

Abstract
Introduction
Bromine-76 radiolabeled analogues of previously reported high affinity A3 adenosine receptor (A3AR) nucleoside ligands have been prepared as potential radiotracers for Positron Emission Tomography (PET).
Methods
The radiosyntheses were accomplished by oxidative radiobromination on the N6-benzyl moiety of trimethyltin precursors. Biodistribution studies of the kinetics of uptake were conducted in awake rats.
Results
We prepared an agonist ligand {[76Br](1′R,2′R,3′S,4′R,5′S)-4-{2-chloro-6-[(3-bromophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol (MRS3581)} in 59% radiochemical yield (RCY) with a specific activity of 19.5 GBq/μmol and an antagonist ligand {[76Br](1R,2R,3S,4R,5S)-4-(6-(3-bromobenzylamino)-2-chloro-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol. (MRS5147)} in 65% RCY with a specific activity of 22 GBq/μmol). The resultant products exhibited the expected high affinity (Ki ~ 0.6 nM) and specific binding at the human A3AR in vitro. Biodistribution studies in the rat showed uptake in the organs of excretion and metabolism. The antagonist MRS5147 exhibited increasing uptake in testes, an organ that contains significant quantities of A3AR, over a 2 h time course, which suggests the presence of a specific A3AR retention mechanism.
Conclusion
We were able to compare uptake of the [76Br]labeled antagonist MRS5147 to [76Br]agonist MRS3581. The antagonist MRS5147 shows increasing uptake in the testes, an A3AR rich tissue, suggesting that this ligand may have promise as a molecular imaging agent.
Keywords: Adenosine A3 receptor, G protein-coupled receptor, nucleoside, purines, receptor binding, Bromine-76
Introduction
The study of adenosine receptors (ARs) has revealed four subtypes: A1, A2A, A2B, and A3.[1] Each of these receptors is a target for therapeutic intervention in various human diseases including asthma, autoimmune inflammatory diseases, cardiovascular diseases, and cancer.[2] The A3AR is expressed at high levels in neutrophils in inflamed tissue[3],[4] and is detectable in certain cancerous tumors, such as human colon carcinoma and melanoma.[5],[6] The presence of the A3AR in these diseases suggest the potential utility of a radiolabeled A3AR ligand for diagnostic applications using molecular imaging techniques. In addition, the A3AR is found in low amounts in rat brain and in higher density in rat testes (based on mRNA presence).[7] We have developed potent, selective agonist ligands of the A3AR for application in inflammatory diseases[8],[9],[10],[11], myeloprotection during chemotherapy,[12] and cancer therapy.[13],[14],[15],[16] The selective A3AR agonist CP-608039 ((2S,3S,4R,5R)-3-amino-5-{6-[5-chloro-2-(3-methylisoxazol-5-ylmethoxy)benzylamino]purin-9-yl]}-4-hydroxytetrahydrofuran-2-carboxylic acid methylamide) was under development as a cardioprotective agent.[17] A3AR agonists are also of interest for the potential treatment of brain ischemia.[18] The first prototypical A3AR agonist IB-MECA [19], compound 1 (Chart 1), is under development for autoimmune inflammatory diseases [8] and was found to be protective by histological criteria and to correct the gene dysregulation in a model of chemically-induced colitis.[20]
Chart 1.

Structures of prototypical (1,2) and later-generation A3AR agonists (3–5) and antagonist (6) based on the (N)-methanocarba ring system.
Positron emission tomography (PET) is a powerful in vivo imaging technique, which utilizes the sensitive detection of tracer levels of receptor-bound ligands that are labeled with an appropriate positron-emitting radionuclide. PET has been applied to the imaging of inflamed tissue via the uptake of radioligands for peripheral benzodiazepine receptors, which are present in monocytes and neutrophils.[21] PET ligands of high affinity and selectivity have also been developed for the A1 and A2AARs [22],[23]. For example, the A1-selective xanthine antagonist [18F]CPFPX and the A2A-selective nonxanthine antagonist [11C]SCH442416 have been developed as centrally-active PET tracers for the imaging ARs in the brain.
The prototypical agonist ligands for the A3AR are IB-MECA 1 (Chart 1) and its 2-chloro analogue 2.[24] These agonists have entered phase II clinical trials for treatment of rheumatoid arthritis and lung cancer, respectively. Structure activity relationship (SAR) studies of nucleoside derivatives with respect to A3AR affinity have progressed from the native ribose ring to its replacement in nucleoside analogues with the (N)-methanocarba (bicyclo[3.1.0]hexane) system. This pseudosugar provides a conformationally rigid ring system that was observed to enhance the human A3AR receptor selectivity over other AR subtypes.[25] A series of doubly halogenated agonist ligands, i.e. compounds 3–5, exhibited nanomolar or sub-nanomolar affinity for both human and rat A3ARs and selectivity for this subtype. The cytoprotective properties of these highly selective (N)-methanocarba A3AR agonists is illustrated in a recent study of protection in a model of traumatic lung injury in the cat that applied compound 3 in vivo[26]. One of these unlabeled compounds, (1′R, 2′R, 3′S, 4′R, 5′S)-4-{2-chloro-6-[(3-bromophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)-bicyclo[3.1.0]hexane-2,3-diol (MRS3581) 4, contains an aromatic bromine atom that we envisioned could be easily labeled with a positron-emitting radionuclide, 76Br (t ½ = 16.2 h).
We had previously prepared two classes of A3AR antagonists that bind potently in rat tissue as well as human tissue. A pyridine class of antagonists included 5-(2-fluoroethyl) 2,4-diethyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate.[27] During the course of our work, the fluorine-18 labeling of this compound was reported.[28] The second class contains structures closely related to the agonist above but lacking the carboxylic methylamide [30]. This carboxylic methyl amide moiety, with its potential as an H-bond donor, was found to be crucial for activation of A3AR [29]. It is this class of 4′-truncated nucleosides, which tend to bind with high affinity to both human and rat A3ARs, from which we selected (1R,2R,3S,4R,5S)-4-(6-(3-bromobenzylamino)-2-chloro-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (6, MRS5147) (6) as our target for radiolabeling.
We set out to develop an A3AR-specific ligand of high affinity useful for PET. This manuscript describes the radiochemical synthesis of two [76Br]-labeled A3AR ligands of closely related structure; one nucleoside from the agonist class and an analogous 4′-truncated nucleoside from the antagonist class. We also conducted preliminary in vitro tissue binding assays and in vivo biodistribution studies in normal rats.
Materials and Methods
General Experimental
1H-NMR spectra were obtained with a Varian Gemini-300 spectrometer (300 MHz) with CDCl3 as solvent. Low-resolution mass spectra were measured with a Finnigan-Thermoquest LCQ with APCI (Atmospheric Pressure Chemical Ionization) interface. Low-resolution and high-resolution FAB (fast atom bombardment) mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix. MRS1523 was purchased from Sigma Aldrich (St. Louis, MO, USA). Authentic (1′R,2′R,3′S,4′R,5′S)-4-{2-chloro-6-[(3-bromophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol (4, MRS3581) and (1R,2R,3S,4R,5S)-4-(6-(3-bromobenzylamino)-2-chloro-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol. (6, MRS5147) were previously prepared in our laboratories.[25], [29]
(1′R,2′R,3′S,4′R,5′S)-4-{2-chloro-6-[(3-(trimethylstannyl)phenylmethyl)-amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol (9)
Compound 5 [25] (0.018 g, 0.033 mmol), PdCl2(PPh3)2 (5 mg), and hexamethyltin (0.032 g, 0.1 mmol) were mixed together in anhydrous dioxane (3 mL), and the resulting reaction mixture was stirred at 70°C for 2 h. The mixture was concentrated under reduced pressure. The purification of the product was effected by PTLC by using CHCl3: MeOH as the eluant to afford the stannyl derivative 9 (0.008 g, 45%) as an oil. 1H NMR (CDCl3, 300 MHz) δ7.79 (s, 1H), 7.51 (s, 1H), 7.44 (t, 1H, J = 4.5 Hz), 7.33 (m, 2H), 6.08 (m, 1H), 6.37 (m, 1H), 5.00 (t, 1H, J = 7.8 Hz), 4.83 (s, 1H), 4.77 (m, 1H), 4.38 (m, 1H), 4.13 (m, 1H), 3.66 (m, 1H), 3.43 (m, 1H), 2.92 (3H, d, J = 5.1 Hz), 2.18 (m, 1H), 2.00 (s, 1H), 1.69 (t, 1H, J = 4.8 Hz), 1.37 (m, 1H),1.26 (m, 1H), 0.29 (s, 9H). HRMS (M + 1)+: calculated 593.1090, found 593.1099. HPLC: Rt = 21.95 min. HPLC system: 5 mM TBAP/CH3CN from 80/20 to 60/40 in 25 min, then isocratic for 2 min; flow rate of 1 mL/min.
[76Br](1′R,2′R,3′S,4′R,5′S)-4-{2-chloro-6-[(3-bromophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol ([76Br]4, MRS3581)
Bromine-76 was prepared from an arsenic metal target using the 75As (3He, 2n) 76Br nuclear reaction. The Br-76 target was processed as previously described after allowing for the decay of the simultaneously produced Br-75 (t ½ = 1.6 h).[30],[31] An aliquot of the aqueous solution of Br-76 (10–20 μL, 18.5–37.0 MBq) was added to a 1 mL reaction vial and the solvent evaporated with argon flow. Trimethyl tin substrate 9 (200 μg) in 50 μL of acetonitrile was added to the vial containing the Br-76 radioactivity and followed by adding 2 μL of 37% peracetic acid in 10 μL of acetonitrile. The vial was sealed and placed on an 80°C heating block and heated for 30 min. At the end the reaction, the reaction mixture was loaded on to a Phenomenex Luna C18 (2) column (250 × 4.6 mm) and eluted with 100 mM ammonium acetate/acetonitrile (60/40) at the flow rate of 1.2 mL/min. The radioactivity peak containing the desired product (tR = 10 min) was collected and analyzed on a separate HPLC system for determination of purity and specific activity.
Preparation of 8: (1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(3-trimethylstannylbenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane (8)
Compound 7 (8.95 mg, 0.018 mmol, prepared as described [28]), PdCl2(PPh3)2 (2.7 mg), and hexamethyltin (11 μL, 0.054 mmol) were mixed together in anhydrous dioxane (2 mL), and the resulting reaction mixture was stirred at 70°C for 2 h. The mixture was concentrated under reduced pressure. The product was purified by flash chromatography by using CHCl3: MeOH (10:1) as the eluant to afford the stannyl derivative 8 (9.3 mg, 90%) as an oil. 1H NMR (300 MHz, CDCl3), 7.81 (s, 1H), 7.53 (s, 1H), 7.34 (m, 2H), 7.33 (m, 1H), 6.49 (br s, 1H), 4.88 (br s, 2H), 4.00 (m, 2H), 3.71 (s, 1H), 3.65 (m, 1H), 3.47 (m, 1H), 2.02 (m, 1H), 1.96 (s, 1H), 1.64 (m, 1H), 1.28 (m, 2H), 0.81 (m, 1H), 0.29 (s, 9H). HRMS (M + 1)+: calculated for C21H27ClIN5O2Sn+ (M+H)+ 535.6338, found 536.0823 HPLC: Rt = 22.12 min. HPLC system: 5 mM TBAP/CH3CN from 80/20 to 60/40 in 25 min, then isocratic for 2 min; flow rate of 1 mL/min.
[76Br](1R,2R,3S,4R,5S)-4-(6-(3-bromobenzylamino)-2-chloro-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol. ([76Br]6, MRS5147)
The same radiobromination procedure was used on trimethylstannane 8 to prepare [76Br]6. The semipreparative HPLC utilized the mobile phase 100 mM NH4OAc/CH3CN (55/45) and the product eluted at 13 min. Specific activity was determined on a separate HPLC under the same chromatographic conditions.
Radioligand binding assays
Membrane preparations
CHO (Chinese hamster ovary) cells expressing the recombinant human or rat A3R were cultured in DMEM (Dulbecco’s modified Eagle’s medium) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 Units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. Cells were harvested by trypsinization. After homogenization and suspension, cells were centrifuged at 500 g for 10 min, and the pellet was re-suspended in 50 mM Tris·HCl buffer (pH 7.4) containing 10 mM MgCl2. The suspension was homogenized with an electric homogenizer for 10 sec, and was then re-centrifuged at 20,000 g for 20 min at 4°C. The resultant pellets were resuspended in buffer containing 3 Units/mL adenosine deaminase, and the suspension was stored at −80°C until the binding experiments. The protein concentration was measured using the Bradford assay.[32]
Binding Assay at the A3AR
Each tube in the competitive binding assay contained 100 μL membrane suspension (20 μg protein), 50 μL [76Br] radioligand 0.5 nM), and 50 μL of increasing concentrations of the test ligands in Tris·HCl buffer (50 mM, pH 7.4) containing 10 mM MgCl2, 1 mM EDTA. Nonspecific binding was determined using 10 μM of 5′-N-ethylcarboxamidoadenosine in the buffer. The mixtures were incubated at 25°C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburg, MD, USA). Filters were washed three times with 9 mL ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. The concentration of [76Br]4 in the competition experiments (0.5 nM) is selected based on its Ki value (0.39 nM) shown previously [25]. At the concentration used, over 60–70% of the total binding was shown to be A3AR specific binding. IC50 values were converted to Ki values as reported.[33] It should be noted that the experimental conditions used in the current assay were similar to those used with the agonist radioligand ([125I]I-AB-MECA). Thus, the ratio of the specific to nonspecific binding could potentially be further improved by optimizing the assay conditions.
In vitro metabolism studies of [76Br]4 in hepatocytes
The cryopreserved hepatocytes from male Sprague Dawley rats and human liver tissue (In Vitro Technologies, Inc., Baltimore, MD) were used in the in vitro metabolism studies. The cells, which were stored in liquid nitrogen, were thawed rapidly at 37°C in a water bath and gradually diluted with cell culture medium (In Vitro Technologies, Inc., Baltimore, MD). After washing the cells with the medium and adjusting the viable cell concentration to 1.0 million per mL, the resulting cell suspension was incubated at 37°C for 15 min prior to the introduction of the test compound. The radioligand [76Br]4 (ca. 370 kBq in 10μl ethanol) and 10 μL unlabeled compound (from a stock solution of 2.0mg/mL in 10% aqueous EtOH) was added to a 1.0 mL suspension of cells. The final concentration of test compound was 20 μg/mL. The suspension was maintained at 37°C; 100 μl of cell suspension was removed and added to 100 μl acetonitrile at 10, 30, 60, 120, 240 min. Each aliquot was centrifuged at 5000 rpm for 5 min. The metabolites in 20 μL supernatant were analyzed by LC-MS and LC with on-line radioactivity detection.
In vitro tissue binding – rat brain and rat testes
All studies using live animals were conducted under protocol approved by the NIH Animal Care and Use Committee. Sprague-Dawley rats were used for the in vitro autoradiography. After sacrificing the rat with carbon dioxide inhalation, the brains and testes were removed and immediately quick-frozen in dry ice. The brain was cut coronally and testes along the long axis into 20 μm slices using a Vibratome Ultrapro 5000(Vibratome, St. Louis, MO, USA). The slices were thaw mounted onto silanated slides, air-dried for 30 min, and then stored at −70 °C until use.. The following solutions were prepared in buffer (50 mM TRIS pH 7.5 and 10 mM MgCl2). One solution contained 37 kBq of [76Br]4 or [76Br]6. For blocking studies, solutions of the antagonist MRS1523 (200 nM) and agonist IB-MECA (100 nM) were prepared in the same buffer, each solution containing 37 kBq of [76Br]4 or [76Br]6. The tissue sections were divided into three groups and each group was incubated in one solution at room temperature for one hour. After incubation, the slides were washed at 4 °C with 1 X PBS with 0.01% Triton X, air dried, and placed on a phosphorimaging plate with a pixel size of 25 μm (Fuji BAS-SR2025). After exposure, the plates were scanned using a Fuji Bio-imaging Analysis System 5000.
In vivo biodistribution study in rats
In the biodistribution studies the Sprague-Dawley male rats (n = 4 or 5) were injected intravenously with 0.37 MBq of 76Br radioligand ([76Br]4 or [76Br]6) and the rats were sacrificed with carbon dioxide at 15, 60, and 120 min. For compound [76Br]4, a 30 min time point was included. The brain, blood and other tissues were taken from each animal and weighed. The radioactive content of the blood and various tissues was counted using a gamma counter. The uptake of the radioactivity was expressed as %ID/g.
Results and Discussion
During the development of PET imaging agents for receptor imaging, one may encounter the choice of agonist or antagonist. The majority of neuroreceptor imaging agents are antagonists. Many G protein-coupled receptors exhibit both high and low affinity states for agonist ligands, while antagonists generally bind with equal affinity to either conformation. The availability of both an agonist and an antagonist for a given receptor may provide Bmax and receptor occupancy more accurately than only one of the ligands may provide. There is ample evidence from our previous publications to suggest that the nucleoside-based agonists and antagonists of the A3AR are competitive inhibitors of each other and thus bind to the same site on the receptor.[34], We had the ability to prepare one agonist and one antagonist radioligand in closely related chemical series. The two structures differ only in the chemical moiety attached at the C-4′ position of the bicyclohexanediol ring.
Chemistry
For the agonist class of compounds, the authentic bromine compound (4) had been prepared for our previous study of the structure activity relationships (SAR).[25] Although several analogues containing fluorine substitution of the N6-benzyl moiety were prepared, all were less potent in activating the A3AR.[25] Because the most active fluorine-containing analogue had a meta configuration, it would not be easily radiolabeled. The 3-bromobenzyl analogue 4 (MRS3581) displayed favorable pharmacological properties, with a Ki value of 0.38 nM at the human A3AR and high selectivity in comparison to other AR subtypes. Therefore, we selected [76Br]4 for development as an agonist PET radioligand employing the positron-emitting bromine-76 radionuclide (t ½ = 16.2 h). Radiobromination can be conducted under electrophilic conditions on an aryl stannane analogue. We prepared the N6-(3-trimethylstannyl benzyl) nucleoside derivative 9 from the corresponding 3-iodo compound 5 by employing standard conditions, using hexamethylditin and dichlorobis(triphenylphosphine) palladium(II) (Scheme 1). Initially, attempts to stannylate the 2′, 3′-isopropylidene-protected form of the nucleoside succeeded. However, the subsequent hydrolysis of the isoproylidene group also led to removal of the stannyl group. For this reason, we introduced the aryltin moiety at the final stage on the preformed and unprotected (N)-methanocarba nucleoside analogue.
Scheme 1.

Synthesis of [76Br] A3AR ligands, agonist [76Br]4 and antagonist [76Br]6, and their stannyl precursors 9 and 8, respectively.
The target antagonist compound, 6, was selected from the structural class lacking the 4′-carbonylmethylamide moiety on the pseudosugar ring [29]. Because the structures are very similar, the chemistry for the preparation of trimethylstannyl precursor (8) for radiobromination was conducted in a similar manner.
Radiochemistry
Radiobromination to obtain the two radiotracer molecules was conducted in the same manner. Bromine-76 was obtained as an aqueous ammonia solution following irradiation of a natural arsenic target.[31] An aliquot of the radiobromine solution was evaporated with argon flow prior to the addition of the trimethylstannyl precursor and peracetic acid (Scheme 1). The evaporation of water prior to addition of substrate and oxidizing agent was critical for achieving the high radiolabeling yield. When the water is present in the reaction mixture, the amount of two unidentified radioactive side products was greatly increased. The final radiochemical was purified by HPLC. The agonist [76Br]4 was obtained in 58.7 ± 16.8% (n=8) radiochemical yield, radiochemical purity of > 98%, and a specific activity of 19.5 ± 7.9 GBq/μmol (n=5) at the end of synthesis. The antagonist [76Br]6 was obtained in 65.5 ± 3.6% (n = 6) radiochemical yield, >98% radiochemical purity, and specific activity of 22 ± 20 GBq/μmol (n =5) at the end of synthesis.
In vitro studies
Membrane Binding Experiments
Binding studies of agonist [76Br]4 and antagonist [76Br]6 to membranes prepared from CHO cells stably expressing the human A3AR were conducted.[35] In competition experiments, both agonists and antagonists showed a binding profile similar to that obtained with other radioligands as demonstrated previously. [1] Briefly, as shown in Figures 1A and 1B, MRS1220 (N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide), a selective antagonist for the human A3AR was shown to be ineffective at rat A3AR. MRS1523 (3-propyl-6-ethyl-5-[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine carboxylate), an A3AR-selective antagonist, and NECA (adenosine-5′-N-ethyluronamide), a nonselective AR agonist, were moderately potent in binding to the rat A3AR, and each was more potent at the human A3AR. 4 (MRS3581) is equally potent at human and rat A3ARs, suggesting that its use in imaging might apply to a variety of animal models. The Ki values of the tested compounds are summarized in Tables 1 and 2.
Figure 1.


Panel A: Inhibition of in vitro binding of the agonist [76Br]4 by various A3AR ligands. Panel B: Inhibition of in vitro binding of antagonist [76Br]6 by various A3AR ligands. The human or rat A3AR receptor was expressed in CHO cells. See legend to Table 1 for compound names.
Table 1.
Affinity of various competing ligands at the human and rat A3ARs expressed in CHO cells using [76Br]4 as radioligand.
| Compounda | Human Ki (nM) | Rat Ki (nM) |
|---|---|---|
| MRS1220 | 2.74 ± 0.77 | >10,000 |
| MRS1523 | 125 ± 13 | 939 ± 98 |
| 4 MRS3581 | 0.63 ± 0.05 | 0.90 ± 0.07 |
| NECA | 20.1 ± 2.5 | 143 ± 18 |
Table 2.
Affinity of competing ligands at human and rat A3ARs expressed in CHO cells using [76Br]MRS5147 6.
| Ligand | Human Ki, nM | Rat Ki, nM |
|---|---|---|
| MRS1220 | 1.1 ± 0.3 | >10,000 |
| MRS1523 | 120 ± 17 | 2830 ± 520 |
| 6 MRS5147 | 0.62 ± 0.16 | 5.2 ± 1.8 |
| NECA | 38 ± 12 | 126 ± 31 |
Metabolism studies
Knowledge of the metabolic fate of the radiolabeled compound is very important for the evaluation of a molecular imaging agent. The imaging technique records the radioactive decay events regardless of the actual chemical species. Applications to brain imaging have the advantage that polar metabolites are generally excluded from the brain because of the blood brain barrier. However, the potential applications of these A3AR ligands are in inflammation and tumor imaging where no barrier to metabolite accumulation is present. Thus, it is most important that the metabolite profile be well characterized and properly evaluated during the course of validation of the radioligand. Incubation of the agonist [76Br]4 with rat and human hepatocytes at various time points and then analysis of the radioactive and non-radioactive components with LC/MS were used to evaluate the metabolic fate of the compound. The results indicated that compound [76Br]4 is very stable in both rat and human hepatocytes. After 4 hours, only very small amounts of metabolites were detected by LC/MS (data not shown).
In vitro tissue slice studies
The rat testes have the highest concentration of the A3AR,[36],[37] and low levels are present in the brain.[7] We conducted dipping studies with freshly prepared slices of whole brain and testes comparing [76Br]4 alone and competed with MRS1523, a selective A3AR antagonist. We also competed with IB-MECA, an agonist. These preliminary studies fail to show any specific uptake in these tissues as neither compound reduced the tissue binding of [76Br]4. Similar studies with the antagonist, [76Br]6, using IB-MECA or MRS5147 (6) as inhibitors also failed to show evidence of specific uptake.
In vivo studies with agonist [76Br]4
In order to evaluate the kinetics of uptake and selective tissue accumulation, we conducted in vivo biodistribution studies in rats. All studies in live animals were conducted under protocol approved by the NIH Animal Care and Use Committee. The biodistribution of [76Br]4 was evaluated after i.v. administration to adult Sprague-Dawley rats. The animals were sacrificed at 15, 30, 60, and 120 min and various tissues harvested for gamma counting. The data are reported in units of percentage injected dose per gram (%ID/g) (Figure 2). An additional study, designed to detect specific tissue accumulation, was conducted with co-administration of the antagonist MRS1523 (Figure 3).
Figure 2.
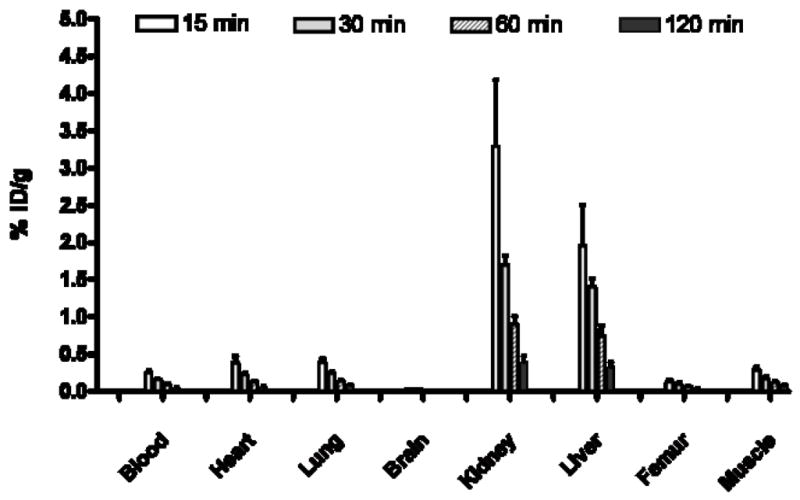
In vivo uptake in rat of A3AR agonist [76Br]4 at 15, 30, 60, and 120 min post injection in various tissues.
Figure 3.
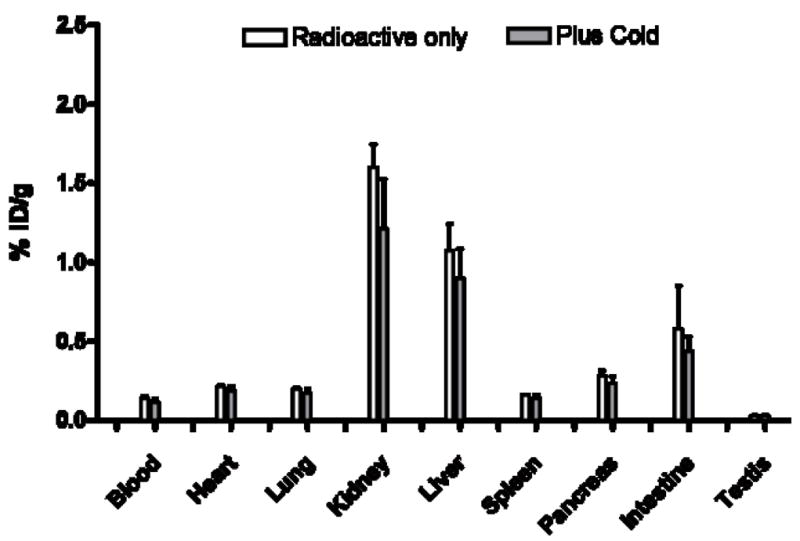
Tissue uptake of [76Br]4 without and with MRS1523, an antagonist, administered at 30 min post-injection in rat.
The biodistribution studies showed the primary uptake to be in liver and kidneys, the organs of excretion and metabolism. The uptake observed on competition with administration of excess non-radioactive ligand showed a trend suggesting some specific binding in the liver and kidney. However, the differences were not statistically significant and may be related to changes in blood flow or excretion rate caused by the larger mass dose. The high uptake in liver may reduce the utility of the radioligand for imaging studies of metastatic colon cancer in the liver, but the resolution of current PET scanners should minimize the effect of very high liver uptake on the ability to observe other adjacent organs. Unfortunately, the lack of demonstration of specific binding in the in vitro tissue dipping experiments in rat testes suggests the need for a more selective and less lipophilic compound. However, final judgment will require evaluation in an animal model that expresses high levels of the A3AR receptor in a tissue that lacks a tissue-to-blood barrier.
In vivo studies with antagonist [76Br]6 (MRS5147)
We conducted similar in vivo studies with the antagonist [76Br]6 measuring the biodistribution at 15, 60, and 120 min. The general biodistribution pattern observed (Figure 4) was similar to that of the agonist with two important differences. Firstly, the uptake of the antagonist [76Br]6 was an order of magnitude lower than agonist [76Br]4. This may, in part, be due to the fact that slightly older animals were utilized in the study of the agonist. However pharmacokinetic differences between the two compounds may be expected, because the presence of the uronamide group in the agonist [76Br]4 might affect its bioavailability and permeation in vivo. Secondly, the uptake in the A3AR-containing testes continued to increase with time after injection. The blood continued to provide an input function over the two hours. In spite of a potential testes-blood barrier[38], uptake of the antagonist increased with time. This suggests that the antagonist may be viable molecular imaging probe for pathological conditions with elevated A3AR.
Figure 4.
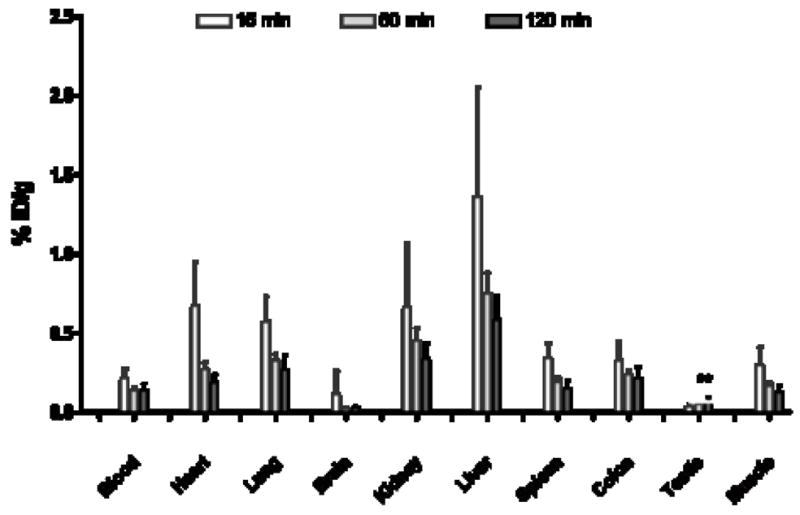
In vivo uptake in rat of the A3AR antagonist [76Br]6 at 15, 60 and 120 min post-injection. Increasing uptake in the testes was observed over time. Both 60 and 120 min uptakes are statistically increased relative to the 15 min time point (p < 0.05)
Conclusions
We have successfully radiolabeled both a high affinity A3AR agonist (MRS3581, 4) and a high affinity A3AR antagonist (MRS5147, 6) with [76Br]bromine- in high radiochemical yield and with high specific activity. The antagonist, [76Br]6, provides evidence of increasing uptake in the A3 receptor-rich testes. Further studies in inflammation models, where the A3AR is known to be up regulated, will be necessary to judge the utility of these positron-emitting high affinity receptor ligands as imaging agents.
Acknowledgments
This work was supported by the Intramural Research Programs of NIBIB and NIDDK, National Institutes of Health, Bethesda, MD. The authors acknowledge the technical assistance of Larry Reyes and Yinghui Shi for in vivo animal experiments and Dr. Larry Szajek, NIH/CC PET Department, for Br-76 isotope production and purification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Reviews Drug Discovery. 2006;5:247–64. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan L, Burbiel JC, Maass A, Muller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerg Drugs. 2003;8:537–76. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 3.Gessi S, Cattabriga EAARGGLLC, et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin Cancer Res. 2004;10:5895–58901. doi: 10.1158/1078-0432.CCR-1134-03. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–5. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 5.Baraldi PG, Tabrizi MA, Romagnoli R, Fruttarolo F, Merighi S, Varani K, et al. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine ligands, new tools to characterize A(3) adenosine receptors in human tumor cell lines. Curr Med Chem. 2005;12:1319–29. doi: 10.2174/0929867054020963. [DOI] [PubMed] [Google Scholar]
- 6.Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P. A3 adenosine receptor activation in melanoma cells - Association between receptor fate and tumor growth inhibition. J Biol Chem. 2003;278:42121–30. doi: 10.1074/jbc.M301243200. [DOI] [PubMed] [Google Scholar]
- 7.Dixon AK, Gubitz AK, Sirinathsinghji DJS, Richardson PJ, Freeman TC. Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol. 1996;118:1461–8. doi: 10.1111/j.1476-5381.1996.tb15561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, et al. Clinical evidence for utilization of the A(3) adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J Rheumatol. 2008;35:41–8. [PubMed] [Google Scholar]
- 9.Madi L, Cohen S, Ochayin A, Bar-Yehuda S, Barer F, Fishman P. Overexpression of A(3) adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis: Involvement of nuclear factor-kappa B in mediating receptor level. J Rheumatol. 2007;34:20–6. [PubMed] [Google Scholar]
- 10.Fishman P, Bar-Yehuda S, Madi L, Rath-Wolfson L, Ochaion A, Cohen S, et al. The PI3K-NF-kappa B signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Research & Therapy. 2006:8. doi: 10.1186/ar1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, et al. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–76. [PubMed] [Google Scholar]
- 12.Bar-Yehuda S, Madi L, Barak D, Mittelman M, Ardon E, Ochaion A, et al. Agonists to the A3 adenosine receptor induce G-CSF production via NF-kappa B activation: A new class of myeloprotective agents. Exp Hematol. 2002;30:1390–8. doi: 10.1016/s0301-472x(02)00962-1. [DOI] [PubMed] [Google Scholar]
- 13.Fishman P, Bar-Yehuda S, Madi L. Targeting the A(3) adenosine receptor (A(3)AR): A new approach for cancer treatment. Drug Dev Res. 2002;56:547. [Google Scholar]
- 14.Fishman P, Bar-Yehuda S. Pharmacology and therapeutic applications of A(3) receptor subtype. Current Topics in Medicinal Chemistry. 2003;3:463–9. doi: 10.2174/1568026033392147. [DOI] [PubMed] [Google Scholar]
- 15.Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, et al. The A(3) adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin Cancer Res. 2004;10:4472–9. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- 16.Fishman P, Bar-Yehuda S, Ohana G, Barer F, Ochaion A, Erlanger A, et al. An agonist to the A(3) adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3 beta and NF-kappa B. Oncogene. 2004;23:2465–71. doi: 10.1038/sj.onc.1207355. [DOI] [PubMed] [Google Scholar]
- 17.Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, et al. Novel N-6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A(3) receptors reduce ischemic myocardial injury. American Journal of Physiology-Heart and Circulatory Physiology. 2003;285:H2780–H7. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- 18.Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y. Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res. 2006;84:1848–55. doi: 10.1002/jnr.21071. [DOI] [PubMed] [Google Scholar]
- 19.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, et al. Structure-Activity-Relationships of N-6-Benzyladenosine-5′-Uronamides as a(3)-Selective Adenosine Agonists. J Med Chem. 1994;37:636–46. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guzman J, Yu JG, Suntres Z, Bozarov A, Cooke H, Javed N, et al. ADOA3R as a therapeutic target in experimental colitis: Proof by validated high-density oligonucleotide microarray analysis. Inflamm Bowel Dis. 2006;12:766–89. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 21.Hardwick MJ, Chen M-K, Baidoo K, Pomper MG, Guilarte TR. In vivo imaging of peripheral benzodiazepine receptors in mouse lungs: a biomarker of inflammation. Mol Imaging. 2005;4:432–8. doi: 10.2310/7290.2005.05133. [DOI] [PubMed] [Google Scholar]
- 22.Bauer A, Langen KJ, Bidmon H, Holschbach MH, Weber S, Olsson RA, et al. F-18-CPFPX PET identifies changes in cerebral A(1) adenosine receptor density caused by glioma invasion. J Nucl Med. 2005;46:450–4. [PubMed] [Google Scholar]
- 23.Moresco RM, Todde S, Belloli S, Simonelli P, Panzacchi A, Rigamonti M, et al. In vivo imaging of adenosine A(2A) receptors in rat and primate brain using [C-11]SCH442416. European Journal of Nuclear Medicine and Molecular Imaging. 2005;32:405–13. doi: 10.1007/s00259-004-1688-5. [DOI] [PubMed] [Google Scholar]
- 24.Gao ZG, Kim SK, Biadatti T, Chen WZ, Lee K, Barak D, et al. Structural determinants of A(3) adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–84. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)-Methanocarba 2,N-6-disubstituted adenine nucleosides as highly potent and selective A(3) adenosine receptor agonists. J Med Chem. 2005;48:1745–58. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matot I, Einav S, Weiniger CF, Pearl RG, Abramovitch R, Joshi BV, et al. Lung injury after in vivo reperfusion - Outcome at 27 hours after reperfusion. Anesthesiology. 2008;109:269–78. doi: 10.1097/ALN.0b013e31817f5b90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li AH, Moro S, Forsyth N, Melman N, Ji XD, Jacobson KA. Synthesis, CoMFA analysis, and receptor docking of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A(3) adenosine receptor antagonists. J Med Chem. 1999;42:706–21. doi: 10.1021/jm980550w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wadsak W, Mien LK, Shanab K, Ettlinger DE, Haeusler D, Sindelar K, et al. Preparation and first evaluation of [F-18]FE@SUPPY: a new PET tracer for the adenosine A(3) receptor. Nucl Med Biol. 2008;35:61–6. doi: 10.1016/j.nucmedbio.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Melman A, Wang B, Joshi BV, Gao Z-G, de Castro S, Heller CL, et al. Selective A3 Adenosine Receptor Antagonists Derived from Nucleosides Containing a Bicyclo[3.0]hexane Ring System. Bioorg Med Chem. 2008;16:8546–56. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szajek LP, Kao CHK, Kiesewetter DO, Sassaman MB, Lang L, Plascjak P, et al. Semi-remote production of Br-76 and preparation of high specific activity radiobrominated pharmaceuticals for PET studies. Radiochim Acta. 2004;92:291–5. [Google Scholar]
- 31.Kao CHK, Sassaman MB, Szajek LP, Ma Y, Waki A, Eckelman WC. The sequential syntheses of [Br-76]FBAU 3′,5′-dibenzoate and [Br-76]FBAU. J Labelled Compd Radiopharm. 2001;44:889–98. [Google Scholar]
- 32.Bradford MM. Rapid and Sensitive Method for Quantitation of Microgram Quantities of Protein Utilizing Principle of Protein-Dye Binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Cheng Y, Prusoff WH. Relationship between Inhibition Constant (K1) and Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic-Reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 34.Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, et al. Discovery of a new nucleoside template for human A(3) adenosine receptor ligands: D-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J Med Chem. 2007;50:3159–62. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]
- 35.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, et al. Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 36.Yaar R, Jones MR, Chen JF, Ravid K. Animal models for the study of adenosine receptor function. J Cell Physiol. 2005;202:9–20. doi: 10.1002/jcp.20138. [DOI] [PubMed] [Google Scholar]
- 37.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:364–74. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 38.Wong CH, Cheng CY. The blood-testis barrier: Its biology, regulation, and physiological role in spermatogenesis. Current Topics in Developmental Biology. 2005;7171:263–96. doi: 10.1016/S0070-2153(05)71008-5. [DOI] [PubMed] [Google Scholar]