

SUMMARY
Smokers are more susceptible than non-smokers to persistent infection by Porphyromonas gingivalis, a causative agent of periodontitis. Patients who smoke exhibit increased susceptibility to periodontitis and are more likely to display severe disease and be refractory to treatment. Paradoxically, smokers demonstrate reduced clinical inflammation. We show that P. gingivalis cells exposed to cigarette smoke extract (CSE) induce a lower pro-inflammatory response (TNF-α, IL-6, IL12 p40) from monocytes and PBMCs than do unexposed bacteria. This effect is reversed when CSE-exposed bacteria are subcultured in fresh medium without CSE. Using microarrays representative of the P. gingivalis genome, CSE-exposure resulted in differential regulation of 6.8% of P. gingivalis genes, including detoxification and oxidative stress-related genes; DNA repair genes; and multiple genes related to P. gingivalis virulence, including genes in the major fimbrial and capsular operons. Exposure to CSE also altered the expression of outer membrane proteins, most notably by inducing the virulence factors RagA and RagB, and a putative lipoprotein co-transcribed with the minor fimbrial antigen. Therefore, CSE represents an environmental stress to which P. gingivalis adapts by altering gene expression and outer membrane proteins. These changes may explain, in part, the altered virulence and host-pathogen interactions that have been documented in vivo in smokers with periodontal disease.
Introduction
Cigarette smokers, and those exposed to secondhand smoke, are more susceptible to multiple infectious diseases than unexposed individuals (Arcavi and Benowitz, 2004; Kum-Nji et al., 2006; Polanska et al., 2006; Keskinoglu et al., 2007; Araco et al., 2008; Jeppesen et al., 2008). Tobacco smoking is considered to be a major environmental risk factor for periodontitis, a common infectious oral disease that results in detachment of the gingival tissue cuff, destruction of the collagen attachment to the root surface of the tooth, and resorption of the supporting alveolar bone. Patients who smoke exhibit not only increased susceptibility to periodontitis but are also more likely than non-smokers to display severe disease and to be refractory to treatment (Palmer et al., 2005). Although periodontitis is caused by a polymicrobial infection of the gingiva, the Gram negative, anaerobic bacterium, Porphyromonas gingivalis, has been strongly associated with diseased sites in the oral cavity and is considered to be a causative agent of chronic periodontitis (Holt and Ebersole, 2005; Socransky and Haffajee, 2005). In addition, P. gingivalis infection elicits systemic inflammation (Dye et al., 2005) and may increase susceptibility to systemic vascular diseases (Gibson et al., 2006; Pussinen et al., 2007). Multiple studies have shown that smokers are more likely to be infected with P. gingivalis, to harbor higher numbers of P. gingivalis, and to exhibit more persistent infection (Zambon et al., 1996; Kamma et al., 1999; Eggert et al., 2001; Haffajee and Socransky, 2001; Grossi et al., 2007) relative to non-smokers.
Despite increased P. gingivalis infection rates and susceptibility to disease progression, smokers consistently display reduced clinical inflammation, measured as angiogenesis, edema, inflammatory index, and/or gingival bleeding compared to non-smokers (Kinane and Chestnutt, 2000; Rezavandi et al., 2002; Nair et al., 2003; Scott and Singer, 2004; Palmer et al., 2005). Smokers also exhibit lower levels of pro-inflammatory cytokines at diseased sites (Bostrom et al., 1999; Shirodaria et al., 2000; Erdemir et al., 2004; Goutoudi et al., 2004; Petropoulos et al., 2004). This represents a clinical conundrum to oral health professionals who must diagnose periodontal diseases, and a scientific enigma to those attempting to understand the mechanisms underlying tobacco-induced and / or exacerbated periodontitis.
It is known that tobacco smoke and specific smoke components and/or metabolites can exert a profound negative influence on the host response (Barbour et al., 1997; Kinane and Chestnutt, 2000; Scott et al., 2001; Johnson and Hill, 2004; Mullally, 2004; Palmer et al., 2005; Rehani et al., 2008). For example, in myelocytic cells we have recently shown that nicotine and/or its primary metabolite, cotinine, suppresses the pro-inflammatory cytokine response to P. gingivalis while promoting the release of the anti-inflammatory cytokine IL-10 (Rehani et al., 2008). Nicotine also suppresses the production of reactive oxygen species in response to Gram negative stimuli, concomitant with a reduced capacity to kill phagocytosed P. gingivalis cells (Xu et al., 2008).
In contrast, there are essentially no data available on how components in cigarette smoke may influence the organisms that comprise the oral microbial biofilm. Thus, it is possible that smoking not only influences the host response but also the organisms in the dental biofilm and that the increased severity of periodontitis in smokers may be attributed to both of these effects. We hypothesized that cigarette smoke extract (CSE) represents an environmental stress to P. gingivalis and that the organism adapts to this stress by altering its pattern of gene expression. In this report, we show that the pro-inflammatory response of primary human peripheral blood mononuclear cells (PBMCs) against P. gingivalis is significantly reduced when the bacteria are pre-treated with CSE and that this effect was reversed when CSE treated bacteria were sub-cultured in fresh growth medium without CSE. The response of P. gingivalis to CSE exposure was examined using both biochemical and molecular biologic approaches. Numerous genes, including those encoding outer membrane proteins and virulence determinants were differentially expressed upon CSE exposure. These results may explain at least in part the altered virulence and host-pathogen interactions associated with cigarette smoking.
Results
P. gingivalis growth in CSE-conditioned medium
To determine if a physiologically relevant dose of CSE (500 ng ml-1 nicotine equivalents [see Experimental Procedures]; (McGuire et al., 1989; Chen et al., 2001; Fraser et al., 2001; Scott et al., 2001) was overtly toxic to P. gingivalis W83 cells, we compared the growth rates of bacteria in CSE-conditioned and non-conditioned medium. As shown in Figure 1, similar growth characteristics were observed, suggesting that P. gingivalis W83 tolerates this level of CSE. Similar results were obtained for bacteria grown in CSE with 4000 ng ml-1 nicotine equivalents. CSE is not overtly toxic at this high concentration. These data are in agreement with Cogo et al, who have recently reported that nicotine and cotinine, the primary nicotine metabolite in humans, does not reduce or stimulate P. gingivalis growth (Cogo et al., 2008).
Figure 1. Typical growth curves of P. gingivalis W83 in CSE-conditioned and unconditioned medium.
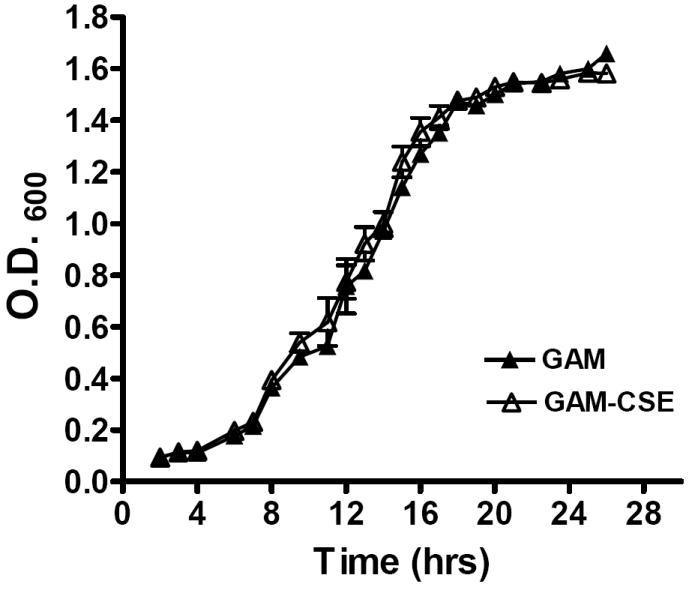
Growth of P. gingivalis was compared in GAM medium and GAM conditioned with CSE (500 ng ml-1 nicotine equivalents). Closed triangles represent growth in GAM. Open triangles represent growth in GAM-CSE. Error bars represent the mean (s.d.) of 3 experiments. There were no significant differences in the growth characteristics of P. gingivalis cultured in GAM or GAM-CSE (p > 0.05).
Induction of TNF-α and other pro-inflammatory cytokine release from innate cells by CSE-exposed P. gingivalis
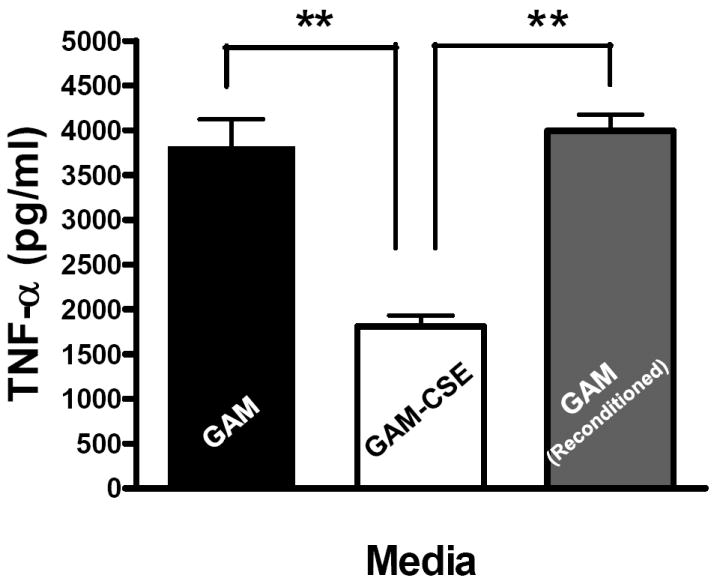
Evidence from in vivo studies in humans that have shown that smokers exhibit reduced clinical inflammation in response to pathogenic plaque bacteria (Scott and Singer, 2004; Palmer et al., 2005) and exhibit decreased gingival crevicular fluid (GCF) concentrations of major pro-inflammatory mediators, such as TNF-α and IL-1 (Bostrom et al., 1999; Shirodaria et al., 2000; Erdemir et al., 2004; Goutoudi et al., 2004; Petropoulos et al., 2004) and increased GCF levels of anti-inflammatory cytokines including IL-10 and TGF-β1 (Goutoudi et al., 2004; Stein et al., 2004). To determine if exposure of P. gingivalis to CSE alters its inflammatory potential, primary human monocytes were challenged with control and CSE-treated bacteria. As shown in Figure 2a, the release of TNF-α was significantly lower when monocytes were incubated with CSE-treated bacteria. Interestingly, when CSE-treated organisms were sub-cultured in fresh growth medium without CSE, their inflammatory potential increased back to the level of untreated bacteria (Figure 2b), suggesting that the CSE-induced effect is reversible. Thus, P. gingivalis appears to reversibly respond to CSE as an environmental stress. The production of multiple pro-inflammatory cytokines (TNF-α, IL-6 and IL-12 p40) by CSE-exposed and control P. gingivalis was also screened in PBMC’s. Production of TNF-α, IL-6 and IL-12 p40 was reduced in PBMCs stimulated with CSE-exposed bacteria compared to untreated P. gingivalis controls and, again, the inflammatory potential of P. gingivalis was restored on reconditioning in fresh GAM (Figure 3).
Figure 2. Reversible suppression of TNF-α release by human monocytes stimulated with CSE-treated P. gingivalis.
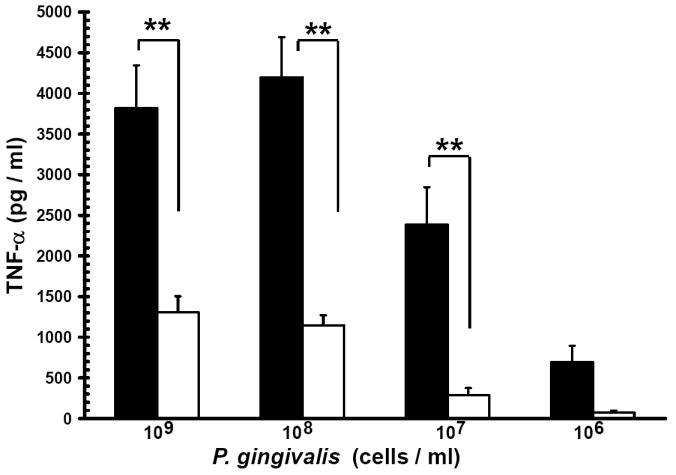
(a) Reduced TNF-α release by human monocytes stimulated with CSE-treated P. gingivalis.
Primary human monocytes (0.5 × 106) were stimulated with 106 to 109 cells of control P. gingivalis (black bars) or GAM-CSE (grey bars) grown P. gingivalis cells (20h). Cell-free supernatants were harvested by centrifugation and levels of TNF-α were determined by ELISA. Error bars represent the mean (s.d.) of 3 experiments.
** Indicates statistical significance at p < 0.01.
(b) Reconditioning of P. gingivalis in GAM rescues pro-inflammatory potential.
Primary human monocytes (0.5 × 106) were stimulated for 20 h with GAM cultured P. gingivalis; CSE-treated P. gingivalis; or P. gingivalis cells that were first grown in GAM-CSE for two passages then reconditioned in untreated GAM for 2 passages (107 P. gingivalis cells). Monocytes stimulated with reconditioned P. gingivalis produced levels of TNF-α that were similar to monocytes that were stimulated with bacteria from control cultures (not exposed to CSE). Error bars represent the mean (s.d.) of 3 experiments.
** Indicates statistical significance at p < 0.01.
Figure 3. Reversible suppression of multiple pro-inflammatory cytokines in human PBMCs stimulated with CSE-treated P. gingivalis.
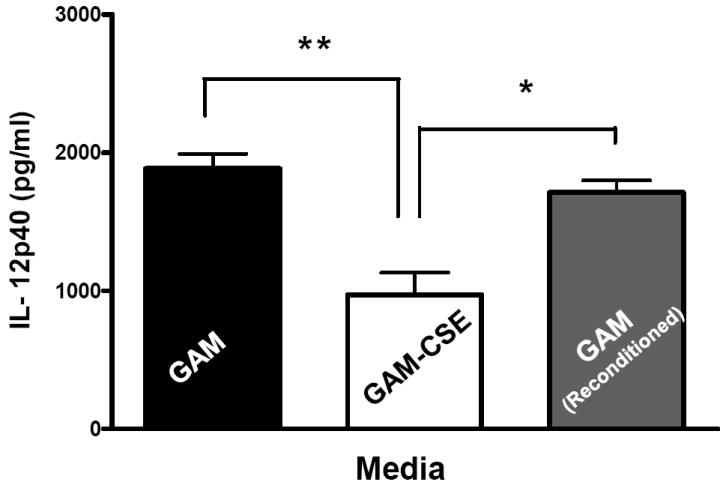
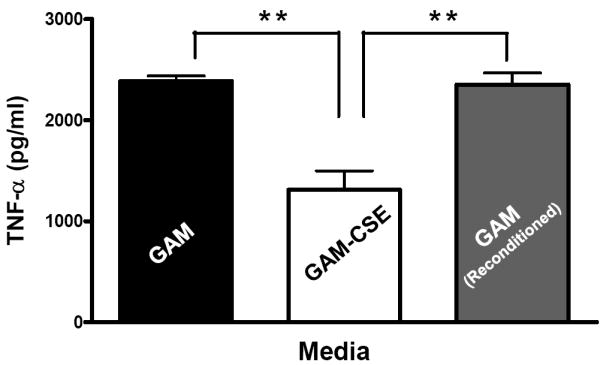
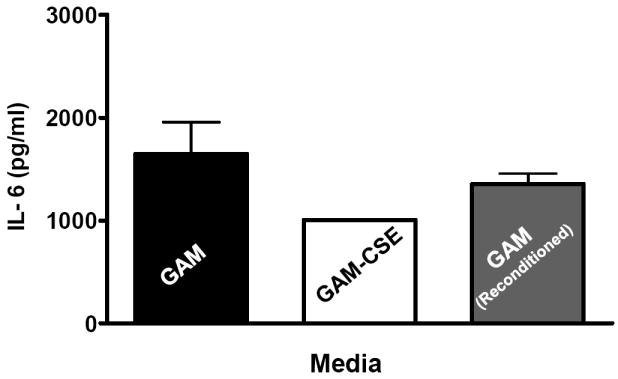
Primary human PBMCs (0.5 × 106) were stimulated for 20 h with GAM cultured P. gingivalis; CSE-treated P. gingivalis; or P. gingivalis cells that were first grown in GAM-CSE for two passages then reconditioned in untreaαted GAM for 2 passages (107 P. gingivalis cells). PBMCs stimulated with reconditioned P. gingivalis produced levels of TNF-α that were similar to PBMCs that were stimulated with bacteria from control cultures (not exposed to CSE). Error bars represent the mean (s.d.) of 3 experiments.
* Indicates statistical significance at p < 0.05.
** Indicates statistical significance at p < 0.01.
P. gingivalis genes up-regulated on CSE-exposure
In order to further characterize the response of P. gingivalis to CSE-exposure, we performed microarray analyses to identify genes that are differentially expressed in CSE-treated versus control bacteria. A total of 104 genes (approximately 4.7% of the P. gingivalis genome) were found to be differentially expressed by 2-fold or more (p < 0.05); 58 genes were induced and 46 were suppressed upon CSE treatment (see Figure 4). Of the 58 genes that were induced by CSE treatment, multiple genes within several predicted operons (based upon the gene annotation of the P. gingivalis genome sequence by TIGR) were identified, including the major fimbrial operon (2 genes), an operon encoding outer membrane antigenic lipoproteins (4 genes); the transfer (tra) gene cluster (4 genes) and an operon encoding an ABC transporter of unknown function [PG0682 – PG0685; 3 genes]. Within the CSE-induced genes, several functional families predominate. One group of genes encode proteins that may be involved in DNA replication and repair (e.g. ruvC [PG1324, holliday junction endonuclease]; PG0817 encoding a putative integrase; and radC [PG0894, DNA repair protein]) or in the insertion and transposition of genetic material (e.g., traE [PG1483]; traF [PG1482]; and traK [PG1478], each encoding proteins associated with a conjugative transposon). Several other transfer genes (traG [PG1481] and traQ [PG1473]) from this same region of the P. gingivalis genome were differentially expressed but fell just below the 2-fold limit used in the analysis. In addition, several of the insertion sequences that are present in the P. gingivalis genome were differentially expressed (ISPg3 [PG0299]; ISPg5 [PG0943]; ISPg6 [PG1061].
Figure 4. P. gingivalis genes differentially expressed following CSE exposure.
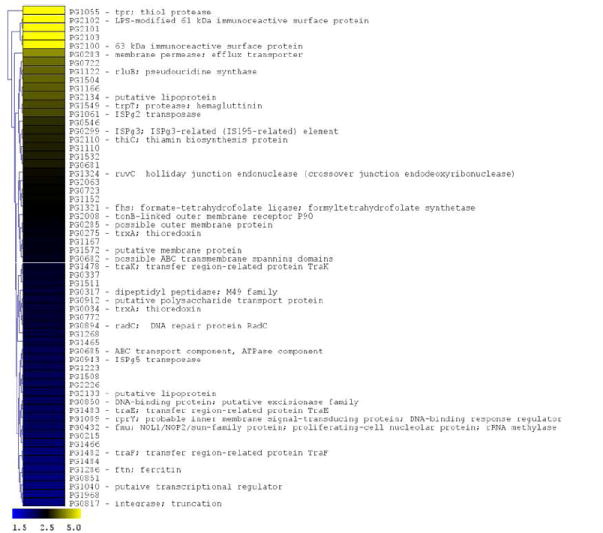
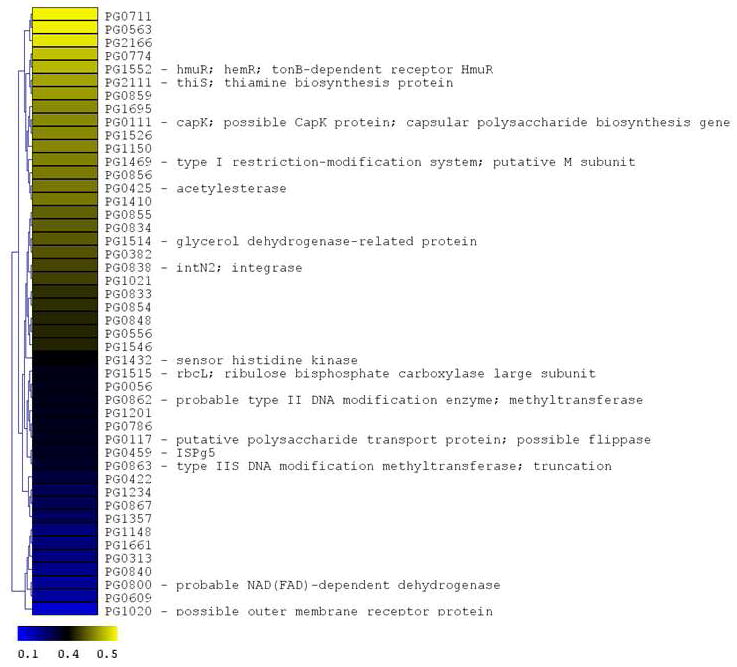
The microarray data of Log2 ratios were uploaded onto MultipleExperiment viewer 4.1v (January 8, 2008 release) software and evaluated by t-test with p value based on ‘t’ distribution with significance (alpha cut-off) set at p <0.01. The subset of significant genes were divided into two groups based on i) upregulated genes (>1.5 fold increase) and ii) down regulated genes (<0.5 fold). The distance was calculated based on Euclidean distance with average linkage and the tree was generated based on hierarchical clustering. CSE-induced genes are shown in Figure 4a (significantly induced genes >1.5 fold compared to baseline; p < 0.01; genes are represented in yellow to blue colour with the lower limit = 1.5; midpoint value = 2.5; and upper limit = 5). CSE-suppressed genes are shown in Figure 4b (significantly down-regulated genes < 0.5 fold compared to baseline; p < 0.01; genes are represented in yellow to blue colour with the lower limit = 0.1; midpoint value = 0.4; and upper limit = 0.5).
A second group of CSE-induced genes encode proteins involved in potential pathogen-host interactions and virulence. These include several proteases (encoded by tpr, trpT and PG0317), an efflux transporter (PG0283) related to the bacterial secretion system I protein HlyD, and a putative polysaccharide transport protein related to O-antigen flippase (PG0912). In addition, two genes encoding putative lipoproteins (PG2133 and PG2134) that are required for the assembly of the major fimbriae of P. gingivalis (Wang et al., 2007) were induced by CSE. These genes are co-expressed with fimA encoding the fimbrial subunit protein. Several other genes encoding cell surface or outer membrane polypeptides were also differentially expressed including the co-expressed major immunoreactive 61 and 63 KDa surface antigens encoded by PG2102 and PG2100, respectively, putative lipoproteins (PG1233, PG1234 and PG0722), and an outer membrane TonB-dependent receptor (PG2008). Other genes were differentially expressed but fell just below the 2-fold limit used in the analysis, including the lpt-1 (ptp1, PG1641) gene that has recently been shown to be a multifunctional regulator of virulence (Maeda et al., 2008).
To verify the results of the microarray experiments, the expression of select genes from the array dataset were analyzed by RT-PCR. As shown in Table 2, the RT-PCR results of the genes that were tested were consistent with the increased expression of these genes observed in the microarray analyses of CSE-treated cells.
Table 2.
Real time PCR analysis of selected CSE-regulated P. gingivalis genes
| Locus name* | Putative identification in TIGR and/or Oralgen database | Microarray (fold [mean, s.d.]) | RT_PCR (fold [mean, s.d.]) |
|---|---|---|---|
| PG1055 | Thiol protease (Tpr) | 13.93, 4.73 | 108.69, 21.43 |
| PG2102 | LPS-modified immunoreactive 61 kDa antigen | 10.58, 1.47 | 36.62, 1.44 |
| PG2100 | Immunoreactive 63 kDa antigen | 6.00, 1.23 | 30.79, 1.47 |
| PG2008 | tonB-linked outer membrane receptor P90 | 2.50, 2.00 | 1.74, 0.09** |
| PG1286 | Ferritin (ftn) | 2.02, 0.85 | 2.23, 0.21 |
| PG1552 | tonB-dependent receptor (HmuR, HemR) | 0.47, 0.29 | 0.72, 0.02*** |
| PG0111 | Putative capsular polysaccaharide biosynthesis gene (CapK) | 0.45, 0.04 | 0.78, 0.02*** |
| PG1432 | Sensor histidine kinase | 0.39, 0.10 | 0.62, 0.05** |
| PG0117 | Putative polysaccharide transport protein | 0.36, 0.14 | 0.50, 0.08 |
The J. Craig Venter Institute annotations
>1.5, <2.0 fold change in gene activity
Reduced activity not reaching threshold (<1.5 fold)
P. gingivalis genes down-regulated on CSE-exposure
Many of the 46 genes (2.1% of the P. gingivalis genome) that were suppressed by two-fold or more (p < 0.05) encode hypothetical proteins (see Figure 4) whose functions have yet to be determined. However, two genes in the capsular biosynthesis locus (Aduse-Opoku et al., 2006) were down-regulated - a putative capsular polysaccharide synthesis gene (capK, PG0111) and a polysaccharide transport protein related to a flippase (PG0117). The expression of two other genes in the capsular biosynthesis locus, encoding a glycosyltransferase (PG0118) and a UDP-N-acetyl-D-mannosaminuronic acid dehydrogenase (wecC, PG0108) were down-regulated just below the 2-fold limit used in the analysis. Also down-regulated in response to CSE was a sensor histidine kinase (fimS) which together with the co-expressed response regulator FimR has been reported to regulate the expression of the major and minor fimbrial operons of P. gingivalis (Wu et al., 2007). Lastly, the expression of hmuR (PG1552) encoding a tonB-dependent hemoglobin receptor HmuR was reduced by approximately 2-fold by CSE treatment. Select down-regulated genes were analyzed by RT-PCR (see Table 1) with consistent results.
Table 1.
Primers employed in real time PCR analysis of selected CSE-regulated P. gingivalis genes
| Gene | Forward primer | Reverse primer |
|---|---|---|
| PG1055; Tpr | CCTACAGATTGGAGGTGGCT | ATAGGCATGGTATGCTGCAA |
| PG2102; 61kDa antigen | ATCGTTTGGTCTGATACGCA | CTGCACGTTCAGCCTGTATT |
| PG2100; 63 kDa antigen | ATTACAAGATGGCTGTGGCA | TGCTGTCATGACTGTCCAAA |
| PG2008; TonB-linked receptor | ATTCTTAGGAACGAGCGCAT | GGGATTCCCTTGATCGAGTA |
| PG1286; Ferritin | AGCGATCAATGACCAAATCA | CACGGCATCGATAGCTTCT |
| PG1552; HemR | GATTTGAAGCCGGAGAAGAG | TAAGGCAGTACCACATTCGC |
| PG0111; CapK | AGGCAACGGAGAAGTATCGT | AAAGCACCATCAATGACGAA |
| PG1432; Sensor histidine kinase | ACAGCTCGAACTGCATCAAC | CTGCATATAAGTGCGGGCTA |
| PG0117; polysaccharide transport protein | TCTCGAACGACCAATAGTGC | CATTCTCTGCAATCGGCTTA |
Primers were designed using the real-time PCR primer design software provided by GenScript (www.genscript.com/ssl-bin/app/primer). Tm of primers was 60 ±2°C (PG1286 forward primer, Tm = 56°C).
CSE alters expression of P. gingivalis outer membrane proteins RagA and RagB
In addition to the microarray analysis described above, a membrane fraction that was enriched for P. gingivalis outer membrane proteins was isolated from CSE-treated and control cells and analyzed by SDS gel electrophoresis. As shown in Figure 5, the Triton X-100 insoluble membrane fraction contained approximately ten major polypeptide bands, of which three were clearly overrepresented in the extract from CSE-treated bacteria. These bands were excised from the gel and analyzed by MALDI-MS after digestion with trypsin. Tryptic peptides were identified for the RagA (43% sequence coverage), RagB (66% coverage) and a putative lipoprotein (PG0179, 53% coverage) that is likely co-expressed with the minor fimbrial antigen of P. gingivalis, based on the TIGR (J. Craig Venter Institute) annotation of the P. gingivalis genome sequence. Interestingly, western blots of RagB proteins revealed that reconditioning of P. gingivalis in GAM, following culture in GAM-CSE, resulted in RagB expression levels returning close to control levels (106%), as determined by densitometry (Figure 6).
Figure 5. Up-regulation of specific outer membrane protein (OMP) in CSE-exposed P. gingivalis.
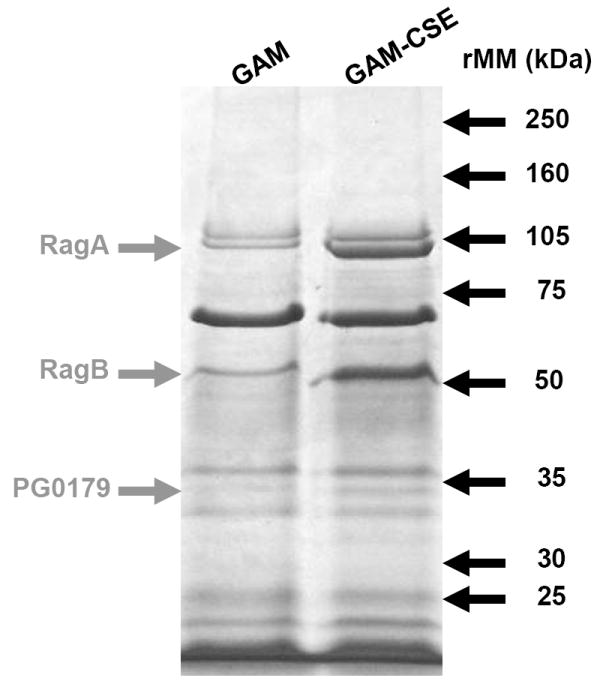
Outer membrane proteins of P. gingivalis grown in GAM and GAM-CSE were isolated by differential extraction with Triton X-100. 20μg total OMP extracts were run through a 4-15% gradient gel. MALDI-MS of excised bands followed by comparison with the ORF database of P. gingivalis W83 from The J. Craig Venter Institute identified the major CSE up-regulated bands, as shown.
Figure 6. CSE-induced RagB upregulation in P. gingivalis is reversible.
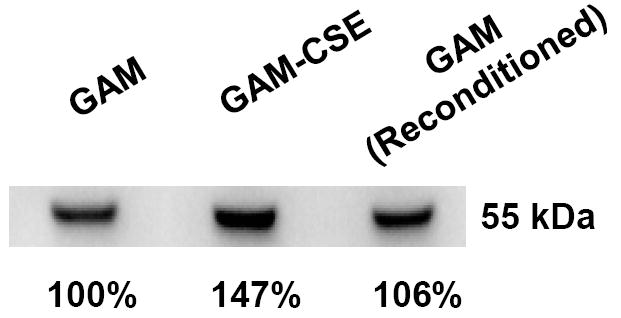
A typical western blot of RagB in lysates of cells sequentially passaged in GAM, GAM-CSE and GAM, respectively, is shown. Culture in GAM-CSE resulted in an increase in relative RagB expression levels (147%). Relative RagB expression returned to near control levels on reconditioning in GAM.
Discussion
Cigarette smoking increases vulnerability to P. gingivalis infection and increases susceptibility to periodontitis, but reduces clinical signs of overt inflammation. The mechanisms underlying this dichotomy remain to be determined. We show that P. gingivalis exposed to CSE induces a significantly lower pro-inflammatory response from monocytes and PMBCs than control bacteria grown without CSE. Furthermore, this effect is reversed when CSE-treated bacteria are sub-cultured in medium without CSE, suggesting that CSE exposure may represent an environmental stress that P. gingivalis is able to specifically respond to. These results are in keeping with several in vivo reports of reduced levels of pro-inflammatory cytokines at diseased sites (Bostrom et al., 1999; Shirodaria et al., 2000; Erdemir et al., 2004; Goutoudi et al., 2004; Petropoulos et al., 2004).
Our results clearly show that the response of P. gingivalis to CSE exposure is varied and genome wide. Approximately 7% of the genes in the P. gingivalis genome were found to be differentially expressed upon exposure to CSE. A number of the differentially expressed genes have been associated with aspects of P. gingivalis virulence. Several functionally-related genes were dysregulated, including multiple genes in the major fimbrial and capsular polysaccharide operons, as well as genes encoding transcriptional regulators; efflux pump and transport proteins; proteases and cell envelope proteins. The major fimbriae have been shown to be important for numerous aspects of P. gingivalis virulence, including bacterial adhesion and invasion of epithelial and endothelial cells (Lamont & Jenkinson, 1998 and 2000). PG2133 and PG2134 encode putative lipoproteins that are co-expressed with FimA, the major fimbrial subunit protein. Wang et al. recently showed that inactivation of these genes results in the expression of significantly shorter fimbriae, suggesting that these putative lipoproteins play an essential role in fimbrial biogenesis. The mutant strains also exhibited reduced invasion and intracellular persistence in macrophages and were less virulent in a mouse model of periodontitis (Wang et al., 2007).
There are at least six different capsular serotypes (K1 to K6) expressed by P. gingivalis and altered virulence has been associated with K+ and K- strains (van Winkelhoff et al., 1993; Laine and van Winkelhoff, 1998; Lamont and Jenkinson, 1998; Califano et al., 1999). While the contribution of P. gingivalis capsule to inflammation is poorly understood, recent evidence shows that capsular polysaccharide from several strains of P. gingivalis, including W83 elicits a substantial pro-inflammatory cytokine response from murine innate cells (d’Empaire et al., 2006). The down regulation of several genes in the capsular biosynthesis operon (PG0111 and PG0117) suggests that capsule synthesis may be reduced upon exposure to CSE, consistent with the reduced inflammatory potential of CSE-treated bacteria.
In addition to the differential expression of genes described above, several other cell surface or outer membrane proteins, i.e., RagA, RagB and PG0179 were shown by biochemical approaches to be present at higher levels after CSE treatment. Interestingly, the genes encoding for these components were not identified as being differentially expressed in the microarray experiments. This suggests that post-transcriptional events may also be involved in the response to CSE, or alternatively may reflect inherent limitations in the microarray approach. Each of the differentially expressed proteins has been associated with aspects of P. gingivalis virulence. The ragAB locus was likely acquired by horizontal gene transfer (Curtis et al., 1999). RagA is a putative tonB-dependent outer membrane receptor whereas RagB is an immunodominant lipoprotein. Both genes have reported to be up-regulated under conditions of thermal stress (Bonass et al., 2000) and both represent significant P. gingivalis virulence factors (Hall et al., 2005; Shi et al., 2007), although their specific contribution to periodontal disease remains to be determined. However, it is interesting that the RagA homolog (designated OmpA) from the closely related bacterium Bacteroides caccae has been associated with inflammatory mucosal (bowel) disease (Wei et al., 2001). PG0179 is a gene that is co-expressed with the minor fimbrial antigen (mfa1). The minor fimbriae play a key role in P. gingivalis autoaggregation (Lin et al., 2006) and are critical mediators of the interspecies interactions between Porphyromonas gingivalis and oral streptococci that facilitate biofilm formation (Lamont et al., 2002; Daep et al., 2006; Daep et al., 2008). Thus, stimulation of the minor fimbrial operon by CSE may facilitate increased P. gingivalis colonization of the periodontia.
Lastly, a number of genes that were differentially expressed encoded proteins involved in DNA replication, DNA repair and the transfer or mobilization of genetic material. Comparison of the recently completed genome sequence of P. gingivalis ATCC 33277 with that of strain W83 indicates that mobile genetic elements have contributed greatly to genomic diversity among P. gingivalis strains (Naito et al., 2008). The up regulation of transfer genes and several insertion sequence elements in CSE-exposed cells suggests that the environmental stress imposed by cigarette smoke may stimulate genetic rearrangements. In addition, considering the highly carcinogenic potential of cigarette smoke, the up-regulation of DNA-repair and -control genes is not surprising. Cigarette smoke is also a major oxidative stressor (Yamaguchi et al., 2007; Lagente et al., 2008). A single cigarette is estimated to contain 1016 oxidant molecules (Pryor et al., 1986). Thus, the up regulation of ferritin [PG1286], an iron sequestrator, may provide protection against free-iron-related oxidative stress (Ratnayake et al., 2000). In addition, the response regulator, RprY (also up regulated after CSE treatment) has recently been shown to be responsive to reactive oxygen species and iron (Duran-Pinedo et al., 2007). It is also interesting to note that several other anti-oxidant genes were differentially expressed (p<0.05) when the threshold for induction in the array experiment was reduced from 2-fold to 1.5-fold. In addition, the up-regulation of efflux pump and other ABC transport systems may confer resistance to potentially harmful chemicals present in CSE. Taken together, these results suggest that CSE may represent a potent environmental stressor that induces a protective response from P. gingivalis.
In summary, smokers are more prone to infection with P. gingivalis and to develop periodontitis, yet exhibit reduced clinical inflammation. Consistent with this, we have found that CSE-exposed P. gingivalis exhibits a reduced capacity to illicit an inflammatory response from immune cells. P. gingivalis responds globally to CSE exposure and multiple virulence associated genes are differentially expressed. These results may explain in part the altered virulence and host-pathogen interactions that occur in smokers with periodontal disease and provide some of the first information illustrating how P. gingivalis responds at the molecular level to cigarette smoke.
Experimental procedures
Materials
P. gingivalis W83 was purchased from the American Type Culture Collection, Manassas, VA; medium supporting P. gingivalis growth (Gifu Anaerobe Medium [GAM]) was bought from Nissui Pharmaceutical, Tokyo; Histopaque 1077, lysozyme and protease inhibitor cocktail came from Sigma, St. Louis, MO; RNA Midi and PCR purification kits from Qiagen, Valencia, CA; random hexamers were from Biosynthesis, Lewiston, TX; RT buffer, DTT and ABI Power SYBR Green Master Mix were from Applied Biosystems, Foster City, CA; 2:3 aminoallyl dUTP:dTTP ratio labeling mix was purchased from Amersham Biosciences, Piscataway, NJ; HRP-linked anti-Rabbit IgG was from Cell Signaling Technology, Danvers, MA; while BCA protein assay kit and SuperSignal West Pico Chemiluminescent Substrate kits were bought from Pierce, Rockford, IL. ELISA kits were purchased from eBioscience, Inc. (San Diego, CA; IL-6 and TNF-α) or BD Biosciences (San Jose, CA; IL-12 p40). RPMI 1640, fetal bovine serum, L-glutamine, HEPES, penicillin G and streptomycin were bought from Invitrogen Life Technologies, Carlsbad, CA. P. gingivalis W83 microarrays were obtained from The J. Craig Venter Institute through the NIDCR Oral Microbe Microarray Initiative (NOMMI). The E38 monoclonal anti-RagB antibody was a kind gift of Dr. Mike A. Curtis of The London, Queen Mary’s School of Medicine and Dentistry, UK.
Bacterial culture and in vitro modeling of tobacco exposure
P. gingivalis W83 was maintained as a frozen stock culture. The use of individual tobacco components/metabolites (e.g. nicotine, cotinine, benzopyrene, pyridines, free radicals, etc) in in vitro experimental models is comparably straight-forward, but may not reflect in vivo complexities. Cigarette smoke contains more than 4000 different compounds, many of which are toxic and/or bioactive. Cigarette smoke extract- (CSE) conditioned GAM (GAM-CSE) was prepared using standard reference cigarettes (2R1, Kentucky Tobacco Research and Development Center). Using a three-way stopcock, cigarette smoke was drawn through GAM by syringe and expelled before a new volume of smoke was drawn through the media. Constituents of tobacco smoke change dramatically at differing combustion temperatures (Bernard, 2005). Therefore, each cigarette was “smoked” in 35 ml “drags.” A “drag” was performed over two seconds, and one was performed every 20 seconds. CSE-conditioned medium was filtered (0.22 μm) and diluted to the required concentration in nicotine equivalents [determined by GLC as we reported previously (Fraser et al., 2001)] and adjusted to pH 7.2. We utilized GAM-CSE at a dose physiologically relevant to P. gingivalis in situ in the sub-gingival environment of the oral cavity, 500 ng ml-1 to 4000 ng ml-1 nicotine equivalents (McGuire et al., 1989; Chen et al., 2001; Fraser et al., 2001; Scott et al., 2001). To examine the response of P. gingivalis to CSE, cells were grown to mid- to late exponential phase (O.D.600nm = 1.0; ≈ 1 × 109 cells ml-1) in GAM or in GAM-CSE under anaerobic conditions (80% N2, 10% H2, 10% CO2) at 37°C in a Coy Laboratories anaerobic chamber.
Isolation and culture of primary human monocytes and PBMCs
Whole, citrated (10%) venous blood was obtained from healthy donors (University of Louisville, Institutional Review Board, Human Subjects Protection Program, study number 503.05). Primary monocytes were isolated by an indirect magnetic monocyte isolation kit (Miltenyi Biotec, Auburn, CA), as we have previously reported (Martin et al., 2003b; Martin et al., 2005). This procedure routinely results in >95% pure CD14+ cells, as shown by flow cytometry, with > 98% viability, as determined by trypan blue exclusion. PBMCs were isolated by separation and collection of the buffy coat and elimination of erythrocyte contamination with Histopaque 1077 density gradients, as previously described (Martin et al., 2003a). This procedure routinely results > 98% PBMC viability, as determined by trypan blue exclusion. Human monocytes and PBMCs were cultured at 37°C and 5% CO2 atmosphere, in complete RPMI (RPMI 1640 supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin G, 100 μg/ml streptomycin) plus or minus stimulating agents, as described below.
Pro-inflammatory cytokine release from P. gingivalis-stimulated innate cells
Primary human monocytes (0.5 × 106 cells per well) were stimulated with 106 – 109 P. gingivalis grown in GAM or GAM-CSE, as detailed in Figure 2a and b. Cell-free supernatants were harvested by centrifugation (3,200 × g, 8 mins) and TNF-α cytokine levels were determined by ELISA. The production of multiple pro-inflammatory cytokines (TNF-α, IL-6 and IL-12 p40) by CSE-exposed and control P. gingivalis was also screened in PBMC’s, as presented in Figure 3.
Porphyromonas gingivalis W83 microarrays
P. gingivalis W83 microarrays were obtained from The J. Craig Venter Institute through the NIDCR Oral Microbe Microarray Initiative (NOMMI). To determine how exposure to CSE influences the activity of the P. gingivalis genome, we compared gene expression in P. gingivalis W83 grown in control (GAM) and conditioned (CSE-GAM) media. Microarrays were performed on triplicate GAM and GAM-CSE cultures. Total RNA from control and experimental cultures was isolated by extraction from washed cells with hot phenol followed by chloroform extraction and isopropanol precipitation. RNA was purified and genomic DNA was removed using a Qiagen Midi kit with on-column DNAse treatment followed by further digestion with RNAse-free DNAse to remove any trace contamination of genomic DNA. The synthesis of labeled cDNA was carried out by methods established by The J. Craig Venter Institute (www.jcvi.org). Briefly, 2-2.5μg of total RNA was mixed with 500ng of random hexamers, incubated at 70°C for 10’ and transferred to ice. A 50 × 2:3 aminoallyl dUTP:dTTP ratio labeling mix containing 25 mM remaining nucleotides, 5 × RT buffer, 3 μl of 0.1 M DTT and 2 μl Superscript II (200U/μl) were added. Samples were then incubated for 10’ at room temperature and transferred to 42°C, overnight. Reactions were stopped with 10 μl of 1N NaOH and 10 μl of 0.5 M EDTA for 15’ at 65°C and neutralized with 25 μl 1M Tris, pH 7.4. Unincorporated aa-dUTP and free amines were removed using a Qiagen QIAquick PCR purification kit protocol with phosphate buffers. The cDNA was then dried down in a speed vacuum and resuspended in 4.5 μl of 0.1 M sodium carbonate buffer, pH 9.0. The appropriate NHS-ester Cy3 or Cy5 dye in DMSO was added (4.5 μl) and the reaction incubated at room temperature in the dark for 2 hours. The reaction was neutralized by adding 35 μl of 100 mM sodium acetate, pH 5.2. The labeled cDNA was purified to remove any uncoupled dye using the Qiagen PCR purification kit with Minelute columns and an extra wash. The labeled cDNA was quantified and evaluated by spectrophotometry and equivalent pmol of Cy3 and Cy5 samples were combined, dried down, and stored at -80°C. Hybridization and subsequent analysis of the arrays will be carried out following protocols established by The J. Craig Venter Institute, essentially as described by (Hegde et al., 2000). Arrays were pre-hybridized at 42°C for 1 hr in 5 × SSC/0.1%SDS/ 1% BSA and washed several times with MilliQ water. Washed arrays were then dipped into isopropanol and dried by centrifugation at 1000 rpm for 10 minutes. For hybridization, Cy3/Cy5 probe mixtures were suspended in 50 μl hybridization buffer (50% formamide, 5 × SSC, 0.1%SDS, 0.1 mM DTT, 0.56 μg/ml sheared salmon sperm DNA), heated then applied to the array under a Lifter™ cover slip. The array was incubated at 42°C in a sealed chamber for 15-20 hr. After hybridization, the array was washed twice in 2 × SSC/0.1% SDS at 55°C, twice in 0.1 × SSC/0.1%SDS, once with 0.1 × SSC, water rinsed, and dried. Arrays were then scanned using an Axon slide reader, scanning in the Cy5 channel and the Cy3 channel. Identification and calculation of spots was carried out using the TIGR Spotfinder software (The J. Craig Venter Institute; http://www.jcvi.org/cms/research/software/). Subsequent normalization of fluorescent data was conducted using TIGR-MIDAS (also freely available from The J. Craig Venter Institute). Subtraction of local background and integration of Cy5 and Cy3 fluorescent intensities was calculated using Excel. Array experiments were carried out in triplicate using independently isolated RNA samples. Up- and down-regulated genes (>1.5, < 0.5, p < 0.01) are presented in Figure 4.
Validation of array data
Differentially expressed genes of interest were INDIVIDUALLY confirmed by quantitative real-time reverse transcription-PCR using an Applied Biosystems 7500 Real-Time PCR system by standard methodology (James et al., 2006). Briefly, forward and reverse primers (1.25 μl, 50 pmol each), ABI Power SYBR Green Master Mix (12.5 μl), and cDNA (10 μl, 20 ng per reaction) were added to a 96 well reaction plate. Samples were quantified using standard cycling conditions (50°C, 2 mins; 95°C 10 mins; followed by forty cycles of 95°C, 15 secs and 60 °C, 1 min). Primer details are provided in Table 1.
Porphyromonas gingivalis W83 outer membrane protein profiling
Outer membrane preparations were obtained from P. gingivalis cells grown in GAM or GAM-CSE. Briefly, the cell pellet was suspended in 10mM Tris, pH 7.5, 1mM EDTA (TE) containing 100 μg/ml lysozyme and protease inhibitor cocktail and was incubated for 15 minutes at 25°C. The cell suspension was sonicated three times on ice and cellular debris was removed by centrifugation at 1000 × g for 10 minutes. The supernatant was subsequently centrifuged at 100,000 × g for 60 minutes. The membrane pellet was suspended in 5 mls TE containing 0.5M NaCl and centrifuged again at 100,000 × g for 60 minutes. After discarding the supernatant, the membrane pellet was suspended in 5 ml MgCl2 containing 1% Triton X-100. Triton X-100 insoluble material was then collected by centrifugation at 100,000 × g. The resulting outer membrane fraction was suspended in TE containing 0.5% SDS and analyzed by PAGE. Outer membrane preparations (20 μg protein) were electrophoresed through 4-15% gradient SDS-PAGE gels and protein bands were visualized by staining with Coomassie brilliant blue R-250. Gel image analysis and densitometry were performed using the Kodak 4000MM Image Station system (Eastman Kodak, New Haven, CT). Protein identification was achieved by excision of the protein bands of interest (see Figure 5) from SDS-PAGE gels, followed by in-gel trypsin digestion, peptide preparation, MALDI-MS analysis, and bioinformatic identification by peptide mass fingerprinting using the core facilities at the University of Louisville core proteomics laboratory.
Expression of RagB in response to CSE
P. gingivalis cells were sequentially passaged in GAM, GAM-CSE and GAM, respectively. Bacterial cells were lysed (10mN Tris, 100mM NaCl and 1mM EDTA) and probed by western blot (20 μg protein per lane). Total protein estimated using a BCA protein assay kit. MAbE38, an anti-RagB antibody, was a kind gift from Mike A. Curtis. Anti-Rabbit HRP-linked IgG was used as the secondary antibody. Probing and visualization of immunoreactive bands was performed by chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate kit as per the manufacturer’s protocol. Gel image analysis and densitometry were performed using the Kodak 4000MM Image Station system.
Statistical analyses
All experiments were repeated a minimum of three times. The statistical approach to the analysis of the microarray data is presented in Figure 4.For all other data, statistical significance between groups was evaluated by ANOVA and the Tukey multiple-comparison test using the InStat program (GraphPad Software, San Diego, CA). Differences between groups were considered significant at the level of p < 0.05.
Supplementary Material
Acknowledgments
This study was kindly supported by an NIEHS pilot grant (DAS and DRD) from funds awarded to the Center for Environmental Genomics and Integrative Biology at the University of Louisville (P30ES014443-01A1, P.I. Kenneth Ramos).
References
- Aduse-Opoku J, Slaney JM, Hashim A, Gallagher A, Gallagher RP, Rangarajan M, et al. Identification and characterization of the capsular polysaccharide (K-antigen) locus of Porphyromonas gingivalis. Infect Immun. 2006;74:449–460. doi: 10.1128/IAI.74.1.449-460.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araco A, Gravante G, Sorge R, Araco F, Delogu D, Cervelli V. Wound infections in aesthetic abdominoplasties: the role of smoking. Plast Reconstr Surg. 2008;121:305e–310e. doi: 10.1097/PRS.0b013e31816b13c2. [DOI] [PubMed] [Google Scholar]
- Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164:2206–2216. doi: 10.1001/archinte.164.20.2206. [DOI] [PubMed] [Google Scholar]
- Barbour SE, Nakashima K, Zhang JB, Tangada S, Hahn CL, Schenkein HA, Tew JG. Tobacco and smoking: environmental factors that modify the host response (immune system) and have an impact on periodontal health. Crit Rev Oral Biol Med. 1997;8:437–460. doi: 10.1177/10454411970080040501. [DOI] [PubMed] [Google Scholar]
- Bernard D. In: Molecular Mechanisms of Tobacco-Induced Diseases. Wang XL, Scott DA, editors. Nova Science; 2005. [Google Scholar]
- Bonass WA, Marsh PD, Percival RS, Aduse-Opoku J, Hanley SA, Devine DA, Curtis MA. Identification of ragAB as a temperature-regulated operon of Porphyromonas gingivalis W50 using differential display of randomly primed RNA. Infect Immun. 2000;68:4012–4017. doi: 10.1128/iai.68.7.4012-4017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom L, Linder LE, Bergstrom J. Smoking and cervicular fluid levels of IL-6 and TNF-alpha in periodontal disease. J Clin Periodontol. 1999;26:352–357. doi: 10.1034/j.1600-051x.1999.260604.x. [DOI] [PubMed] [Google Scholar]
- Califano JV, Schifferle RE, Gunsolley JC, Best AM, Schenkein HA, Tew JG. Antibody reactive with Porphyromonas gingivalis serotypes K1-6 in adult and generalized early-onset periodontitis. J Periodontol. 1999;70:730–735. doi: 10.1902/jop.1999.70.7.730. [DOI] [PubMed] [Google Scholar]
- Chen X, Wolff L, Aeppli D, Guo Z, Luan W, Baelum V, Fejeskov O. Cigarette smoking, salivary/gingival crevicular fluid cotinine and periodontal status. A 10-year longitudinal study. J Clin Periodontol. 2001;28:331–339. doi: 10.1034/j.1600-051x.2001.028004331.x. [DOI] [PubMed] [Google Scholar]
- Cogo K, Montan MF, Bergamaschi Cde C, DA E, Rosalen PL, Groppo FC. In vitro evaluation of the effect of nicotine, cotinine, and caffeine on oral microorganisms. Can J Microbiol. 2008;54:501–508. doi: 10.1139/w08-032. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Hanley SA, Aduse-Opoku J. The rag locus of Porphyromonas gingivalis: a novel pathogenicity island. J Periodontal Res. 1999;34:400–405. doi: 10.1111/j.1600-0765.1999.tb02273.x. [DOI] [PubMed] [Google Scholar]
- d’Empaire G, Baer MT, Gibson FC., 3rd The K1 serotype capsular polysaccharide of Porphyromonas gingivalis elicits chemokine production from murine macrophages that facilitates cell migration. Infect Immun. 2006;74:6236–6243. doi: 10.1128/IAI.00519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daep CA, Lamont RJ, Demuth DR. Interaction of Porphyromonas gingivalis with oral streptococci requires a motif that resembles the eukaryotic nuclear receptor box protein-protein interaction domain. Infect Immun. 2008 doi: 10.1128/IAI.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daep CA, James DM, Lamont RJ, Demuth DR. Structural characterization of peptide-mediated inhibition of Porphyromonas gingivalis biofilm formation. Infect Immun. 2006;74:5756–5762. doi: 10.1128/IAI.00813-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Nishikawa K, Duncan MJ. The RprY response regulator of Porphyromonas gingivalis. Mol Microbiol. 2007;64:1061–1074. doi: 10.1111/j.1365-2958.2007.05717.x. [DOI] [PubMed] [Google Scholar]
- Dye BA, Choudhary K, Shea S, Papapanou PN. Serum antibodies to periodontal pathogens and markers of systemic inflammation. J Clin Periodontol. 2005;32:1189–1199. doi: 10.1111/j.1600-051X.2005.00856.x. [DOI] [PubMed] [Google Scholar]
- Eggert FM, McLeod MH, Flowerdew G. Effects of smoking and treatment status on periodontal bacteria: evidence that smoking influences control of periodontal bacteria at the mucosal surface of the gingival crevice. J Periodontol. 2001;72:1210–1220. doi: 10.1902/jop.2000.72.9.1210. [DOI] [PubMed] [Google Scholar]
- Erdemir EO, Duran I, Haliloglu S. Effects of smoking on clinical parameters and the gingival crevicular fluid levels of IL-6 and TNF-alpha in patients with chronic periodontitis. J Clin Periodontol. 2004;31:99–104. doi: 10.1111/j.0303-6979.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- Fraser HS, Palmer RM, Wilson RF, Coward PY, Scott DA. Elevated systemic concentrations of soluble ICAM-1 (sICAM) are not reflected in the gingival crevicular fluid of smokers with periodontitis. J Dent Res. 2001;80:1643–1647. doi: 10.1177/00220345010800070901. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–121. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- Goutoudi P, Diza E, Arvanitidou M. Effect of periodontal therapy on crevicular fluid interleukin-1beta and interleukin-10 levels in chronic periodontitis. J Dent. 2004;32:511–520. doi: 10.1016/j.jdent.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Grossi SG, Goodson JM, Gunsolley JC, Otomo-Corgel J, Bland PS, Doherty F, Comiskey J. Mechanical therapy with adjunctive minocycline microspheres reduces red-complex bacteria in smokers. J Periodontol. 2007;78:1741–1750. doi: 10.1902/jop.2007.070118. [DOI] [PubMed] [Google Scholar]
- Haffajee AD, Socransky SS. Relationship of cigarette smoking to the subgingival microbiota. J Clin Periodontol. 2001;28:377–388. doi: 10.1034/j.1600-051x.2001.028005377.x. [DOI] [PubMed] [Google Scholar]
- Hall LM, Fawell SC, Shi X, Faray-Kele MC, Aduse-Opoku J, Whiley RA, Curtis MA. Sequence diversity and antigenic variation at the rag locus of Porphyromonas gingivalis. Infect Immun. 2005;73:4253–4262. doi: 10.1128/IAI.73.7.4253-4262.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, et al. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29:548–550. 552–544, 556. doi: 10.2144/00293bi01. passim. [DOI] [PubMed] [Google Scholar]
- Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- James CE, Hasegawa Y, Park Y, Yeung V, Tribble GD, Kuboniwa M, et al. LuxS involvement in the regulation of genes coding for hemin and iron acquisition systems in Porphyromonas gingivalis. Infect Immun. 2006;74:3834–3844. doi: 10.1128/IAI.01768-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DL, Nielsen SD, Ersboll AK, Valerius NH. Maternal Smoking during Pregnancy Increases the Risk of Postnatal Infections in Preterm Neonates. Neonatology. 2008;94:75–78. doi: 10.1159/000113535. [DOI] [PubMed] [Google Scholar]
- Johnson GK, Hill M. Cigarette smoking and the periodontal patient. J Periodontol. 2004;75:196–209. doi: 10.1902/jop.2004.75.2.196. [DOI] [PubMed] [Google Scholar]
- Kamma JJ, Nakou M, Baehni PC. Clinical and microbiological characteristics of smokers with early onset periodontitis. J Periodontal Res. 1999;34:25–33. doi: 10.1111/j.1600-0765.1999.tb02218.x. [DOI] [PubMed] [Google Scholar]
- Keskinoglu P, Cimrin D, Aksakoglu G. The Impact of Passive Smoking on the Development of Lower Respiratory Tract Infections in Children. J Trop Pediatr. 2007 doi: 10.1093/tropej/fmm037. [DOI] [PubMed] [Google Scholar]
- Kinane DF, Chestnutt IG. Smoking and periodontal disease. Crit Rev Oral Biol Med. 2000;11:356–365. doi: 10.1177/10454411000110030501. [DOI] [PubMed] [Google Scholar]
- Kum-Nji P, Meloy L, Herrod HG. Environmental tobacco smoke exposure: prevalence and mechanisms of causation of infections in children. Pediatrics. 2006;117:1745–1754. doi: 10.1542/peds.2005-1886. [DOI] [PubMed] [Google Scholar]
- Lagente V, Planquois JM, Leclerc O, Schmidlin F, Bertrand CP. Oxidative stress is an important component of airway inflammation in mice exposed to cigarette smoke or lipopolysaccharide. Clin Exp Pharmacol Physiol. 2008;35:601–605. doi: 10.1111/j.1440-1681.2007.04848.x. [DOI] [PubMed] [Google Scholar]
- Laine ML, van Winkelhoff AJ. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol. 1998;13:322–325. doi: 10.1111/j.1399-302x.1998.tb00714.x. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, El-Sabaeny A, Park Y, Cook GS, Costerton JW, Demuth DR. Role of the Streptococcus gordonii SspB protein in the development of Porphyromonas gingivalis biofilms on streptococcal substrates. Microbiology. 2002;148:1627–1636. doi: 10.1099/00221287-148-6-1627. [DOI] [PubMed] [Google Scholar]
- Lin X, Wu J, Xie H. Porphyromonas gingivalis minor fimbriae are required for cell-cell interactions. Infect Immun. 2006;74:6011–6015. doi: 10.1128/IAI.00797-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Tribble GD, Tucker CM, Anaya C, Shizukuishi S, Lewis JP, et al. A Porphyromonas gingivalis Tyrosine Phosphatase is a Multifunctional Regulator of Virulence Attributes. Mol Microbiol. 2008 doi: 10.1111/j.1365-2958.2008.06338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Michalek SM, Katz J. Role of innate immune factors in the adjuvant activity of monophosphoryl lipid A. Infect Immun. 2003a;71:2498–2507. doi: 10.1128/IAI.71.5.2498-2507.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003b;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- McGuire JR, McQuade MJ, Rossmann JA, Garnick JJ, Sutherland DE, Scheidt MJ, Van Dyke TE. Cotinine in saliva and gingival crevicular fluid of smokers with periodontal disease. J Periodontol. 1989;60:176–181. doi: 10.1902/jop.1989.60.4.176. [DOI] [PubMed] [Google Scholar]
- Mullally BH. The Influence of Tobacco Smoking on the Onset of Periodontitis in Young Persons. Tobacco Induced Diseases. 2004;2:53–65. doi: 10.1186/1617-9625-2-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair P, Sutherland G, Palmer RM, Wilson RF, Scott DA. Gingival bleeding on probing increases after quitting smoking. J Clin Periodontol. 2003;30:435–437. doi: 10.1034/j.1600-051x.2003.20039.x. [DOI] [PubMed] [Google Scholar]
- Naito M, Hirakawa H, Yamashita A, Ohara N, Shoji M, Yukitake H, et al. Determination of the Genome Sequence of Porphyromonas gingivalis Strain ATCC 33277 and Genomic Comparison with Strain W83 Revealed Extensive Genome Rearrangements in P. gingivalis. DNA Res. 2008 doi: 10.1093/dnares/dsn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Wilson RF, Hasan AS, Scott DA. Mechanisms of action of environmental factors--tobacco smoking. J Clin Periodontol. 2005;32(Suppl 6):180–195. doi: 10.1111/j.1600-051X.2005.00786.x. [DOI] [PubMed] [Google Scholar]
- Petropoulos G, McKay IJ, Hughes FJ. The association between neutrophil numbers and interleukin-1alpha concentrations in gingival crevicular fluid of smokers and non-smokers with periodontal disease. J Clin Periodontol. 2004;31:390–395. doi: 10.1111/j.1600-051x.2004.00489.x. [DOI] [PubMed] [Google Scholar]
- Polanska K, Hanke W, Ronchetti R, van den Hazel P, Zuurbier M, Koppe JG, Bartonova A. Environmental tobacco smoke exposure and children’s health. Acta Paediatr Suppl. 2006;95:86–92. doi: 10.1080/08035320600886562. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Dooley MM, Church DF. The inactivation of alpha-1-proteinase inhibitor by gas-phase cigarette smoke: protection by antioxidants and reducing species. Chem Biol Interact. 1986;57:271–283. doi: 10.1016/0009-2797(86)90002-5. [DOI] [PubMed] [Google Scholar]
- Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2007;193:222–228. doi: 10.1016/j.atherosclerosis.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Ratnayake DB, Wai SN, Shi Y, Amako K, Nakayama H, Nakayama K. Ferritin from the obligate anaerobe Porphyromonas gingivalis: purification, gene cloning and mutant studies. Microbiology. 2000;146(Pt 5):1119–1127. doi: 10.1099/00221287-146-5-1119. [DOI] [PubMed] [Google Scholar]
- Rehani K, Scott DA, Renaud D, Hamza H, Williams LR, Wang H, Martin M. Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim Biophys Acta. 2008;1783:375–382. doi: 10.1016/j.bbamcr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Rezavandi K, Palmer RM, Odell EW, Scott DA, Wilson RF. Expression of ICAM-1 and E-selectin in gingival tissues of smokers and non-smokers with periodontitis. J Oral Pathol Med. 2002;31:59–64. doi: 10.1046/j.0904-2512.2001.joptest.doc.x. [DOI] [PubMed] [Google Scholar]
- Scott DA, Singer DL. Suppression of overt gingival inflammation in tobacco smokers - clinical and mechanistic considerations. Int J Dent Hyg. 2004;2:104–110. doi: 10.1111/j.1601-5037.2004.00079.x. [DOI] [PubMed] [Google Scholar]
- Scott DA, Palmer RM, Stapleton JA. Validation of smoking status in clinical research into inflammatory periodontal disease. J Clin Periodontol. 2001;28:715–722. doi: 10.1034/j.1600-051x.2001.280801.x. [DOI] [PubMed] [Google Scholar]
- Shi X, Hanley SA, Faray-Kele MC, Fawell SC, Aduse-Opoku J, Whiley RA, et al. The rag locus of Porphyromonas gingivalis contributes to virulence in a murine model of soft tissue destruction. Infect Immun. 2007;75:2071–2074. doi: 10.1128/IAI.01785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirodaria S, Smith J, McKay IJ, Kennett CN, Hughes FJ. Polymorphisms in the IL-1A gene are correlated with levels of interleukin-1alpha protein in gingival crevicular fluid of teeth with severe periodontal disease. J Dent Res. 2000;79:1864–1869. doi: 10.1177/00220345000790110801. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000. 2005;38:135–187. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- Stein SH, Green BE, Scarbecz M. Augmented transforming growth factor-beta1 in gingival crevicular fluid of smokers with chronic periodontitis. J Periodontol. 2004;75:1619–1626. doi: 10.1902/jop.2004.75.12.1619. [DOI] [PubMed] [Google Scholar]
- van Winkelhoff AJ, Appelmelk BJ, Kippuw N, de Graaff J. K-antigens in Porphyromonas gingivalis are associated with virulence. Oral Microbiol Immunol. 1993;8:259–265. doi: 10.1111/j.1399-302x.1993.tb00571.x. [DOI] [PubMed] [Google Scholar]
- Wang M, Shakhatreh MA, James D, Liang S, Nishiyama S, Yoshimura F, et al. Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- Wei B, Dalwadi H, Gordon LK, Landers C, Bruckner D, Targan SR, Braun J. Molecular cloning of a Bacteroides caccae TonB-linked outer membrane protein identified by an inflammatory bowel disease marker antibody. Infect Immun. 2001;69:6044–6054. doi: 10.1128/IAI.69.10.6044-6054.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lin X, Xie H. Porphyromonas gingivalis short fimbriae are regulated by a FimS/FimR two-component system. FEMS Microbiol Lett. 2007;271:214–221. doi: 10.1111/j.1574-6968.2007.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Scott JE, Liu KZ, Bishop HR, Renaud DE, Palmer RM, et al. The influence of nicotine on granulocytic differentiation - inhibition of the oxidative burst and bacterial killing and increased matrix metalloproteinase-9 release. BMC Cell Biol. 2008;9:19. doi: 10.1186/1471-2121-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Nasu F, Harada A, Kunitomo M. Oxidants in the gas phase of cigarette smoke pass through the lung alveolar wall and raise systemic oxidative stress. J Pharmacol Sci. 2007;103:275–282. doi: 10.1254/jphs.fp0061055. [DOI] [PubMed] [Google Scholar]
- Zambon JJ, Grossi SG, Machtei EE, Ho AW, Dunford R, Genco RJ. Cigarette smoking increases the risk for subgingival infection with periodontal pathogens. J Periodontol. 1996;67:1050–1054. doi: 10.1902/jop.1996.67.10s.1050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.