

Abstract
The identification and physical isolation of epithelial stem cells is critical to our understanding of their growth regulation during homeostasis, wound healing, and carcinogenesis. These stem cells remain poorly characterized because of the absence of specific molecular markers that permit us to distinguish them from their progeny, the transit amplifying (TA) cells, which have a more restricted proliferative potential. Cell kinetic analyses have permitted the identification of murine keratinocyte stem cells (KSCs) as slowly cycling cells that retain [3H]thymidine ([3H]Tdr) label, termed label-retaining cells (LRCs), whereas TA cells are visualized as rapidly cycling cells after a single pulse of [3H]Tdr, termed pulse-labeled cells (PLCs). Here, we report on the successful separation of KSCs from TA cells through the combined use of in vivo cell kinetic analysis and fluorescence-activated cell sorting. Specifically, we demonstrate that murine dorsal keratinocytes characterized by their high levels of α6 integrin and low to undetectable expression of the transferrin receptor (CD71) termed α6briCD71dim cells, are enriched for epithelial stem cells because they represent a minor (≈8%) and quiescent subpopulation of small blast-like cells, with a high nuclear:cytoplasmic ratio, containing ≈70% of label-retaining cells, the latter being a well documented characteristic of stem cells. Conversely, TA cells could be enriched in a phenotypically distinct subpopulation termed α6briCD71bri, representing the majority (≈60%) of basal keratinocytes that are actively cycling, and importantly contain ≈70% of [3H]Tdr pulse-labeled cells. Importantly, immunostaining of dorsal skin revealed the presence of CD71dim cells in the hair follicle bulge region, a well documented location for KSCs.
In common with other rapidly renewing tissues such as the bone marrow and the intestinal epithelium, stratified epithelia are in a process of constant regeneration. Terminally differentiated cells lost continuously from the skin surface are replaced by an intricate and highly regulated proliferative process. In vivo cell kinetic studies in murine epidermis have elegantly demonstrated that this process is achieved by two distinct subpopulations of keratinocyte progenitors: (i) keratinocyte stem cells (KSCs), which represent a minor subpopulation of relatively quiescent or slow-cycling cells, defined by their great proliferative potential and an unlimited capacity for self-renewal; and (ii) transit amplifying (TA) cells—the progeny of the stem cells, with a limited proliferative capacity, identified as a pool of rapidly proliferating cells that are lost from the basal layer to terminal differentiation within 4–5 days (1–6). In addition, a third subpopulation of basal keratinocytes representing postmitotic differentiating (PMD) cells in the early stages of keratinization can also be identified (7–10). Despite their central role in cell renewal, and presumably in wound repair and neoplasia, epidermal stem cells remain relatively poorly characterized. Cell kinetic studies have permitted the identification of murine KSCs in situ, as cells that retain tritiated thymidine over extended periods because of their slow-cycling nature [termed [3H]thymidine ([3H]Tdr) label-retaining cells (LRCs)] in the interfollicular epidermis, hair follicles, and oral mucosa of adult mice (1–6, 11). More recently, it has been shown that LRCs from ear epidermis could be enriched from a suspension of primary basal keratinocytes by selecting for cells that rapidly adhered to a variety of adhesive matrices including polyd-lysine (12). Notably, LRCs represented on average approximately 50% of the adherent cells, indicating the presence of an equal number of nonstem cells. This study represents an important step toward the isolation of KSCs.
In the hemopoietic system, multilineage reconstituting stem cells can be separated from committed progenitor cells by fluorescence activated cell sorting (FACS), based on differences in their expression of cell surface markers (13–17). Clearly the development of a similar strategy for the isolation of KSCs would greatly facilitate their characterization. Early studies utilizing antibodies to the β1 integrin demonstrated that human basal keratinocytes from neonatal foreskin epidermis, expressing high levels of this cell surface adhesion molecule, were enriched for KSCs, given their higher plating efficiency and ability to generate an epithelial sheet when grafted onto nude mice (18, 19). Subsequent studies from our laboratory demonstrated a further enrichment for putative human KSCs, using long-term proliferative output and cycling status as assays for stem cells (20). In that study, we utilized two cell surface molecules to fractionate primary keratinocytes: the adhesion molecule α6, also a member of the integrin family, and a proliferation-associated cell surface marker recognized by mAb, 10G7 (21). Specifically, we demonstrated that the basal layer of foreskin epidermis could be fractionated into three phenotypically and functionally discrete populations: (i) α6bri10G7dim cells comprising a minor population (≈4%) of quiescent basal keratinocytes at the time of isolation from the epidermis as determined by cell cycle analysis, exhibiting the greatest long-term proliferative capacity of any basal cells, thought to represent the putative epidermal stem cell population; (ii) α6bri10G7bri cells, an actively cycling population showing lower long-term proliferative output designated the putative TA fraction; and (iii) α6dim cells, a largely quiescent postmitotic differentiating fraction with relatively poor long-term proliferative output, expressing the keratinocyte differentiation markers keratin 10 and involucrin, designated the PM-D fraction (20). These data represented an important step in resolving keratinocyte progenitors, but did not provide concrete verification of the human KSC population because of the absence of a rigorous reconstitution assay for epidermal stem cells. Because murine epidermal [3H]Tdr LRCs detected in vivo are widely accepted to be KSCs, we set out to fractionate murine dorsal keratinocytes by cell surface markers and FACS techniques and, more importantly, to determine whether this approach could be used to enrich for LRCs.
Materials and Methods
Chicken Embryo Fibroblast (CEF) and CEF Human Transferrin Receptor (CEFHTR) Cells.
CEFs expressing the human transferrin receptor (CEFHTR), also known as CD71 (22), and uninfected CEFs, kindly provided by Ian Trowbridge (Salk Institute, San Diego), were grown in DMEM containing 1% chicken serum and 1% FCS and were stained with a known antibody to CD71, B3/25.4 (23), also supplied by Ian Trowbridge, mAb 10G7, and isotype-matched negative control mAbs 1B5 and 1D4.5 (20) for flow cytometric analysis.
Murine Adult Epidermal Keratinocytes.
Dorsal keratinocytes isolated from female BALB/c mice aged between 7 and 9 weeks were used for flow cytometric analysis of cell surface markers (α6 and CD71), for cell cycle analysis, and for pulse-labeling experiments (in mice killed 1 h after a single injection of [3H]Tdr). Dorsal keratinocytes isolated from 14-week-old CD-1 mice containing [3H]Tdr LRCs were used in phenotyping experiments. Methods for generating LRCs and PLCs were as described (24). The epidermal cell isolation procedure used (24) yields both hair follicle and interfollicular epidermal cells.
Immunofluorescent Staining for α6 and CD71.
Cells were processed for two-color staining for α6 and CD71 for FACS by incubating with mAb GoH3 (anti-α6 integrin rat monoclonal; IgG2a) at 10 μg/ml (Serotec), and mAb R17 208.2 (a rat anti-mouse CD71 IgM, provided by Bob Hyman, Salk Institute) used as hybridoma supernatant for 1 h, after blocking in 4-ml blocking buffer (4% normal human serum/0.4% BSA/5% FCS in HBSS) for 20 min at 4°C. Cells were washed twice with DMEM containing 10% FCS and incubated with isotype-specific secondary antibodies, i.e., anti-rat IgG FITC (1:80) to detect α6 and anti-rat IgM biotin (1:125) (Jackson ImmunoResearch). A third-layer reagent, streptavidin–phycoerythrin (1:80), was used to detect CD71. mAbs B220 and a rat anti-mouse CD8a mAb (IgM; kindly provided by Bob Hyman) were used as isotype-matched negative controls. All incubations were performed at 4°C and the cells were fixed after staining for flow cytometry. Alternately, for FACS experiments, cells were resuspended in HBSS with 5% FCS to achieve a concentration of 107 cells per ml, containing 1 μg/ml of the viability dye 7-amino-actinomycin D (7-AAD; Molecular Probes) to exclude nonviable cells. Cell sorting was performed on a FACStarplus (Becton Dickinson), and the viability of sorted cells determined to be ≈95% by trypan blue exclusion.
Cell Cycle Analysis.
Sorted cells were collected into DMEM-10 and kept on ice. Per fraction, 105 cells were collected and processed for cell cycle analysis. Cells were fixed and permeabilized in 70% ethanol (−20°C) at 4°C for 20 min, washed with HBSS/5% FCS, and treated with 40 μg/ml RNase for 20 min at 37°C. Cells were then incubated in 3 μg/ml propidium iodide for 5 min at room temperature and placed on ice before flow cytometric analysis on an Coulter EPICS XL.
Measurement of Cell Size, Cytoplasmic, and Nuclear Area.
Cytospins of fractionated keratinocytes were stained with Giemsa and microscopic images of these cells were subjected to automated computer image analysis at a magnification of ×20 to determine cell diameter and area. The nuclear and total cell areas were measured and the cytoplasmic area was calculated from these parameters. These data were obtained with analysis software developed for video pro (Leading Edge Proprietary, Ltd., Adelaide, Australia). Approximately 100 cells per fraction were analyzed and the results were subjected to a paired t test.
Autoradiography for LRCs and PLCs.
Cytospins were prepared from fractionated keratinocytes derived from [3H]Tdr-labeled CD-1 mice containing LRCs or from BALB/c mice containing PLCs and processed for autoradiography as previously described (24). Three separate LRC sorting experiments (LRC-1, LRC-2, and LRC-3) were performed on groups of three CD-1 mice, whereas two separate PLC sorting experiments (PLC-1 and PLC-2) were performed on groups of four mice. The percentage of LRCs or PLCs was determined by counting a total of 500 cells per fraction for each experiment.
Immunohistochemical Staining for CD71.
Cryostat sections of dorsal skin from 8.5-week-old female mice were fixed in acetone for 10 min at −20°C, blocked in 10% normal goat serum for 20–30 min, and stained with a 1:250 dilution of a rat mAb to murine CD71 (clone R17 217.1.4; Caltag Laboratories, South San Francisco, CA) directly conjugated to FITC for 1 h. Sections were counterstained with propidium iodide.
Results
Identification of 10G7 Antigen as the Transferrin Receptor CD71.
To extend the cell surface phenotype ascribed to candidate human KSCs into murine epithelia, it was necessary to identify the proliferation-related antigen recognized by mAb 10G7. Flow cytometric analysis of a range of nonepithelial cell types, including hemopoietic progenitors and their differentiated progeny demonstrated that the 10G7 antigen was widely expressed by actively proliferating committed progenitors. Subsequent immunoprecipitation studies from these cells revealed that mAb 10G7 recognized an abundant cell surface protein with a molecular mass of about 200 kDa under nonreducing conditions and 95kDa under reducing conditions, leading us to infer that mAb 10G7 may recognize the transferrin receptor of the same molecular weight, known to be expressed on all proliferating cell types. This finding was confirmed by the demonstration that mAb 10G7 bound to CEFs expressing human CD71 (Fig. 1), but not to untransfected controls. Thus, the phenotype of putative human KSCs can now be confirmed as α6briCD71dim.
Figure 1.

mAb 10G7 recognizes the human transferrin receptor (HTR) CD71. Flow cytometric analysis of CEF + HTR cells showing their reactivity with a known anti-CD71 antibody (B3/25.4) and mAb 10G7. mAb 1D4.5 is an isotype-matched negative control for mAb 10G7.
Phenotyping of Murine Dorsal Keratinocytes.
Primary keratinocytes were isolated from adult murine dorsal skin and their identity confirmed by determining their expression of Keratin 14 (K14), a marker of undifferentiated epidermal cells of the basal layer and within the hair follicle. K14-positive cells represented 85.4 ± 3.0% of the total number of isolated cells in replicate experiments (n = 3). The α6 integrin is also expressed by dorsal keratinocytes and has been shown to have a functional role in mediating adhesion of basal keratinocytes to the basement membrane via hemi-desmosomes (25–28). Two-color flow cytometric analysis of isolated cells revealed that all cells expressing high levels of α6 integrin also expressed K14, indicating that the selection of α6bri cells yields keratinocytes and excludes nonepithelial cells (data not shown). Subsequent, two-color flow cytometric analysis of α6 and CD71 consistently revealed three phenotypically discrete populations of cells (n = 25; Fig. 2): (i) a major subpopulation representing 58.6 ± 8.2% (n = 3) of the total isolated cells exhibiting high levels of both α6 and CD71 (termed α6briCD71bri); (ii) a minor population representing about 8.1 ± 2.0% of the cells, characterized by high level expression of α6 and low levels of CD71 (termed α6briCD71dim); and (iii) a less distinct third population of cells representing about 15.1 ± 2.5%, expressing relatively low levels of α6 (termed α6dim). In all our cell preparations, a population of α6 negative cells was also consistently observed. These nonepithelial cells were excluded from subsequent analysis by selection of α6-positive cells.
Figure 2.

Two-color flow cytometric analysis of α6 integrin and CD71 expression on primary murine dorsal keratinocytes. α6 was detected with FITC (x axis, FL1) and CD71 with phycoerythrin (y axis, FL2). Three phenotypically distinct fractions of α6 positive keratinocytes were consistently discernable (n = 25) as indicated: a, α6briCD71bri cells making up the majority of basal keratinocytes; b, α6briCD71dim cells representing a discrete but minor proportion of basal keratinocytes; and c, α6dim cells that appear as a less discrete population. A number of α6 negative nonepithelial cells were also detected.
α6briCD71dim Cells Are a Quiescent Population of Small Cells, with a High Nuclear:Cytoplasmic Ratio.
To establish the cycling status of these three phenotypically distinct murine epidermal fractions, flow cytometric analysis was performed on the sorted subpopulations stained with propidium iodide as previously described (20). These data demonstrated that approximately 3% of all basal keratinocytes (unfractionated total basal keratinocytes) are engaged in DNA synthesis (Fig. 3a), in close accord with published data (29, 30). Notably, the α6briCD71bri fraction was significantly enriched for actively cycling cells, as compared with the α6briCD71dim (P = 0.036) and α6dim fractions (P = 0.019), which contained significantly fewer cells in S and G2/M. In addition, the α6briCD71dim fraction also contained the greatest number of G0/G1 cells, consistent with it being the most quiescent subpopulation of keratinocytes.
Figure 3.

Cell cycle, cell size, and morphological characteristics of fractionated murine primary dorsal keratinocytes. (a) Cell cycle analysis from two independent sorting experiments demonstrated that the α6briCD71bri fraction was enriched for actively cycling basal keratinocytes, with the fewest number of cells in G0/G1; conversely, the α6briCD71dim fraction was the most quiescent subpopulation with the least number of cells in S and G2/M, but the highest frequency of cells in G0/G1. The α6dim cells represented an intermediate population that was not significantly different from the α6briCD71dim fraction (P = 0.126). (b and c) A comparison of the forward- and side-scatter dot plots of these two populations of basal keratinocytes shows that the quiescent α6briCD71dim cells (b) are smaller in size as compared with the actively cycling α6briCD71bri cells (c). (d and e) Giemsa-stained cytospin preparations of these fractions confirm these size differences as well as demonstrating that the α6briCD71dim population has a high nuclear:cytoplasmic ratio (d) as compared with the α6briCD71bri population (e). These properties indicate enrichment for putative stem cells in the α6briCD71dim fraction.
The α6briCD71dim and α6briCD71bri populations also exhibited distinct differences in cell size and cytological characteristics. The quiescent α6briCD71dim population (Fig. 3b) contained significantly smaller cells than did the α6briCD71bri fraction (Fig. 3c) as determined by differences in the forward light scatter properties of the two populations. Moreover, microscopic examination of cytospin preparations of these cells revealed that the quiescent α6briCD71dim cells exhibited a “blast-like” appearance with a high nuclear to cytoplasmic ratio consistent with undifferentiated or primitive cells (Fig. 3d) as compared with the α6briCD71bri cells, which had a basophilic appearance indicative of transcriptionally active cells (Fig. 3e). These differences in the morphological appearance of the two populations were verified by using computer image analysis to quantitate cell size and nuclear versus cytoplasmic areas of Giemsa-stained cytospin preparations of about 100 cells per fraction. The results shown in Table 1 demonstrate that the α6briCD71dim cells are smaller compared with the α6briCD71bri cells as determined by their area and diameter. Notably, the larger cell size of the actively cycling α6briCD71bri fraction can be attributed directly to a two-fold increase in the cytoplasmic area of these cells, compared with the quiescent α6briCD71dim cells (Table 1). These data indicated that the α6briCD71dim fraction had characteristics typical of a stem cell population comprising small, immature cells with few cytoplasmic organelles as reported for hemopoietic progenitors (31).
Table 1.
Cell size and nuclear and cytoplasmic areas of fractionated keratinocytes
| Parameter | α6briCD71dim | α6briCD71bri | P values |
|---|---|---|---|
| Cell area, μm2 | 69.68 ± 16.36 | 99.68 ± 9.91 | <0.0001 |
| Nuclear area, μm2 | 42.27 ± 5.33 | 42.52 ± 3.18 | 0.9932 |
| Cytoplasmic area, μm2 | 27.42 ± 14.28 | 57.16 ± 12.04 | <0.0001 |
| Cell diameter, μm | 9.38 ± 1.07 | 11.24 ± 0.57 | <0.0001 |
α6briCD71dim, n = 104; α6briCD71bri, n = 97.
Enrichment for [3H]Tdr LRCs or Stem Cells in the α6briCD71dim Fraction.
We then set out to determine the location of LRCs within the three phenotypically fractionated populations of murine keratinocytes, i.e., α6briCD71dim, α6briCD71bri, and α6dim. Newborn mice were given multiple injections of [3H]Tdr as previously described (24) and killed at 14 weeks of age. Keratinocytes were isolated from these [3H]Tdr LRC-containing mice and fractionated into α6briCD71dim, α6briCD71bri, and α6dim subpopulations by FACS as before. Cytospins were prepared from the collected cells and processed for autoradiography, and the frequency of LRC per fraction was determined microscopically. The results of three independent sorting experiments (Table 2) showed that on average 11.30 ± 0.99% of the total unfractionated cells represented LRCs (mean ± SD of LRC-1, -2, and -3). Importantly, the majority of these LRCs (71.95 ± 14.02%) were in the relatively quiescent α6briCD71dim fraction (Fig. 4), indicating significant enrichment for murine KSCs. Smaller numbers of LRCs were also found in the actively cycling α6briCD71bri fraction (12.6 ± 5.3%), as well as in the α6dim fraction (15.45 ± 9.35%) as shown in Fig. 4 and Table 2.
Table 2.
Distribution of LRCs and PLCs in primary fractionated keratinocytes
| Experiment | α6briCD71dim | α6briCD71bri | α6dim | Unfractionated |
|---|---|---|---|---|
| LRC-1 | 87 | 13 | 16 | 54 |
| LRC-2 | 78 | 15 | 10 | 53 |
| LRC-3 | 113 | 18 | 32 | 62 |
| PLC-1 | 4 | 24 | 6 | 15 |
| PLC-2 | 6 | 20 | 3 | 26 |
Numbers represent frequency in 500 cells per fraction.
Figure 4.

Enrichment for [3H]Tdr LRCs in the α6briCD71dim and PLCs in the α6briCD71bri compartments. The results displayed represent the percentage of LRCs/PLCs found in the three phenotypically distinct keratinocyte fractions, expressed as a proportion of the total labeled cells displayed as mean ± SD.
Enrichment for [3H]Tdr PLCs or TA Cells in the α6briCD71bri Fraction.
In vivo, murine epidermal TA cells are readily labeled after a single injection of [3H]Tdr, because they represent a population of actively dividing cells and are therefore termed pulse-labeled cells (PLCs). Cell kinetic studies indicate that 1 h after a pulse of [3H]Tdr, approximately 2–5% of basal keratinocytes are labeled and are subsequently lost from the epidermis within a week to differentiation (5, 8, 29, 30, 32). We therefore sought to determine whether the α6briCD71bri fraction shown by our cell cycle analysis to consist of actively proliferating cells contained PLCs. Keratinocytes were isolated from mice given a single injection of [3H]Tdr, fractionated by cell surface phenotype as described above, and subjected to autoradiography. The distribution of PLCs among the fractionated keratinocytes from two separate sorting experiments (PLC-1 and PLC-2) are shown in Table 2. Consistent with published studies (5, 8, 29, 30, 32) and with our cell cycle data (Fig. 3a and ref. 20), on average 4.1 ± 1.6% of total unfractionated keratinocytes were labeled with [3H]Tdr. Importantly, the α6briCD71bri fraction contained the majority (69.73 ± 1.09%) of the total overall number of these PLCs, whereas the α6briCD71dim fraction previously shown to be enriched for LRCs and the α6dim fraction contained 16.24 ± 6.28% and 13.97 ± 5.13% of total PLCs, respectively. These data demonstrate that the α6briCD71bri fraction is significantly enriched for TA cells as shown in Fig. 4 and Table 2.
Localization of CD71dim Cells to the Hair Follicle Bulge Region.
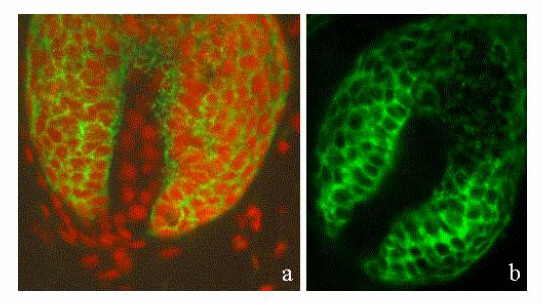
Cell kinetic studies have demonstrated unequivocally that LRCs are found at two specific sites in the dorsal epithelium, i.e., interspersed as single cells within the basal layer of the epidermis and clustered within the bulge region of the hair follicle (1–6, 11). Consistent with these studies, autoradiographic analysis of dorsal skin sections revealed that LRCs could be found principally in the first generation follicle, commonly called the bulge, and occasionally in the basal layer of the interfollicular epidermis (data not shown). We reasoned that if the α6briCD71dim cells were indeed stem cells, then immunohistochemical staining should reveal decreased staining for CD71 in the bulge region of the hair follicle and in rare cells within the epidermis. The data shown in Fig. 5 demonstrate the differential expression of CD71 in the hair follicle. Notably the interfollicular epidermis and upper portion of the hair follicle including the sebaceous gland exhibited relatively bright CD71 staining (Fig. 5 a and b and Supplementary Fig. 8), with the strongest expression detected in the bulb region at the base of hair follicles in early or mid-anagen (Fig. 5 a and c; see supplementary Fig. 6, published on the PNAS web site, www.pnas.org). Importantly, the bulge region consistently showed low to negative staining for CD71 (Fig. 5 a and b; see also supplementary Figs. 7 and 8). Differential expression of CD71 in the basal layer of the epidermis was not detected. Bright staining for α6 was observed throughout the basal layer of the interfollicular epidermis and in the outer root sheath of the hair follicle (data not shown).
Figure 5.

The bulge region of the hair follicle is CD71dim. (a) Immunofluorescence micrographs of CD71 staining (green) in dorsal skin illustrating several early anagen hair follicles (arrowheads), with bright staining for CD71 at the base of the follicle. Note the longitudinal section through a mid-anagen hair follicle showing relatively low to negative CD71 expression in the bulge region (block arrows), directly below the sebaceous glands (arrow). (b and c) Dual immunofluorescence for CD71 (green) and nuclei stained with propidium iodide (red) illustrating the presence of CD71dim cells in the bulge region (b) and CD71bri cells in the bulb region (c). Dermal papilla is indicated by the star.
Discussion
Despite the central importance of stem cells in cell renewal within the epidermis, our ability to directly study this minor subpopulation of keratinocyte progenitors has been hampered by the lack of markers that allow us not only to identify these cells but also to isolate them in a viable state. We have reported here that cells that satisfy many of the criteria assigned to keratinocyte stem cells reside within a population of cells with the surface phenotype α6briCD71dim, the most important of which is their definition as slow-cycling [3H]Tdr LRCs, a widely accepted definition of murine KSCs in vivo. In addition, dorsal keratinocytes with the phenotype α6briCD71dim comprised a minor population (≈8%) of small, unspecialized cells with a large nuclear:cytoplasmic ratio. The localization of CD71dim cells to the hair follicle bulge, a well documented site for slow-cycling stem cells (11, 33) further strengthens our data. It has been shown unequivocally that the interfollicular epidermis is a self-renewing tissue organized into functionally discrete epidermal proliferative units (EPUs), comprising a central stem cell (recognizable as an LRC) and its progeny of approximately 10 basal cells, giving rise to a column of differentiated keratinocytes directly above it (8). These interfollicular stem cells should also be CD71dim but were not easily identifiable, presumably because they occur as single cells interspersed among other basal cells.
Interestingly, we observed that not all cells within the α6briCD71dim fraction at 14 weeks postlabeling were LRCs, the latter representing ≈1.4% of total isolated keratinocytes. The incidence of stem cells in the interfollicular epidermis alone is estimated at 1–10% of the basal layer from cell kinetic studies, not taking into account the stem cell population known to reside in the hair follicle bulge region (11). Thus, clearly in our experiments, not all epidermal stem cells are represented as LRCs. In particular, although interfollicular epidermal LRCs comprise ≈4% of the basal layer at 8 weeks postlabeling (29), our autoradiographic analysis showed that at 14 weeks, LRCs resided primarily in the hair follicle bulge region and very rarely in the interfollicular epidermis. Thus, interfollicular epidermal stem cells likely to be present within the α6briCD71dim fraction are likely to be represented as non-LRCs. In addition, it is highly likely that not all stem cells were proliferating during the [3H]Tdr labeling period in the newborn mice. Nevertheless, these factors alone cannot account for only ≈18% of the cells within the α6briCD71dim fraction representing LRCs at 14 weeks postlabeling, and there remains the possibility that a hierarchy of keratinocyte progenitors exists within the α6briCD71dim fraction that may appear as LRCs at earlier time points, an inference that is supported by detailed cell kinetic studies reporting rates of loss of LRCs from the epidermis over time (6, 29). This hypothesis is also supported by recent data that demonstrate the presence of rare LRCs in hair follicles as long as 1 year after labeling (33). This notion is consistent with the “diminishing stem-ness spiral model” proposed by Potten and Loeffler (34), which suggests a continuum between stem cells and TA cells, accompanied by a gradual loss in self-renewal capacity and increasing probability of differentiation.
We have also ascribed the murine epidermal TA population with the phenotype α6briCD71bri and have shown that PLCs reside within this subpopulation. Not all cells within this fraction were labeled with [3H]Tdr because only a small proportion of this population is in S-phase at a given time as reported previously (5, 8, 29, 30, 32). The incidence of TA cells reported here (≈60%) is also consistent with cell kinetic studies reviewed by Potten and Morris (35).
We have previously shown that α6 integrin is expressed on both KSCs and TAs (20). The use of CD71 to fractionate proliferative cells into quiescent and cycling compartments is not unprecedented and has been validated by previous studies in the hemopoietic system that demonstrated that candidate human hemopoietic stem cells could be separated from their immediate progeny, the committed progenitors, by their low to undetectable expression of CD71 (36, 37).
In conclusion, our studies provide the means to identify and enrich for viable KSCs and TAs. The ability to recognize these populations phenotypically will permit investigation of the molecular differences between these keratinocyte progenitors and provide a basis for the identification of genes with a critical role in epidermal growth, differentiation, and self-renewal of KSCs. Further, the data have important implications for the study of epidermal carcinogenesis, given that stem cells are likely to be a target for carcinogens resulting in the development of carcinomas (24, 38). Finally, the accessibility of skin makes human KSCs an ideal vehicle for genetic manipulation and gene therapy for the treatment of both skin disorders and systemic deficiencies. The ability to identify and isolate these cells represents an important prerequisite for the development of these approaches.
Supplementary Material
Acknowledgments
We thank Sharon Paton for technical assistance and Amy Li for technical advice. We are grateful to Sandy MacIntyre for assistance with cell sorting and to Alan Bishop for his expertise with FACS analysis. We thank Dr. Paul Simmons for valuable discussions and critical reading of this manuscript. This work was supported by grants from the National Health and Medical Research Council, Australia, to P.K. (981881) and from the National Institutes of Health to R.J.M. (CA 45293) and by funds from the Lankenau Foundation. H.T. is the recipient of an Australian Postgraduate Award.
Abbreviations
- KSC
keratinocyte stem cell
- TA
transit amplifying
- LRC
label-retaining cell
- PLC
pulse-labeled cell
- FACS
fluorescence activated cell sorting
- CEF
chicken embryo fibroblasts
- [3H]Tdr
[3H]thymidine
References
- 1.Bickenbach J R. J Dent Res. 1981;60:1611–1620. doi: 10.1177/002203458106000311011. [DOI] [PubMed] [Google Scholar]
- 2.Potten C S, editor. Stem Cells: Their Identification and Characterization. London: Churchill Livingstone; 1983. pp. 200–232. [Google Scholar]
- 3.Morris R J, Fischer S M, Slaga T J. J Invest Dermatol. 1985;84:277–281. doi: 10.1111/1523-1747.ep12265358. [DOI] [PubMed] [Google Scholar]
- 4.MacKenzie I C, Bickenbach J R. Cell Tissue Res. 1985;242:551–556. doi: 10.1007/BF00225420. [DOI] [PubMed] [Google Scholar]
- 5.Potten C S. Int J Radiat Biol. 1986;49:257–278. doi: 10.1080/09553008514552541. [DOI] [PubMed] [Google Scholar]
- 6.Bickenbach J R, McCutecheon J, MacKenzie I C. Cell Tissue Kinet. 1986;19:325–333. doi: 10.1111/j.1365-2184.1986.tb00684.x. [DOI] [PubMed] [Google Scholar]
- 7.Christophers E. J Invest Dermatol. 1971;56:165–169. doi: 10.1111/1523-1747.ep12260765. [DOI] [PubMed] [Google Scholar]
- 8.Allen T D, Potten C S. J Cell Sci. 1974;15:291–319. doi: 10.1242/jcs.15.2.291. [DOI] [PubMed] [Google Scholar]
- 9.Schweizer J, Kinjo M, Furstenberger G, Winter H. Cell. 1984;37:159–170. doi: 10.1016/0092-8674(84)90311-8. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie I C, Mackenzie S L, Rittman G A. Differentiation (Berlin) 1989;41:127–138. doi: 10.1111/j.1432-0436.1989.tb00740.x. [DOI] [PubMed] [Google Scholar]
- 11.Cotsarelis G, Sun T-T, Lavker R M. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 12.Bickenbach J R, Chism E. Exp Cell Res. 1998;244:184–195. doi: 10.1006/excr.1998.4163. [DOI] [PubMed] [Google Scholar]
- 13.Civin C L, Strauss L C, Brovall C, Frackler M J, Schwartz J F, Shaper J H. J Immunol. 1984;133:57–165. [PubMed] [Google Scholar]
- 14.Spangrude G J, Heinfeld S, Weissman I L. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 15.Berenson R J, Bensinger W I, Hill R S, Andrews R G, Garcia-Lopez J, Kalamasz D F, Still B J, Spitzer G, Buckner C D, Bernstein I D, Thomas E D. Blood. 1991;77:1717–1722. [PubMed] [Google Scholar]
- 16.Terstappen L W, Huang S, Safford M, Lansdorp P M, Loken M R. Blood. 1991;77:1218–1227. [PubMed] [Google Scholar]
- 17.Baum C M, Weissman I L, Tsukamoto A S, Buckle A M, Peault B. Proc Natl Acad Sci. 1992;89:2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones P H, Watt F M. Cell. 1993;73:713–724. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 19.Jones P H, Harper S, Watt F M. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 20.Li A, Simmons P J, Kaur P. Proc Natl Acad Sci. 1998;95:3902–3907. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaur P, Paton S, Furze J, Wrin J, Olsen S, Danks J, Scurry J. J Invest Dermatol. 1997;109:194–199. doi: 10.1111/1523-1747.ep12319332. [DOI] [PubMed] [Google Scholar]
- 22.Schwarting R, Stein H. In: Leukocyte Typing IV. Knapp W, Dorken B, Gilks W, Rieber E, Schmidt R E, Stein H, von dem Borne A, editors. New York: Oxford Univ. Press; 1989. pp. 455–460. [Google Scholar]
- 23.Trowbridge I S, Omary M B. Proc Natl Acad Sci USA. 1981;78:3039–3043. doi: 10.1073/pnas.78.5.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris R J, Fischer S M, Slaga T J. Cancer Res. 1986;46:3061–3066. [PubMed] [Google Scholar]
- 25.Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer L M, Falcioni R, Kennel S J, Aplin J D, Baker J, Loizidou M, et al. J Cell Biol. 1991;113:907–917. doi: 10.1083/jcb.113.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowling J, Yu Q-C, Fuchs E. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Nat Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- 28.Van-der-Neut R, Krimpenfort P, Calafat J, Neissen C M, Sonnenberg A. Nat Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- 29.Morris R J, Potten C S. Cell Proliferation. 1994;27:279–289. doi: 10.1111/j.1365-2184.1994.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 30.Morris R J, Fischer S M, Klein-Szanto A J P, Slaga T J. Cell Tissue Kinet. 1990;23:587–602. doi: 10.1111/j.1365-2184.1990.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 31.Van Bekkum D W, van den Engh G J, Wagemaker G, Bol S J L, Visser J W M. Blood Cells. 1979;5:143–159. [PubMed] [Google Scholar]
- 32.Morris R J, Argyris T. Cancer Res. 1983;43:4935–4942. [PubMed] [Google Scholar]
- 33.Morris R J, Potten C S. J Invest Dermatol. 1999;112:470–475. doi: 10.1046/j.1523-1747.1999.00537.x. [DOI] [PubMed] [Google Scholar]
- 34.Potten C S, Loeffler M. Development (Cambridge, UK) 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 35.Potten C S, Morris R J. J Cell Sci Suppl. 1988;10:45–62. doi: 10.1242/jcs.1988.supplement_10.4. [DOI] [PubMed] [Google Scholar]
- 36.Sutherland H J, Eaves C J, Eaves A C, Lansdorp P M. In: Leukocyte Typing IV. Knapp W, Dorken B, Gilks W R, Rieber P, Schmidt R E, Stein H, von dem Borne A E, editors. New York: Oxford Univ. Press; 1989. pp. 910–912. [Google Scholar]
- 37.Lansdorp P M, Dragowska W. J Exp Med. 1992;175:1501–1509. doi: 10.1084/jem.175.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris R J, Coulter K, Tryson K, Steinberg S R. Cancer Res. 1997;57:3436–3443. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}