t1/e = 31.6 ± 3.46 s (n = 4), and t1/e = 40.1 ± 8.5 s (n = 7), respectively, each ∼2-fold slower than in control cells. Acid-quenching measurements (data not shown) revealed that the reacidification time scale was not significantly perturbed in the absence of AP-2. Thus, AP-2 appears to play a critical role in controlling the kinetics of endocytosis of at least four of the major SV cargo proteins.
Figure 3.

Poststimulus endocytosis of four major synaptic vesicle proteins slowed in the absence of AP-2. A, Representative ensemble average traces of endocytosis from various SV constructs. Cells were transfected with pHlourin-tagged synaptic vesicle proteins, vG–pH, synaptophysin (Sphy–pH), synaptotagmin (Stag–pH), VAMP-2 (SpH) with/without shRNA-targeting μ2. Neurons were stimulated at 10 Hz (100 action potentials). B, The time to decay to 1/e of the peak value was significantly greater for each construct in the AP-2KD compared with control (SpH only, n = 4; SpH with shRNA μ2, n = 7; vG–pH only, n = 13; vG–pH with shRNA μ2, n = 18; Stag-pH only, n = 4; Stag-pH with shRNA μ2, n = 4; Sphy-pH only, n = 5; Sphy-pH with shRNA μ2, n = 16).
Endocytosis during activity is severely impaired the absence of AP-2
To determine the impact of AP-2 depletion on endocytosis during repetitive stimulation, we compared vG–pH responses to 30 s stimulation at 10 Hz in the presence and absence of bafilomycin. In the absence of bafilomycin, vG–pH reports the net accumulation of this reporter on the synaptic and axonal surface arising from the balance of exocytosis and endocytosis. In the presence of bafilomycin, as reacidification of vesicles is blocked, vG–pH fluorescence reports the net amount of exocytosis. Comparison of the two signals allows one to calculate the total amount of endocytosis during a given stimulus period (Sankaranarayanan and Ryan, 2001). Under conditions where only vG–pH is transfected, the total amount of endocytosis at the end of a 300 AP at 10 Hz is equivalent to ∼25% of the entire recycling pool (Fig. 4A,C), similar to previous results (Ferguson et al., 2007; Mani et al., 2007). In the absence of AP-2, the amount of endocytosis during stimulation is ∼5-fold lower (Fig. 4B,C). Thus, both ongoing endocytosis during stimulation as well as poststimulus endocytic recovery of SV components require AP-2 for normal function.
Figure 4.
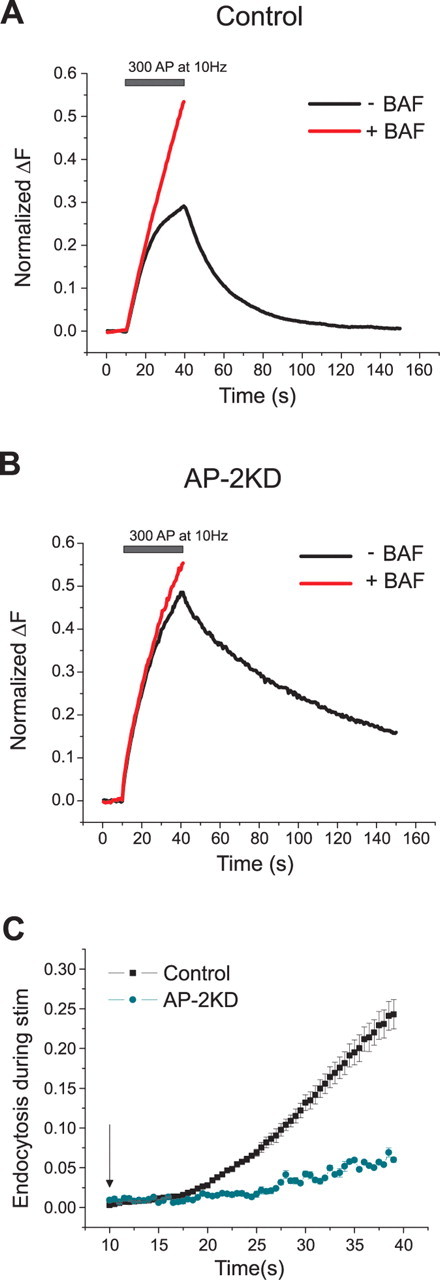
Endocytosis during stimulation is severely decreased in the absence of AP-2. A, B, Representative ensemble average vG–pH traces from control (A) and AP-2KD (B) neurons stimulated in the presence (red) or absence (black) of bafilomycin (Baf) with 300 action potentials (10 Hz) normalized to the maximum fluorescence value obtained with 900 action potential stimulation in Baf (the total recycling pool). C, Endocytosis during stimulation in control neurons corresponds to 24.3 ± 1.8% (n = 6), whereas in AP-2KD neurons, it was 5.9 ± 0.3% (n = 8), respectively, of the recycling pool. Stimulation start marked by arrow. Endocytosis during activity was calculated as (ΔFBaf − ΔF300 non baf) and normalized to the maximal plateau value achieved during stimulation in Baf.
Expression shRNA-resistant μ2 rescues defects of knockdown
To verify that perturbation of presynaptic endocytosis was due specifically to loss of AP-2 and not off-target shRNA effects, we used a shRNA-resistant plasmid encoding μ2 to restore function in the AP-2KD cells. In this case, triple transfection of vG–pH, shRNA-targeting μ2, and shRNA-resistant plasmid encoding μ2 was used. Expression of shRNA-resistant plasmid for μ2 restored the surface fraction levels to that obtained with vG–pH alone (Fig. 5A), 4.5 ± 1.5% (n = 7). Similarly, poststimulus endocytosis was nearly completely restored under these conditions (Fig. 5B,D), as was endocytosis during persistent activity (Fig. 5C). These rescue effects of endocytosis were in good agreement with recoveries of AP-2 expression levels (Fig. 5E). Similarly, the ∼3-fold reduction in vesicle pool size was mostly restored under these conditions as well (Fig. 5F). Thus, the major endocytic defects produced by expression of shRNA targeted to μ2 are likely to be directly related to the severe depletion of AP-2 and not more generic off-target effects of shRNA treatment.
Figure 5.
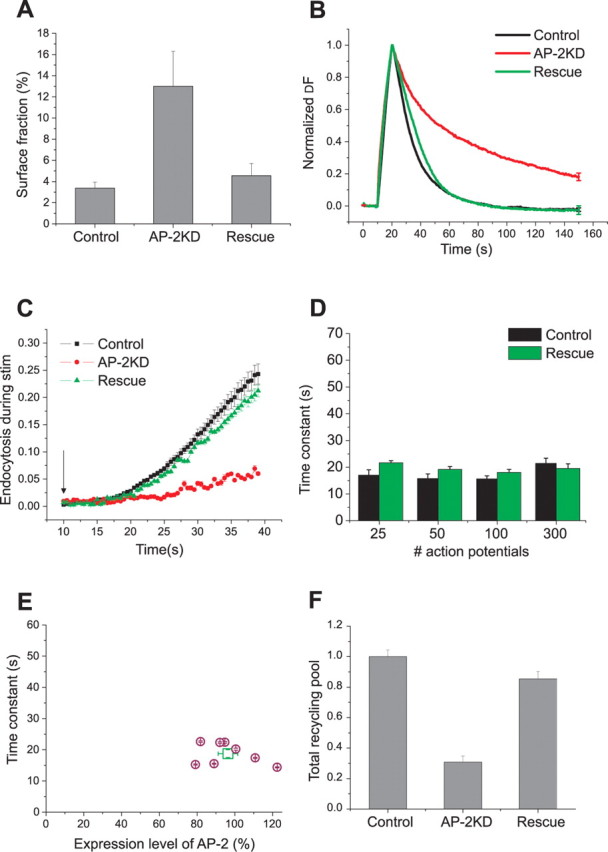
Expression of shRNA-resistant μ2 rescues endocytic phenotype in AP-2KD neurons. A, Expression of shRNA-resistant μ2 in AP-2KD cells restores the surface fraction to control levels. Surface fraction was determined as in Figure 2 (A, B) by application of NH4Cl, pH 7.4, giving a value 4.6 ± 1.2%. n = 7 in the rescued cells, similar to controls (3.38 ± 0.6%, n = 10). B, Representative ensemble average examples from control (black), AP-2KD (red), and rescue (green) neuron responses to 100 action potential stimulation. For rescue of AP-2KD, neurons were transfected with shRNA-targeting μ2, shRNA resistant μ2, and vG–pH; n = 8. C, shRNA-resistant μ2 expression in AP-2KD neurons rescued endocytosis during stimulation. Endocytosis during stimulation (300 action potentials at 10 Hz, start of stimulus marked by arrow) was determined as in Figure 4 as (ΔFBaf − ΔF300 no baf) and showed that the total amount of endocytosis was 21.2 ± 1.1% (n = 7) similar to controls (Fig. 4C). D, Poststimulus endocytosis is restored by expression of shRNA-resistant μ2 in the AP-2KD for various stimulation conditions (25, 50, 100, and 300 action potential stimuli at 10 Hz, respectively; n = 8). E, Comparison of endocytosis time constant with the degree of AP-2 expression obtained in rescue experiments. After live-cell imaging, neurons were fixed and labeled with anti-α-adaptin antibody and expression quantified as in Figure 1. The average value is shown (square symbol) in addition to the individual values for each neuron (n = 8). F, Recovery of the defect in recycling pool was achieved by expression of shRNA-resistant μ2 in the AP-2KD neurons. Recycling pool size was measured as in Figure 2 (C, D). The recycling pool size compared with neurons transfected with vG–pH alone was 85.4 ± 4.9% (n = 6).
In the absence of AP-2, endocytosis kinetics displays two distinct components
Under control conditions, we previously showed that for modest stimulation (<100 AP) endocytosis is well described by a single exponential process with a time constant of ∼15 s. Plotting the ensemble average, vG–pH fluorescence decay on a semilog plot (Fig. 6A) for cells transfected only with the reporter shows a characteristic single straight line slope. In contrast, a similar plot for the fluorescence decay derived from AP-2KD neurons showed two clear decay components (Fig. 6B). We verified that the appearance of two kinetic components was invariant with stimulus number by examining how the amplitude and value of each component varied as a function of stimulus number (Fig. 6C). In general, we found that the faster decay constant (τfast = 11 ± 1.7 s) was significantly faster than the time constant obtained in the presence of AP-2 (τcontrol = 14.8 ± 0.2 s), and the slower time constant was much slower (τslow = 93.2 ± 4.6 s). This complexity in endocytic behavior was not simply a consequence of removing AP-2 during a critical window of synapse development, as this phenotype was still apparent when neurons were transfected at 12 d in vitro (DIV) and examined at 15 DIV (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material). To determine whether the appearance of this more complex endocytosis was a general property of individual boutons, we analyzed the fluorescent decays for a large number of synapses from many cells. Notably, we discovered that even within a single cell, two types of single bouton kinetic behavior are evident, demonstrating that these disparate kinetic features are bouton and not cell specific. Figure 6D shows an example of two individual bouton responses to 100 action potentials stimulation obtained from the same axon. One shows a fluorescence decrease which has more than one component of decay, whereas another is adequately fit by a single exponential decay. Similar analysis across 427 boutons derived from 11 cells indicates that ∼36% of boutons can be fit by a single exponential, and the remainder of the boutons requires a two component fit. Individual bouton analysis derived from neurons transfected with only vG–pH indicate that under these conditions, all boutons can be adequately fit by a single exponential. Analysis of the distribution of the values of these time constants for all boutons is shown in Figure 6E–G and reveals that in the absence of AP-2, two types of endocytic phenotypes arise, both significantly different from the control endocytosis behavior. Similar analysis of other three other synaptic vesicle cargo proteins revealed an almost identical kinetic behavior and distribution (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 6.
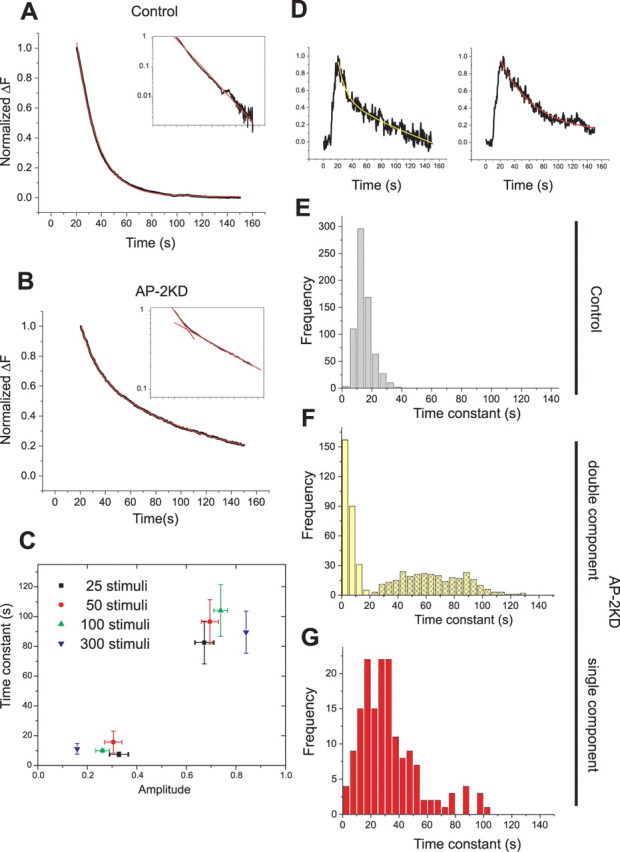
In the absence of AP-2, endocytosis has complex internalization kinetics. A, B, Representative average traces of endocytosis from control (A) and AP-2KD (B) neurons after stimulation. Ensemble average vG–pH fluorescence traces from individual neurons were fit with a single exponential decay (red) in the control neuron (A) or double exponential decay (red) in the AP-2KD neuron (B). Traces displayed as semilog plot (inset) to illustrate the appearance of a second component. C, In AP-2KD neurons, the weighting amplitudes (amp) and values of the two time constants were determined for various stimulation conditions (n = 7–8 cells for each condition): τ25AP fast = 7.5 ± 1.5 s, amp25AP fast = 32.8 ± 3.9%; τ25AP slow = 82.6 ± 14.4 s, amp25AP slow = 67.2 ± 3.9%; τ50AP fast = 15.7 ± 7.4 s, amp50AP fast = 30.5 ± 3.6%; τ50AP slow = 96.5 ± 14.9 s, amp50AP slow = 69.5 ± 3.6%; τ100AP fast = 10.0 ± 1.0 s, amp100AP fast = 26.2 ± 3.5%; τ100AP slow = 104.1 ± 17.4 s, amp100AP slow = 73.8 ± 3.5%; τ300AP fast = 11.2 ± 3.6 s, amp300AP fast = 15.9 ± 2.8%; τ300AP slow = 89.6 ± 14.1 s, amp300AP slow = 84.1 ± 2.8%, respectively). D, Individual boutons from the same AP2-KD neuron show different internalization kinetics. The vG–pH trace of an individual bouton on the left shows a double-exponential decay, whereas the trace from an individual bouton on the right is adequately fit with a single exponential decay. Analysis over several hundred boutons from many cells shows that 36% of boutons were adequately fit by single exponential decays. E–G, The distribution of time constants of single bouton analysis from control (E, n = 680) and AP-2KD (F, double exponential, n = 273; G, single exponential, n = 154) neurons.
Endocytosis in the absence of AP-2 is brefeldin sensitive
We reasoned that the appearance of two distinct kinetic components in the absence of AP-2, both significantly different from that obtained in control conditions, might arise from alternate compensatory mechanisms. Although under wild-type conditions endocytosis in this system has previously shown to be insensitive to disruption of the actin cytoskeleton (Sankaranarayanan et al., 2003), in bipolar cells actin seems to play a role in membrane recovery under conditions that stimulate bulk endocytosis (Holt et al., 2003). We tested for a possible role for actin in controlling endocytosis in the absence of AP-2 using the G-actin sequestering agent latrunculin A. These experiments, however, failed to show any change in the sensitivity of endocytosis kinetics to actin depolymerization attributable to the absence of AP-2 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Recently, Voglmaier et al. (2006) provided evidence that an alternate endocytic pathway that is sensitive to the ARF–GEF inhibitor BFA becomes operational during very prolonged action potential firing. For the briefer bursts of stimuli used here, endocytosis kinetics in wild-type synapses are completely insensitive to BFA (Fig. 7A–C). However, in the absence of AP-2 application of BFA shifts endocytosis from a dual-time constant decay to a single component decay (Fig. 7D–F). Interestingly, the average time constant for endocytosis in the AP-2KD in the presence of brefeldin-A (Fig. 7H) τBFA-KD = 42 ± 1.4 s is very similar to the average value of the endocytosis time constant for the ∼36% of boutons that showed a single component decay in the AP-2KD (Fig. 6G), τKD− single = 44 ± 4.3 s. We, therefore, explicitly examined the sensitivity of AP-2KD boutons that displayed single component decays to BFA application. These experiments showed that these boutons had little sensitivity to BFA treatment (Fig. 7G,H). Thus, in the absence of AP-2, a brefeldin-sensitive pathway appears to become operational for a subset of synapses that results in complex internalization kinetics.
Figure 7.

Endocytosis is brefeldin-A-sensitive in the majority of synapses in the absence of AP-2. A–C, Endocytosis in control neurons is insensitive to BFA. vG–pH fluorescence recovery of neurons transfected with vG–pH and stimulated with 100 action potentials at 10 Hz before (black) and after (red) 30 min treatment of BFA (10 μg/ml). B, C, Semilog plots of the same traces before (B) and after (C) BFA treatment are shown. D–F, Endocytosis in AP-2KD neurons is sensitive to brefeldin-A. vG–pH fluorescence recovery of neurons transfected with vG–pH and shRNA-targeting μ2 and stimulated with 100 action potentials at 10 Hz before (black) and after (red) 30 min treatment of BFA (10 μg/ml). Semilog plots of the same traces before (E) and after (F) BFA treatment are shown. After BFA treatment, only a single component persists in the AP-2KD neurons. Time constant in control cells (A–C) is not significantly different after BFA treatment (Con; τBefore = 12.4 ± 0.9 s, τAfter = 12.9 ± 1.5 s; n = 3). In AP-2KD (D–F), only a single intermediate valued time constant is required to fit the endocytic recovery after BFA treatment (AP-2KD; τ-BFA fast = 11.2 ± 1.9 s, τ-BFA slow = 84.8 ± 6.7 s, τ+BFA = 45.1 ± 6.7 s; n = 7). G, H, The distribution of time constants obtained from single boutons in AP-2KD neurons before (G) and after (H) BFA treatment.
Multicomponent endocytosis and brefeldin sensitivity arise from compensation by AP-1
Brefeldin-A is a fungal metabolite discovered originally because of its potency in disrupting the integrity of the Golgi apparatus (Fujiwara et al., 1988). Subsequently, it was shown to have more pleiotropic effects on additional cellular organelles and their trafficking (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Wood et al., 1991). The only identified and molecularly well defined mechanism of BFA action is its ability to block a Golgi-associated GEF for ARF1, thereby blocking ARF1 activation and the assembly of COPI (coatomer protein I) coats on the Golgi complex (Donaldson et al., 1992; Helms and Rothman, 1992). The target of BFA in endosomal systems has not been well defined, although recruitment of AP-1 and AP-3 and subsequent ability to recruit clathrin-coats on membranes has previously been shown to be driven by ARFs that are sensitive to BFA (Liang and Kornfeld, 1997; Drake et al., 2000). We, therefore, tested the possibility that in the absence of AP-2, an alternate adaptor was substituting in endocytic sorting. In polarized epithelia, vesicle budding from recycling endosomes has been shown to be brefeldin sensitive and depend on AP-1 but not AP-3 (Pagano et al., 2004). We, therefore, investigated the possible role of AP-1 in synaptic vesicle endocytosis. Transfection of neurons with a shRNA targeting the γ subunit of AP-1 results in ∼70% depletion of this subunit (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Cotransfection of this shRNA into neurons with vG–pH leaves endocytosis kinetics intact (Fig. 8A). However, depletion of both AP-1 and AP-2 by simultaneous transfection of μ2 and γ-targeting shRNAs and vG–pH resulted in endocytosis kinetics that had a single component of endocytic decay (Fig. 8B) similar to endocytosis in the presence of BFA in AP-2KD neurons (Fig. 6). Furthermore, application of BFA to AP-1/AP-2KD synapses showed that these synapses now had little sensitivity to BFA (Fig. 8B). Interestingly, detailed analysis of the kinetics of endocytosis at individual boutons in AP-2KD and AP-1/AP-2KD synapses showed that they were nearly indistinguishable from those obtained from either the subset of synapses originally showing two kinetic components in AP-2KD and then treated with BFA or the subset showing a single BFA-insensitive component (Fig. 8C,D). These results strongly support the idea that in a subset of synapses in the absence of AP-2, AP-1 acts as a partial-functional substitute resulting in complex internalization kinetics and BFA sensitivity.
Figure 8.

Brefeldin-A sensitivity and complex kinetics in the absence of AP-2 arise from compensation by AP-1. A, Representative ensemble average of endocytosis from control (black) and AP-1KD (red) neurons. Neurons were stimulated at 10 Hz (100 action potentials). The average time constant for vG–pH fluorescence decay in control (τcontrol = 15.6 ± 1.2 s) and AP-1KD neurons (τAP-1KD = 16.4 ± 1.3 s; n = 7) are not significantly different. B, Representative vG–pH trace of endocytosis in the presence or in the absence of BFA in double (AP-1/AP-2) KD neurons. Semilog plot of poststimulus endocytosis are shown in the inset (inset: top, −BFA; bottom, +BFA). Time constant of endocytosis obtained from individual cell ensemble average in the presence or in the absence of BFA (n = 5) in double KD neurons has minor sensitivity to BFA (τBefore = 38.2 ± 4.3 s; τAfter = 43.9 ± 5.1 s). C, D, Representative ensemble average vG–pH fluorescence recovery traces (C) and average time constant (D) of endocytosis from single bouton analysis from BFA treated or untreated AP-2KD and double (AP-1/AP-2) KD neurons. The data for AP-2KD were sorted into the average value of the time constant in BFA for boutons that showed either two (Two) or a single (One) exponential component before BFA application. This analysis shows that these conditions all have similar average endocytosis kinetics (AP-2KD; τtwo BFA+ = 46.2 ± 2.5 s, τone BFA− = 49.9 ± 2.8 s, τone BFA+ = 51.4 ± 2.2 s: Double KD; τBFA− = 40.0 ± 1.7 s, τ BFA+ = 42.7 ± 1.5 s.). For each category, the average was derived from between 108 and 131 boutons from four to seven different cells.
Discussion
The mechanisms of synaptic vesicle recycling have been of significant interest since the original ultrastructural observations of extracellular tracer uptake at the neuromuscular junction (NMJ) (Heuser and Reese, 1973). As the molecular identity of synaptic vesicles became more defined in the last decade, the question of how specific synaptic vesicle proteins are recycled, and in particular how they are resorted into synaptic vesicles, has become particularly interesting. One possibility is that once a synaptic vesicle is made, it is simply reused repeatedly without loss of molecular identity, as would potentially occur in a scenario where kiss-and-run predominates. However, recent experiments demonstrated that removal of clathrin severely perturbs endocytosis in both rodent CNS synapses (Granseth et al., 2006) and Drosophila NMJs (Heerssen et al., 2008), suggesting the majority of vesicle retrieval occurs through a clathrin-mediated mechanism. Additionally, we and others have shown that during vesicle recycling both VAMP-2 (Fernández-Alfonso and Ryan, 2006; Wienisch and Klingauf, 2006) and synaptotagmin I (Fernández-Alfonso et al., 2006) exchange with counterparts residing on the cell surface during recycling. Thus, the question of how SV cargo proteins are recognized and resorted into endocytic vesicles is of significant interest. Mutations in both vGlut1 (Voglmaier et al., 2006) as well as synaptotagmin I (Poskanzer et al., 2006; Jung et al., 2007) lead to endocytic defects, which in the latter case is thought to be mediated through the adaptor-like protein stonin 2.
Here, we have addressed the question of whether the classical clathrin adaptor protein complex AP-2 functions during synaptic vesicle recycling. At the surface of most cells, AP-2 is thought to act as a key molecular bridge, interacting with the cytoplasmic tails of cell surface transmembrane proteins as well as the molecular machinery of the clathrin coat, thus helping to concentrate specific cargo molecules into endocytic pits (Bonifacino and Traub, 2003). Additionally, because of the plethora of interactions between AP-2 and proteins generally considered to be part of the endocytic machinery, AP-2 has also been described as a “hub” protein central to clathrin-mediated endocytosis (Schmid and McMahon, 2007; Rodemer and Haucke, 2008). Our analysis shows that for all stimulation conditions tested, a drastic reduction in the amount of AP-2 present in neurons significantly perturbs the kinetics of endocytosis of at least four SV proteins at nerve terminals. Given the fact that AP-2 is only known to participate in clathrin-mediated endocytosis, this is consistent with the idea that synaptic vesicle recycling in this system is mediated via clathrin. However, the reduction of endocytosis kinetics of all four of the SV proteins tested suggests that AP-2's role is not likely to be solely one of cargo recognition, especially because endocytosis persists even when AP-2 levels have been reduced to ∼1/25th (or ∼4%) of normal levels. The loss of AP-2, however, led to unexpected changes in the nature of endocytosis at these nerve terminals. Under control conditions, the kinetics of endocytosis appears to be limited by a single rate-determining step (Balaji and Ryan, 2007), and the distribution of endocytosis time constants across individual synapses shows limited variability (Fig. 6E). In the absence of AP-2, however, single bouton analysis revealed significant heterogeneity in endocytic behavior, with the majority showing two distinct kinetic components. Although the simplest interpretation would be to assign each component to a different molecular pathway, our double knockdown and BFA experiments revealed that both components appear to rely on the function of AP-1, an adaptor complex that does not normally function in endocytosis at the plasma membrane. Our experiments do not attempt to understand the normal function, if any, of AP-1 at nerve terminals. AP-1 is generally thought to participate in membrane traffic to or from early endosomes. It is possible that AP-1 is normally present to support cargo sorting at synaptic intermediate endosomes (either as part of synaptic vesicle recycling or other membrane trafficking events), and in the absence of AP-2, it associates with the plasma membrane and clathrin-coat complexes. Alternatively, AP-1 could be recruited from nonsynaptic sites in the absence of AP-2. The heterogeneity we observed across boutons suggests that the ability of AP-1 to substitute in AP-2's absence varies from synapse to synapse.
Previous experiments have implied that sensitivity to BFA at nerve terminals was likely mediated by the adaptor complex AP-3. AP-3 was originally implicated in synaptic vesicle biogenesis in PC-12 cells, which is sensitive to BFA (Faúndez et al., 1998; Blumstein et al., 2001). Genetic ablation of the neuronal specific isoform of AP-3 results in specific GABAergic synaptic transmission deficits (Nakatsu et al., 2004), whereas mocha mice that lack both ubiquitous and neuronal forms of AP-3 have a number of synaptic transmission abnormalities (Kantheti et al., 1998; Scheuber et al., 2006; Voglmaier et al., 2006), which may result from improper export of synaptic vesicle proteins from the cell soma (Danglot and Galli, 2007). It remains to be seen, however, if the AP-3 pathway is the sole target of BFA at nerve terminals, because the sensitivity of either mocha mice or the neuronal AP-3 knockout to BFA treatment has not been reported. In our experiments, we are only sensitive to disruption by BFA in the absence of AP-2. Although our experiments indicate that functional substitution by AP-1 can account for all our data, it does not rule out an additional possible role for AP-3.
Our experiments reveal a significant defect in endocytosis kinetics when AP-2 levels have been decreased to at most ∼4% of control levels. Although it is possible that this residual AP-2 could be sufficient to drive the remaining endocytic function, the fact that the kinetics of endocytosis under these conditions are relatively insensitive to the degree of stimulation argues against this interpretation. The lowest stimulus level for which we could reliably detect pHluorin signals in these experiments was for 25 action potentials, and similar phenotypes were observed at 300 AP. This corresponds to exocytosis of ∼13 and ∼65% of the recycling pool, respectively (see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Thus, to explain the data based on residual levels of AP-2, one would have to posit that it is capable of handling an ∼5-fold range of exocytosis with similar kinetics.
One can interpret continued function of endocytosis after the loss of a AP-2 in one of three ways. The first is that AP-2 has no role in endocytosis of SV components. However, given that endocytosis kinetics were impacted by the loss of AP-2, this can be ruled out. The second interpretation is that in AP-2's absence, compensation by another mechanism, which does not normally operate, accounts for the residual functionality. Here, AP-1 appears to be doing this, and although we have managed to eliminate AP-1's ability to perform this compensation, we cannot rule out that other such mechanisms are taking place accounting for continued endocytosis in the absence of both AP-1 and AP-2. The third interpretation is that AP-2 was not originally acting alone in the sorting of SV cargo proteins, and AP-2's removal left residual functionality via other mechanisms that normally operate in conjunction with AP-2. One such candidate is stonin 2, a protein with homology to μ2 that has been shown to interact genetically with synaptotagmin and can control its cell surface localization (Jung et al., 2007). In the future, it will be important to understand how the hierarchy and network of possible interactions of SV cargo proteins with the endocytic machinery leads to efficient and presumably stoichiometrically accurate synaptic vesicle identity and to identify other possible compensatory pathways.
Note added in proof.
Since the submission of this paper, a complementary study examining neuronal function after loss of the μ2 subunit in Caenorhabditis elegans was published (Gu et al., 2008).
Footnotes
This work was supported by grants from the National Institutes of Health (T.A.R.). The vGlut–pHluorin construct was kindly provided by Susan Voglmeier and Robert Edwards (University of California, San Francisco), the synaptophysin–pHluorin construct was kindly provided by Yongling Zhu (Salk Institute), and the synapto–pHluorin and synaptotagmin–pHluorin constructs were kindly provided by James Rothman (Yale University). The μ2 construct was a gift from Alexander Sorkin (University of Colorado). We thank members of the Ryan laboratory as well as Jeremy Dittman and Tim McGraw for useful discussions and Ricky Kwan for excellent technical support.
References
- Atluri PP, Ryan TA. The kinetics of synaptic vesicle reacidification at hippocampal nerve terminals. J Neurosci. 2006;26:2313–2320. doi: 10.1523/JNEUROSCI.4425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Morgan JR, Villalba-Galea CA, Jin S, Prasad K, Lafer EM. Clathrin and synaptic vesicle endocytosis: studies at the squid giant synapse. Biochem Soc Trans. 2006;34:68–72. doi: 10.1042/BST0340068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji J, Ryan TA. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc Natl Acad Sci U S A. 2007;104:20576–20581. doi: 10.1073/pnas.0707574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji J, Armbruster M, Ryan TA. Calcium control of endocytic capacity at a CNS synapse. J Neurosci. 2008;28:6742–6749. doi: 10.1523/JNEUROSCI.1082-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumstein J, Faundez V, Nakatsu F, Saito T, Ohno H, Kelly RB. The neuronal form of adaptor protein-3 is required for synaptic vesicle formation from endosomes. J Neurosci. 2001;21:8034–8042. doi: 10.1523/JNEUROSCI.21-20-08034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Danglot L, Galli T. What is the function of neuronal AP-3? Biol Cell. 2007;99:349–361. doi: 10.1042/BC20070029. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A. 2006;103:11399–11404. doi: 10.1073/pnas.0600784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Drake MT, Zhu Y, Kornfeld S. The assembly of AP-3 adaptor complex-containing clathrin-coated vesicles on synthetic liposomes. Mol Biol Cell. 2000;11:3723–3736. doi: 10.1091/mbc.11.11.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faúndez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93:423–432. doi: 10.1016/s0092-8674(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, O'toole E, Flavell R, Cremona O, Miesenböck G, Ryan TA, De Camilli P. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Fernández-Alfonso T, Ryan TA. The efficiency of the synaptic vesicle cycle at central nervous system synapses. Trends Cell Biol. 2006;16:413–420. doi: 10.1016/j.tcb.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Fernández-Alfonso T, Kwan R, Ryan TA. Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron. 2006;51:179–186. doi: 10.1016/j.neuron.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem. 1988;263:18545–18552. [PubMed] [Google Scholar]
- Gad H, Ringstad N, Löw P, Kjaerulff O, Gustafsson J, Wenk M, Di Paolo G, Nemoto Y, Crun J, Ellisman MH, De Camilli P, Shupliakov O, Brodin L. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron. 2000;27:301–312. doi: 10.1016/s0896-6273(00)00038-6. [DOI] [PubMed] [Google Scholar]
- González-Gaitán M, Jäckle H. Role of Drosophila alpha-adaptin in presynaptic vesicle recycling. Cell. 1997;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Gu M, Schuske K, Watanabe S, Liu Q, Baum P, Garriga G, Jorgensen EM. Mu2 adaptin facilitates but is not essential for synaptic vesicle recycling in Caenorhabditis elegans. J Cell Biol. 2008;183:881–892. doi: 10.1083/jcb.200806088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V, De Camilli P. AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science. 1999;285:1268–1271. doi: 10.1126/science.285.5431.1268. [DOI] [PubMed] [Google Scholar]
- Haucke V, Wenk MR, Chapman ER, Farsad K, De Camilli P. Dual interaction of synaptotagmin with mu2- and alpha-adaptin facilitates clathrin-coated pit nucleation. EMBO J. 2000;19:6011–6019. doi: 10.1093/emboj/19.22.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen H, Fetter RD, Davis GW. Clathrin dependence of synaptic-vesicle formation at the Drosophila neuromuscular junction. Curr Biol. 2008;18:401–409. doi: 10.1016/j.cub.2008.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature. 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Cooke A, Wu MM, Lagnado L. Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J Neurosci. 2003;23:1329–1339. doi: 10.1523/JNEUROSCI.23-04-01329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis by brefeldin A in MDCK cells. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Jung N, Wienisch M, Gu M, Rand JB, Müller SL, Krause G, Jorgensen EM, Klingauf J, Haucke V. Molecular basis of synaptic vesicle cargo recognition by the endocytic sorting adaptor stonin 2. J Cell Biol. 2007;179:1497–1510. doi: 10.1083/jcb.200708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Liang JO, Kornfeld S. Comparative activity of ADP-ribosylation factor family members in the early steps of coated vesicle formation on rat liver Golgi membranes. J Biol Chem. 1997;272:4141–4148. doi: 10.1074/jbc.272.7.4141. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, Ryan TA. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron. 2007;56:1004–1018. doi: 10.1016/j.neuron.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunari T, Nakatsu F, Shioda N, Love PE, Grinberg A, Bonifacino JS, Ohno H. Clathrin adaptor AP-2 is essential for early embryonal development. Mol Cell Biol. 2005;25:9318–9323. doi: 10.1128/MCB.25.21.9318-9323.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F, Okada M, Mori F, Kumazawa N, Iwasa H, Zhu G, Kasagi Y, Kamiya H, Harada A, Nishimura K, Takeuchi A, Miyazaki T, Watanabe M, Yuasa S, Manabe T, Wakabayashi K, Kaneko S, Saito T, Ohno H. Defective function of GABA-containing synaptic vesicles in mice lacking the AP-3B clathrin adaptor. J Cell Biol. 2004;167:293–302. doi: 10.1083/jcb.200405032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A, Crottet P, Prescianotto-Baschong C, Spiess M. In vitro formation of recycling vesicles from endosomes requires adaptor protein-1/clathrin and is regulated by rab4 and the connector rabaptin-5. Mol Biol Cell. 2004;15:4990–5000. doi: 10.1091/mbc.E04-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, Fetter RD, Davis GW. Discrete residues in the c(2)b domain of synaptotagmin I independently specify endocytic rate and synaptic vesicle size. Neuron. 2006;50:49–62. doi: 10.1016/j.neuron.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Rodemer C, Haucke V. Clathrin/AP-2-dependent endocytosis: a novel playground for the pharmacological toolbox? Handb Exp Pharmacol. 2008;(186):105–122. doi: 10.1007/978-3-540-72843-6_5. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–135. doi: 10.1038/nn1002. [DOI] [PubMed] [Google Scholar]
- Scheuber A, Rudge R, Danglot L, Raposo G, Binz T, Poncer JC, Galli T. Loss of AP-3 function affects spontaneous and evoked release at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2006;103:16562–16567. doi: 10.1073/pnas.0603511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid EM, McMahon HT. Integrating molecular and network biology to decode endocytosis. Nature. 2007;448:883–888. doi: 10.1038/nature06031. [DOI] [PubMed] [Google Scholar]
- Shim J, Lee J. Molecular genetic analysis of apm-2 and aps-2, genes encoding the medium and small chains of the AP-2 clathrin-associated protein complex in the nematode Caenorhabditis elegans. Mol Cells. 2000;10:309–316. [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wienisch M, Klingauf J. Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat Neurosci. 2006;9:1019–1027. doi: 10.1038/nn1739. [DOI] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- Wu LG, Ryan TA, Lagnado L. Modes of vesicle retrieval at ribbon synapses, calyx-type synapses, and small central synapses. J Neurosci. 2007;27:11793–11802. doi: 10.1523/JNEUROSCI.3471-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Davletov BA, Südhof TC, Anderson RG. Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 1994;78:751–760. doi: 10.1016/s0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]