

Abstract
The development of inhibitors of Dishevelled (Dvl) PDZ protein-protein interactions attracts attention due to a possible application in drug discovery and development. Using nuclear magnetic resonance (NMR) spectroscopy, we found that a tripeptide VVV binds to the PDZ domain of Dvl, which is a key component involved in Wnt signaling. Using a computational approach calculating the binding free energy of the complexes of the Dvl PDZ domain and each of the tripeptides VXV (X: any amino acid residue except Pro), we found that a tripeptide VWV had the highest binding affinity. Consistent with the computational result, experimental results showed that the binding of the tripeptide VWV to the Dvl PDZ domain was stronger than that of the tripeptide VVV. The binding affinity of the tripeptide VWV was comparable to that of the organic molecule NSC668036, which was the first identified Dvl PDZ inhibitor. The three-dimensional structure of the complex Dvl1 PDZ/VWV was determined to investigate the role of the energetically favorable W(−1) residue in binding. These interactions were also explored by using molecular dynamic simulation and the molecular mechanics Poisson-Boltzmann surface area method. Taken together, these two tripeptides may be used as modulators of Wnt signaling or as a scaffold to optimize an antagonist for targeting Dvl1 PDZ protein-protein interaction.
Keywords: binding free energy, protein-ligand interaction, ligand computational screening, NMR, fluorescence spectroscopy
1. Introduction
Dishevelled (Dvl) proteins function in cell differentiation and apoptosis through Wnt signaling pathways.1,2 Overexpression of Dvl showing activation of canonical Wnt signaling has been reported in several types of cancer cells and tissues.3–5 These findings suggest that discovery of small molecules or peptides modulating the function of Dvl proteins in Wnt signaling would be valuable in understanding biological processes and developing a potential drug.3 Dvl proteins have three domains that are highly conserved in the animal kingdom, including the DIX, PDZ, and DEP domains.1,6–10 Among these domains, the Dvl PDZ domain, which is a protein-protein interaction module, has been identified as a potential target for drug discovery and development.9,11 The Dvl PDZ domain is ~100 amino acids long and comprises six β-strands (βA to βF) and two α-helices (αA and αB); the Dvl1 PDZ domain recognizes an internal sequence and an extreme C-terminal tail of target proteins through a binding groove formed between the αB-helix and βB-strand.9,12,13 A peptide bound to Dvl1 PDZ-domain forms an additional β-strand structure.12 Notably, the Dvl1 PDZ-bound peptide attenuates the Wnt/β-catenin signaling induced by Wnt1.9 This discovery inspired us and others to explore small-molecule inhibitors of Dvl PDZ protein-protein interaction.14–16
In addition to the development of small molecules, we are also investigating short peptides targeting the Dvl PDZ domain for a possible application in peptide-based therapeutics.17 Despite the disadvantages of peptide drugs, such as their high price and low bioavailability, they are of continued interest because they are more target-specific and less toxic than small molecules.17 Natural peptides and synthetics derived from bioactive peptides have actually been used for the prevention and treatment of diseases.17,18
Tripeptides are drawing especially great attention in drug discovery because they are easy to modify and small (M.W. < 500 Da), which is consistent with typical small-molecule bioavailable oral drugs.19–24 In addition, the conformational properties of tripeptides have been explored to allow an understanding of the protein folding and intrinsic properties of amino acid residues in protein structures.25–30 Recent studies revealed that even short peptides could adopt a specific conformation in gas phase or in water solution, implying that they might serve as a model as well as a molecular scaffold for future drug design.25–30
Using nuclear magnetic resonance (NMR) spectroscopy, we found that the tripeptide VVV interacts directly with the Dvl PDZ domain. After confirming the binding, we optimized a peptide to enhance the binding affinity to the Dvl PDZ domain using an approach that combines computational biology, peptide chemistry, and structural biology. To understand the binding mechanism, we determined the three-dimensional structure of the complex and Dvl1 PDZ/tripeptide VWV using NMR-derived information. We also conducted molecular dynamic (MD) simulations and the molecular mechanics Poisson-Boltzmann surface area method (MM-PBSA) calculation to reveal the difference between the two peptides in their binding affinities to the Dvl PDZ domain.
2. Results and Discussion
2.1. Tripeptide VVV recognizes the Dvl1 PDZ domain
To discover and optimize a Dvl PDZ-binding peptide, we chose the tripeptide VVV as a model because it has been reported to predominantly sample a β-strand structure in water solution25,31 and it resembles the PDZ-binding motif, such as S/T-X-Φ and Φ-X-Φ (X: any amino acid; Φ: hydrophobic residue).32 To probe the interaction between the tripeptide VVV and the Dvl1 PDZ domain, we used NMR spectroscopy. The interaction of a protein and a short peptide can be easily detected by the chemical shift perturbations or peak broadening upon adding the unlabeled peptide to the 15N-labeled targeted protein.33 Fig. 1A shows the 1H-15N-HSQC spectra of the 15N-labeled Dvl1 PDZ domain with titration of the unlabeled tripeptide VVV, indicating a direct interaction. The large chemical shift perturbations resulting from the binding of the tripeptide VVV to the Dvl1 PDZ domain were observed for residues Ile264 and Ser265 in the βB-strand and the residues Arg322 and Val325 on the αB-helix structure of the Dvl1 PDZ domain (Fig. 1B). These peaks showed the continuous changes in chemical shifts in the 1H-15N-HSQC spectra as the concentration of the tripeptide VVV was increased, indicating that its interaction is in the fast exchange range on the NMR time scale. The worm representation of the backbone structure of the Dvl1 PDZ domain based on the chemical shift perturbation data in Fig. 1C demonstrates that the tripeptide VVV binds to the αB/βB-binding groove of the Dvl1 PDZ domain as expected.
Figure 1.

Tripeptide VVV interacts with the Dvl1 PDZ domain. (a) 15N-HSQC spectra of 15N-labeled Dvl1 PDZ domain alone (0.3 mM, blue) and with varying concentrations of tripeptide VVV. (b) The extended 15N-HSQC spectra of the Dvl1 PDZ domain as a function of the tripeptide VVV concentration (blue: free, gold 1:4; green: 1:7; purple 1:11, red 1:22). (c) The worm representation of the backbone structure of the Dvl1 PDZ domain. The thickness and color of the worm is proportional to the weighted sum of the proton and amide chemical shift perturbations upon binding of peptide VVV (red, high; blue, low). The program Insight II was used was used to prepare Fig. 1c.
2.2. Evaluation of the binding free energy of the complex of Dvl1 PDZ and model tripeptide VXV
The observation of the direct interaction between the tripeptide VVV and the Dvl PDZ domain prompted us to investigate whether other similar tripeptides could bind to the Dvl PDZ domain. Because the tripeptide VVV resembles the typical PDZ-binding motif, investigating the role of a particular amino acid residue in binding was expected to provide valuable information to optimize a peptide bound to the Dvl1 PDZ domain. Because the P(−1) residue in the PDZ-binding peptide (position 0 referring to the extreme C-terminal residue) has been found to play a role in enhancing binding in several cases,34–36 we virtually evaluated the binding affinities of Dvl PDZ with model tripeptides VXV (X: any amino acid residue except Pro) using the ICM empirical binding energy function (ICM Pro ver. 3.2).37 In this study, we used x-ray crystallographic data of the structure of the Xenopus Dishevelled PDZ (Xdsh PDZ)/Dapper peptide (SGSLKLMTTV) complex (PDB code: 1L6O:A).12 We used the coordinates of the last three amino acid residues (TTV) for the Dapper peptide in the complex structure to generate the model tripeptides. We assumed that all tripeptides bound to the PDZ domain adopt the β-strand that resemble the bound conformation of the Dapper peptide.12 The side chain of each modeled tripeptide in the complex was optimized to escape a possible collapse of the side chain between Xdsh PDZ and the VXV tripeptide before calculating the binding free energy of the complex. Fig. 2 shows the relative binding free energy (ΔΔGbinding) of the Xdsh PDZ and model tripeptide VXV with respect to the tripeptide VVV (ΔGbinding is −21.8±3.3 kcal/mol). Notably, the tripeptide VWV had the highest binding energy to the PDZ domain of Xdsh. Although the ICM empirical binding energy function has been validated for several cases,37 we wondered whether this would be the case for our model system. To confirm the theoretical result, we used an NMR-binding assay.
Figure 2.

Tripeptide VWV had the highest binding energy. The relative binding energies (ΔΔGbinding) of Xenopus Dsh PDZ and model tripeptides VXV with respect to the tripeptide VVV. The energies were calculated by using the ICM empirical binding energy function (X represents any amino acid residue except Pro).
2.3. Tripeptide VWV indeed binds to the Dvl PDZ domain
We chemically synthesized the tripeptide VWV and explored its interaction with the Dvl1 PDZ domain using NMR spectroscopy. Fig. 3A shows the fingerprint region of the 1H-15N-HSQC spectra of the 15N-labeled Dvl1 PDZ domain with varying concentrations of unlabelled tripeptide VWV. Surprisingly, the residues I264, R322, and V325 began to disappear upon stepwise addition of the tripeptide VWV and reappeared at the saturated concentration. This indicates that the complex formation is in the intermediate exchange range on the NMR time scale. The two largest chemical shift perturbations were found in residues I264 (Δδtotal = 0.565 ppm) on the βB-strand and R322 (Δδtotal = 0.497 ppm) on the αB-helix of Dvl1 PDZ at the saturated concentration. They are much larger than the chemical shift perturbations in the same residues caused by the binding of the VVV peptide (Figs. 1B and 3A), indicating that the VWV peptide binds to the PDZ domain tighter than the VVV peptide.
Figure 3.

Direct interaction of the tripeptide VWV and the Dvl1 PDZ domain. (a) The extended 15N-HSQC spectra of the Dvl1 PDZ domain at various concentration of tripeptide VWV (blue: free, cyan 1:1, green 1:3, purple 1:5, red 1:8). (b) The worm representation of the backbone structure of the Dvl1 PDZ domain. The thickness of the worm structure was normalized by the chemical shift perturbation datum of the I264 residue in Fig. 1.
We next determined the binding affinity (KD) of tripeptides using fluorescence spectroscopy (Table 3). In this study, we made a fluorescence-labeled PDZ domain, 2-((5(6)-tetramethylrhodamine)carboxylamino)ethylethanethiosulfonate (TMR)-PDZ domain of Dvl1 (Fig. 4).14 The fluorescence intensity of the TMR-PDZ domain at 597 nm was monitored while the tripeptide VVV or VWV was added. The KD value was calculated from a reciprocal plot of fluorescence intensity quenching against the concentration of the peptide. The result showed that the binding affinity of the tripeptide VWV was 2 μM and that of the tripeptide VVV was 71 μM for the TMR-PDZ domain, which supports the ICM theoretical result that modification of the P(−1) position in the tripeptide can increase the binding affinity for the Dvl1 PDZ domain. Notice that the KD values of the tripeptides are much lesser than that of the organic molecule NSC668036, which was the first identified antagonist for targeting Dvl1 PDZ protein interactions.14 Using the same binding assay, the KD value of NSC668036 and TMR-PDZ was found to be 237±31 μM.14
Table 3.
The binding affinity of the complex from MD simulations (MM-PBSA calculation)a
| Thermodynamic parameters | Dvl1 PDZ/VVV | Dvl1 PDZ/VWV |
|---|---|---|
| ΔH | −23.48(0.39)d | −30.07(0.41) |
| Eele(solute) | −70.53(1.10) | −74.76(1.00) |
| Eele(solvent) | 77.20(0.93) | 84.32(0.81) |
| Eele+PB = Eele(solute)+Eele(solvent) | 6.67 | 9.56 |
| Evdw | −25.80(0.19) | −34.69(0.23) |
| Htrans/rot | −1.78 | −1.78 |
| Eno_pol | −4.35(0.31) | −4.94(0.01) |
| ΔTS | −19.74(0.74) | −24.70(1.13) |
| bΔG | −5.52(0.84) | −7.15(1.20) |
| cΔG | −5.56 | −7.64 |
In kcal/mol,
MM-PBSA calculation.
From fluorescence experiment, KD of the TMR-PDZ and tripeptide. ΔGo= 2.303 RT(log KD), R=1.987 cal·K−1·mol−1, T=293 K.
Standard errors of mean in parenthesis.
Figure 4.

A reciprocal plot of fluorescence intensity quenching (ΔF) of the TMR-PDZ(C338) domain of Dvl1 against the concentration (ΔS) of the tripeptides (a) VVV and (b) VWV.
2.4. Structures of the complex between the Dvl PDZ domain and the VWV peptide
To understand the greater affinity of the tripeptide VWV than of the tripeptide VVV for binding to the Dvl1 PDZ domain, we determined the three-dimensional structure of the Dvl1 PDZ/VWV complex. The assigned intermolecular nuclear Overhauser effects (NOEs) between tripeptide VWV and the Dvl1 PDZ domain are summarized in Fig. 5A and Table 1. In the complex structure calculation, a total of 26 experimental restraints were used, including 22 intermoleulcar NOEs between the peptide and the protein and 4 intramolecular NOEs from the tripeptide VWV bound to the PDZ domain. The 20 lowest energy conformations were selected from the 100 water-refined complexes for further structural analysis (Fig. 5B). The statistical result of the complex is summarized in Table 2. Not surprisingly, the tripeptide VWV was fitted into the hydrophobic pocket of the Dvl1 PDZ domain and formed an additional β-strand with respect to the βB-structure (Fig. 5B). The lowest energy conformation of the PDZ/VWV complex is shown in Fig. 5C: the side chains of P(0) and P(−2) contacted the hydrophobic residues (color code: yellow) within the binding site of the Dvl1 PDZ domain. The side chain of the P(−1) residue in the tripeptide was oriented to the βB- and βC-strand regions of the Dvl1 PDZ domain. The amino acid residues of the Dvl1 PDZ domain in proximity (<4.0 Å) to tripeptide V(-2)-W(-1)-V(0) were as follows: the residues Leu262, Gly263, Ile264, Val325, Arg322, Leu321, and Val318 were close to the residue V(0); the residues Ile266 and Val318 were close to the residue Val(−2); and Ser265 and Met284 were close to the W(−1) residue (Fig. 5C). The solution structure of the PDZ/VWV complex was in a good agreement with the complex structure used for the ICM calculations (data not shown).
Figure 5.

Structures of the Dvl1 PDZ/VWV complex. (a) Summary of intermolecular NOEs between the tripeptide VWV and the Dvl1 PDZ domain. (b) The ensemble of the 20 lowest energy conformations of the Dvl1 PDZ/VWV complex. (c) The lowest energy conformation of the Dvl1 PDZ/VWV complex. Surface representation shows the binding interface between the Dvl1 PDZ domain and the VWV tripeptide. The hydrophobic amino acid residues in the Dvl1 PDZ domain surface model are drawn in yellow, the positively charged residues in blue, the negatively charged residues in red, and the uncharged polar residues in gray. Tripeptide is shown in the stick model (blue: VWV). The program Pymol was used to prepare the figures.
Table 1.
Intermolecular NOEs between the Dvl1 PDZ domain and the tripeptide V1(-2)-W2(-1)-V3(0). Data were obtained from a 3D 13C-F1-half-filtered F2-edited NOESY-HSQC experiment (mixing time = 200 ms) at 10 °C in 0.1 M phosphate buffer, 0.5 mM EDTA, pH 7.5.
| Assignments | ω1 | ω2 | ω3 | Relative NOE Intensity a |
|---|---|---|---|---|
| V1Hβ-I266Cδ1-Hδ1 | 1.965 | 10.416 | 0.417 | w |
| V1Hβ-I266CB-Hβ | 1.932 | 36.188 | 1.553 | w |
| V1Hβ-I266CG2-Hγ2 | 1.967 | 15.579 | 0.464 | m |
| V1Hγ2-I266CB-Hβ | 0.659 | 36.254 | 1.554 | w |
| V1Hγ2-V318Cα-Hα | 0.650 | 24.194 | 3.212 | m |
| V1Hγ1-V318Cγ1-Hγ1 | 0.635 | 20.565 | 0.936 | m |
| W2Hring-S265Cβ-S265Hβ2 | 7.282 | 22.563 | 3.589 | m |
| W2Hring-S265Cβ-S265Hβ3 | 7.289 | 22.591 | 3.399 | m |
| W2Hring-S265Cβ-S265Hβ2 | 7.027 | 22.600 | 3.591 | s |
| W2Hring-S265Cβ-S265Hβ3 | 7.018 | 22.576 | 3.397 | s |
| W2Hring-M284Cε-Hε | 7.288 | 14.116 | 1.467 | w |
| W2Hring-M284Cε-Hε | 7.032 | 14.101 | 1.467 | m |
| V3Hβ-V318Cγ1-Hγ1 | 1.787 | 20.584 | 0.937 | m |
| V3Hγ2-L262Cβ-Hβ | 0.628 | 39.300 | 1.623 | m |
| V3Hγ2-I264Cβ-Hβ2 | 0.633 | 39.104 | 1.850 | m |
| V3Hγ2-I264Cδ1-Hδ1 | 0.628 | 10.031 | 0.376 | m |
| V3Hγ2-I266Cγ1-Hγ11 | 0.639 | 22.744 | 1.246 | s |
| V3Hγ2-I266Cγ1-Hγ12 | 0.634 | 22.763 | 0.916 | m |
| V3Hγ2-I266Cγ2-Hγ2 | 0.633 | 15.568 | 0.464 | m |
| V3Hγ1-R322Cβ-Hβ | 0.665 | 27.324 | 1.838 | w |
| V3Hγ2-V318Cγ2-Hγ2 | 0.632 | 19.344 | 0.303 | w |
| V3Hγ2-V325Cγ1-Hγ1 | 0.629 | 19.122 | 0.883 | m |
NOE cross-peak intensities were classified as strong, medium, weak, and very weak, and assigned to restraints of 1.8–3.0Å, 1.8–4.0 Å, 1.8–5.0 Å, 1.8–6.0 Å, respectively, with appropriate pseudoatom corrections.
Table 2.
Structural statistics of the 20 lowest energy structures of Dvl1 PDZ/VWV complex
| PDZ-VWV | |
|---|---|
| Number of unambiguous interaction restraints | |
| Intramolecular NOEs | 4 |
| Intermolecular NOEs | 22 |
| RMSD deviation from the averaged coordinates a | |
| All backbone (Å) | 0.31 ± 0.03 |
| All heavy atom (Å) | 0.53 ± 0.04 |
| Overall energy of the 20 lowest energy conformations | |
| Etotal (kcal/mol) | −117.9 ± 3.2 |
| Evdw (kcal/mol) | −30.8 ± 1.6 |
| Eelec (kcal/mol) | −87.1 ±1.3 |
| ENOE (kcal/mol) | 0.03 ± 0.01 |
| Buried Surface of the 20 lowest energy conformations (Å)2 | 653.6 ± 15.6 |
Calculated from the program MOLMOL.
The structure of the PDZ/VWV complex implies that the difference in the binding affinity for the tripeptides VVV and VWV may result from the hydrophobic interaction between the side chain of the Ser265 or Met284 residues and the Trp side chain in the P(−1) position of the tripeptide VWV. However, it is difficult to quantitatively explain the linkage between the binding affinity of the tripeptide and the Dvl1 PDZ domain with the structural information.
2.5 Molecular dynamics simulation and MM-PBSA calculation for the complexes of Dvl1 PDZ with tripeptides VVV and VWV
To further understand the difference in the binding energies for the two complexes, we performed an MD simulation using AMBER software.38 The starting structures of the complexes used were the lowest energy conformation as described in the Materials and Methods section. MD simulations were performed in explicit water for 2 ns. To analyze the stability of the MD simulations, we plotted the root-mean-square deviation (RMSD) values relative to the initial structures of the PDZ backbone atoms during the 2 ns MD simulation against time (Fig. 6). The complexes of the Dvl1 PDZ domain with the VVV or VWV tripeptide reached convergence quickly and remained stable thereafter. We then calculated the binding free energy of the interaction between the Dvl1 PDZ domain and the tripeptides for the last 1 ns with the MM-PBSA method.39,40 The individual energy terms that contribute to binding free energy are listed in Table 3. The predicted binding free energy (ΔG) of the Dvl1 PDZ/VVV complex was −5.52±0.80 kcal/mol and that of the Dvl1 PDZ/VWV complex was −7.15±1.20 kcal/mol, indicating that the binding of the tripeptide VWV to the Dvl1 PDZ domain is stronger than that of the tripeptide VVV. The relative binding free energies obtained from MM-PBSA calculations were in good agreement with experimental results (ΔG = −5.56 kcal/mol for TMR-PDZ/VVV and ΔG = −7.64 kcal/mol for TMR-PDZ/VWV; Table 3). Besides ranking the binding free energies correctly, MM-PBSA allowed us to break down the total binding free energy into individual components, which enabled us to understand the binding interactions in detail. In Table 3, the Eelec+PB is the total electrostatic contribution, including solute-solute and solute-solvent interactions, and Evdw is the van der Waals contribution to the binding. As seen in Table 3, the Evdw favors interaction of the Dvl1 PDZ domain with tripeptide VWV by 8.89 kcal/mol, whereas the electrostatic contribution resists the binding between the Dvl1 PDZ and VWV by 2.89 kcal/mol. The differences between the binding free energy of the two complexes therefore are largely controlled by the van der Waals contribution. This correlates well with the observation that the indole ring of the Trp(−1) residue in the tripeptide was close to the side chain of the S265 and M284 residues. The entropy contribution (TΔS) disfavors the binding of the PDZ/VWV due to orientational and positional restraints imposed by the larger side chain of the Trp residue; however, the overall binding free energy (ΔGoverall) still promotes the binding of the PDZ/VWV complex over that of the PDZ/VVV complex.
Figure 6.
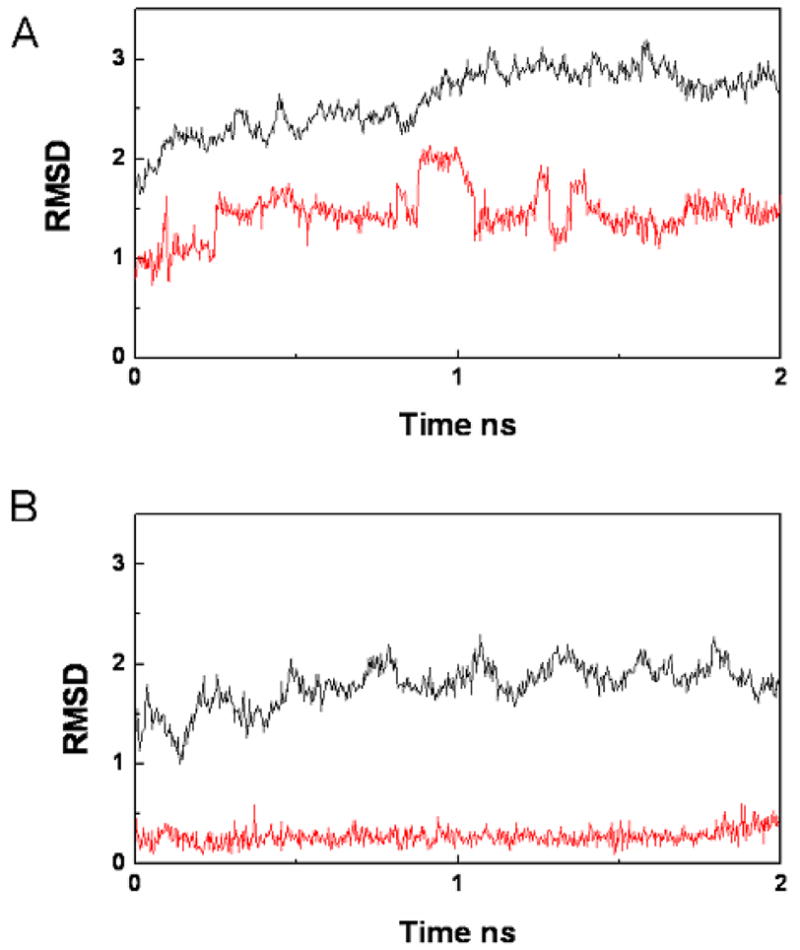
The backbone root-mean-square deviation (RMSD) relative to the initial structures of the backbone atoms and ligand atoms in the (a) Dvl1 PDZ/VVV and (b) Dvl1 PDZ/VWV complexes during a 2-ns molecular dynamics simulation. The black lines represent the backbone RMSDs of the PDZ proteins, and the red line represents the backbone RMSD of the peptides.
3. Conclusion
In this study, we identified the tripeptides VVV and VWV as Dvl1 PDZ-binding partners using NMR spectroscopy and other methods. The experimental and theoretical results provided evidence that a single modification at the P(−1) residue in the Dvl1 PDZ-binding peptide can increase the binding affinity through the hydrophobic interaction contribution. Because the binding ability of the tripeptide VWV was comparable to that of the organic compound NSC668036, the first Dvl1 PDZ antagonist identified by the virtual screening and NMR method,14 the result implies that the tripeptide VWV may serve as a modulator of Dvl PDZ protein interactions or as a molecular scaffold for the design of a new peptidomimetic compound.41 It can also be proposed that short peptides containing the motif VWV at the C-terminus are potential Dvl PDZ inhibitors. Further studies of the effect of the tripeptide VWV on Wnt signaling should yield valuable information regarding the function of Dvl in embryonic development and malignant diseases. Because numerous PDZ domains are found in the animal kingdom, we cannot rule out the possibility of the tripeptide binding to other PDZ domains.42–44 To obtain a more selective inhibitor of Dvl PDZ protein-protein interaction, processes similar to those described here are under way to optimize the residues at the −2 and −3 positions of the PDZ-binding ligands that also may influence PDZ-binding affinities significantly.
4. Materials and Methods
4.1 Purification of the 13C, 15N-labeled mouse Dvl1 PDZ domain for NMR spectroscopy
The detailed methods for protein production have been described elsewhere.9 In brief, the cDNA of the mouse Dishevelled1 (Dvl1) PDZ domain (amino acids 251–341) was sub-cloned into the pET28a vector. To improve the protein solubility and to prevent dimerization, Cys338 was replaced with Ala for NMR study. The protein was expressed in BL21(DE3)-codon plus RP Esherichia coli cells. The transformed E. Coli BL21(DE3) cells were grown at 37 °C in MOPS-containing medium supplemented with 15NH4Cl (1g/L) and/or 13C-glucose (3g/L) as the sources of nitrogen and carbon. Protein expression was induced by the addition of 1 mM isopropyl-1-thio-β-D-galactoside (IPTG) until the optical density at 600 nm of the cells was about 0.5. Cells were lysed by microfluidization, and the protein was purified with a HiTrap Chelating HP column of NiCl2 (Amersham Pharmacia Biotech). The column was washed with 20 mM imidazole, followed by 200 mM imidazole (pH 7.8) with 300 mM NaCl. The Dvl1 PDZ domain was further purified by gel filtration on Sephadex G75 in 0.1 M phosphate buffer, pH 7.5, with 0.5 mM EDTA.9
4.2 Peptide synthesis and purification
Tripeptide VVV (NSC35938) was obtained from the National Cancer Institute and used without purification. The tripeptide VWV was chemically synthesized by the Hartwell Center for Bioinformatics & Biotechnology at St. Jude Children’s Research Hospital and purified by reverse-phase high-performance liquid chromatography. [M+H]+(calc) = 402.5, [M+H]+(observed) = 403.4 for the VWV peptide.
4.3 NMR spectroscopy and NMR titration experiment
The NMR experiments were performed at 25 °C using a Varian INOVA 600 MHz spectrometer and a Bruker Avance 800 MHz spectrometer equipped with a triple resonance, triple axis actively shielded gradient cold probe. All NMR spectra were processed with the program NMRpipe45 and analyzed using the program Sparky 3 (T.D. Goddard and D.G. Kneller, University of California, San Francisco). The interaction between protein and peptide was directly monitored by collecting the 15N-HSQC spectra of ~0.3 mM 15N-labeled protein at varying concentrations of tripeptides in 0.1 M phosphate buffer (pH 7.5) at 298 K. Chemical shift perturbations (Δδtotal) of 1H and 15N resonances were obtained and weighted according to equation (1).
4.4 The binding free energy for the Dvl1 PDZ/tripeptide complexes by ICM software
The binding free energy between model tripeptide VXV with the Dvl1 PDZ domain was calculated by using the ICM empirical binding function (X: any amino acid residue). The starting structures of the Dvl1 PDZ/tripeptide complex were generated using the coordinates from the X-ray crystallographic data of the complex structure of Xenopus Dishevelled PDZ (Xdsh PDZ) and Dapper peptide (SGSLKLMTTV) (PDB code: 1L6O) using the program SYBYL 7.3 (Tripos Inc.). Hydrogen atoms were added to the model complex structure using SYBYL. Before calculating the binding free energy of the complex, the side chain of each modeled tripeptide in the complex was optimized to escape a possible collapse of the side chain between Xdsh PDZ and its binding partner using the default method provided by the ICM software. The binding free energy of the model tripeptide VXV and Xdsh PDZ was calculated using equation (2).
Here, ΔGvw is Van der Waals energy, which is defined as non-bonded interatomic pairwise interactions; ΔGhb is hydrogen bonding energy, which is a different form of the “vw” term for hydrogen bonding donors and acceptors; ΔGto is torsional energy, or dihedral angle deformation energy; ΔGel is electrostatic energy; and ΔGsf is the surface energy, which is surface tension as a product of atomic accessibilities of the total solvent accessible area. We calculated the binding free energy of the complexes twice independently and averaged the values.
4.5 Fluorescence spectroscopy
A FluoroLog-3 spectrofluorometer (Jobin-Yvon, Inc.) was used for the binding assay. The fluorescently labeled PDZ domain TMR-PDZ(C338) was generated to obtain the binding affinity (KD) of the tripeptides VVV and VWV as described previously.14 Briefly, the TMR-PDZ protein of Dvl was dialyzed in 0.1 M potassium phosphate (pH 7.5) with 0.5 mM EDTA at least 3 times with a 3,500 Da cutoff membrane. The fluorescently labeled TMR-PDZ was confirmed by SDS gel and mass spectroscopy. The excitation wavelength of TMR-PDZ(C338) was 552 nm (slit width = 5 nm), and the maximum emission fluorescence at 597 nm (slit width = 5 nm) was recorded during the titration of model peptides. Titration experiments were performed at 25 °C in phosphate buffer (0.1 M potassium phosphate; 0.5 mM EDTA, pH 7.5). Typically, the peptide solutions (5 to 200 μM) were injected into 2.0 mL of 40 nM TMR-PDZ(C338) domain. The fluorescence data were analyzed using the ORIGIN program (Microcal). The KD value was calculated from a reciprocal plot of fluorescence intensity quenching (ΔF) against the concentration (ΔS) of peptides.
ΔF is the change of fluorescence intensity and [S] is the peptide concentration.
4.6 Structure calculation
The 13C,15N-double-labeled PDZ domain of Dvl1 with the unlabeled tripeptide VWV was used for structure determination at 10 °C in 0.1 M phosphate buffer (pH 7.5). To avoid intermediate exchange, excess unlabeled VWV peptide was added. Tripeptide VWV resonances were assigned by using 2D [F1,F2]-double filtered TOCSY and NOESY experiments with a mixing time of 200 ms. To obtain the distance restraints, a 3D 13C-F1-half-filtered and F2-edited NOESY-HSQC experiment with the 13C,15N-labeled PDZ domain of Dvl1 and the unlabeled tripeptide VWV with a mixing time of 200 ms was recorded (Table 1). Based on the previous assignment of the backbone and side chains of the Dvl1 PDZ domain,9 the intermolecular NOEs between the tripeptide VWV and the Dvl1 PDZ domain were assigned. NOE cross-peak intensities were classified as strong, medium, weak, or very weak and were assigned restraints of 1.8–3.0 Å, 1.8–4.0 Å, 1.8–5.0 Å, and 1.8–6.0 Å, respectively, with appropriate pseudoatom corrections. A total of 26 intermolecular and intramolecular NOEs were identified in the complex of the Dvl1 PDZ/VWV peptide. Based on the NOEs, we then calculated the structure of the Dvl1 PDZ/VWV complex by using the simulated annealing program CNS within the HADDOCK platform.46 The coordinates of the Dvl1 PDZ domain were taken from the X-ray structure of the Xenopus PDZ domain (PDB code: 1L6O), and the amino acid residues were modified to the mouse Dvl1 PDZ domain using SYBYL.14 Two thousand initial structures were calculated. Among the final 100 water-refinement structures for the complex, we selected the 20 lowest energy conformations to represent the solution structure of the complex. The calculated structures were analyzed by the programs PROCHECK47 and MOLMOL.48
4.6 Molecular Dynamics Simulation
MD simulation was conducted by using the Sander program in AMBER8 with the parm99 force field.38 MD simulations were performed on a Linux cluster at the Hartwell Center at St. Jude. The starting complex structure of the Dvl1 PDZ/VWV used was the lowest energy conformation as shown in Fig. 5C. Because both tripeptides VVV and VWV bound at the same site on the PDZ domain of Dvl1, we predicted that the tripeptide VVV bound to the Dvl1 PDZ domain would adopt a conformation similar to that of the VWV tripeptide. We used the coordinates of the lowest energy conformation of the PDZ/VWV complex structure as a template to generate the starting structure of the Dvl1 PDZ/VVV complex for an MD simulation: the Trp(−1) residue replaced Val(−1) residues. After neutralization with Na+ ions, complexes were solvated in a periodic rectangular TIP3P water box with each side 9 Å away from the edge of the system.49 The MD run included several steps as follow: (1) systems were minimized by a 1000-step steepest descent minimization followed by a 9000-step conjugated gradient minimization; (2) systems were heated from 100K to 300K with 5 kcal/mol harmonic restrains via a 50-ps NVT MD simulation; (3) the restraints were gradually reduced to zero via a 50-ps NPT MD simulation; and (4) trajectories were collected from a 2-ns NPT production run. The MD simulations were performed with a time step of 1 fs, and the SHAKE algorithm was used to restrain all bonds involving hydrogen. The production trajectories were saved every 2.5 ps. After simulation, the ptraj module was used to extract trajectories for further analysis.
To calculate the binding free energy, we used the MM-PBSA module implemented in the AMBER8 package without any modification; the method combines the molecular mechanical energies with the continuum solvent approaches.39,40,50 We used 200 snapshots extracted from the last 1 ns to calculate ΔH and used 20 snapshots extracted from the last 1 ns for estimating the entropy ΔTS by normal-mode analysis. The binding free energy of complex was calculated using equations (4~7) as below.
Hgas is the molecular mechanical energy in the gas phase; Htrans/rot is the translational and rotational enthalpy, which equals 3RT; Evdw is the van der Waals contribution; Eelec is the electrostatic contribution; Gsolvation represents the free energy of solvation; and TS is the solute entropic contribution at temperature T. Gsolvation consists of two parts, the polar solvation energy GPB and the nonpolar solvation energyGsur. Gsolvation arises from the electrostatic potential between the solute and solvents, and Gsur is determined by the solvent-accessible area (A) and two empirical parameters, γ and b, which equal 0.00542 and 0.92, respectively.
Acknowledgments
We thank Drs. Weixing Zhang and Charles Ross for support with the NMR instruments and computer resources; Dr. Patrick Rodrigues and Mr. Robert Cassell at the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital for peptide synthesis. We thank David Galloway from Scientific Editing for editorial help. This work is supported by the American Lebanese Syrian Associated Charities, by the Cancer Center Support Grant (CA21765) from the National Cancer Institute and by National Institutes of Health grant GM081492.
Abbreviations
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single-quantum coherence
- NOE
nuclear Overhauser enhancement spectroscopy
- AIR
ambiguous interaction restraint
- MM-PBSA
molecular mechanics Poission-Boltzmann surface area
- RMSD
root-mean-square deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wallingford JB, Habas R. Development. 2005;132(20):4421–4436. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- 2.Malbon CC, Wang HY. Curr Top Dev Biol. 2006;72:153–166. doi: 10.1016/S0070-2153(05)72002-0. [DOI] [PubMed] [Google Scholar]
- 3.Uematsu K, Kanazawa S, You L, He B, Xu Z, Li K, Peterlin BM, McCormick F, Jablons DM. Cancer Res. 2003;63(15):4547–4551. [PubMed] [Google Scholar]
- 4.Uematsu K, He B, You L, Xu Z, McCormick F, Jablons DM. Oncogene. 2003;22(46):7218–7221. doi: 10.1038/sj.onc.1206817. [DOI] [PubMed] [Google Scholar]
- 5.Mizutani K, Miyamoto S, Nagahata T, Konishi N, Emi M, Onda M. Tumori. 2005;91(6):546–551. doi: 10.1177/030089160509100616. [DOI] [PubMed] [Google Scholar]
- 6.Sussman DJ, Klingensmith J, Salinas P, Adams PS, Nusse R, Perrimon N. Dev Biol. 1994;166(1):73–86. doi: 10.1006/dbio.1994.1297. [DOI] [PubMed] [Google Scholar]
- 7.Wong HC, Mao J, Nguyen JT, Srinivas S, Zhang W, Liu B, Li L, Wu D, Zheng J. Nat Struct Biol. 2000;7(12):1178–1184. doi: 10.1038/82047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capelluto DG, Kutateladze TG, Habas R, Finkielstein CV, He X, Overduin M. Nature. 2002;419(6908):726–729. doi: 10.1038/nature01056. [DOI] [PubMed] [Google Scholar]
- 9.Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, Mlodzik M, Shi DL, Zheng J. Mol Cell. 2003;12(5):1251–1260. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarz-Romond T, Fiedler M, Shibata N, Butler PJ, Kikuchi A, Higuchi Y, Bienz M. Nat Struct Mol Biol. 2007;14(6):484–492. doi: 10.1038/nsmb1247. [DOI] [PubMed] [Google Scholar]
- 11.Wang NX, Lee HJ, Zheng JJ. Drug News Perspect. 2008;21(3):137–141. [PMC free article] [PubMed] [Google Scholar]
- 12.Cheyette BN, Waxman JS, Miller JR, Takemaru K, Sheldahl LC, Khlebtsova N, Fox EP, Earnest T, Moon RT. Dev Cell. 2002;2(4):449–461. doi: 10.1016/s1534-5807(02)00140-5. [DOI] [PubMed] [Google Scholar]
- 13.London TB, Lee HJ, Shao Y, Zheng J. Biochem Biophys Res Commun. 2004;322(1):326–332. doi: 10.1016/j.bbrc.2004.07.113. [DOI] [PubMed] [Google Scholar]
- 14.Shan J, Shi DL, Wang J, Zheng J. Biochemistry. 2005;44(47):15495–15503. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]
- 15.Fujii N, You L, Xu Z, Uematsu K, Shan J, He B, Mikami I, Edmondson LR, Neale G, Zheng J, Guy RK, Jablons DM. Cancer Res. 2007;67(2):573–579. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]
- 16.Shan J, Zheng JJ. J Comput Aided Mol Des. 2008 doi: 10.1007/s10822-008-9236-1. [DOI] [Google Scholar]
- 17.Bhutia SK, Maiti TK. Trends Biotechnol. 2008;26(4):210–217. doi: 10.1016/j.tibtech.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Hong F, Ming L, Yi S, Zhanxia L, Yongquan W, Chi L. Peptides. 2008;29(6):1062–1071. doi: 10.1016/j.peptides.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Rifai Y, Elder AS, Carati CJ, Hussey DJ, Li X, Woods CM, Schloithe AC, Thomas AC, Mathison RD, Davison JS, Toouli J, Saccone GT. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G1094–G1099. doi: 10.1152/ajpgi.00534.2007. [DOI] [PubMed] [Google Scholar]
- 20.Kannengiesser K, Maaser C, Heidemann J, Luegering A, Ross M, Brzoska T, Bohm M, Luger TA, Domschke W, Kucharzik T. Inflamm Bowel Dis. 2008;14(3):324–331. doi: 10.1002/ibd.20334. [DOI] [PubMed] [Google Scholar]
- 21.Charnley M, Moir AJ, Douglas CW, Haycock JW. Peptides. 2008;29(6):1004–1009. doi: 10.1016/j.peptides.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Yao Z, Lu R, Jia J, Zhao P, Yang J, Zheng M, Lu J, Jin M, Yang H, Gao W. Peptides. 2006;27(6):1167–1172. doi: 10.1016/j.peptides.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 23.Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, Metcalf WW. Nat Chem Biol. 2007;3(8):480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HJ, Park HM, Lee KB. Biophys Chem. 2007;125:117–126. doi: 10.1016/j.bpc.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Eker F, Cao X, Nafie L, Schweitzer-Stenner R. J Am Chem Soc. 2002;124(48):14330–14341. doi: 10.1021/ja027381w. [DOI] [PubMed] [Google Scholar]
- 26.Schweitzer-Stenner R, Eker F, Perez A, Griebenow K, Cao X, Nafie LA. Biopolymers. 2003;71(5):558–568. doi: 10.1002/bip.10534. [DOI] [PubMed] [Google Scholar]
- 27.Eker F, Griebenow K, Schweitzer-Stenner R. J Am Chem Soc. 2003;125(27):8178–8185. doi: 10.1021/ja034625j. [DOI] [PubMed] [Google Scholar]
- 28.Eker F, Griebenow K, Cao X, Nafie LA, Schweitzer-Stenner R. Proc Natl Acad Sci U S A. 2004;101(27):10054–10059. doi: 10.1073/pnas.0402623101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eker F, Griebenow K, Cao X, Nafie LA, Schweitzer-Stenner R. Biochemistry. 2004;43(3):613–621. doi: 10.1021/bi035740+. [DOI] [PubMed] [Google Scholar]
- 30.Motta A, Reches M, Pappalardo L, Andreotti G, Gazit E. Biochemistry. 2005;44(43):14170–14178. doi: 10.1021/bi050658v. [DOI] [PubMed] [Google Scholar]
- 31.Shi Z, Chen K, Liu Z, Ng A, Bracken WC, Kallenbach NR. Proc Natl Acad Sci U S A. 2005;102(50):17964–17968. doi: 10.1073/pnas.0507124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaccaro P, Dente L. FEBS Lett. 2002;512(1–3):345–349. doi: 10.1016/s0014-5793(02)02220-2. [DOI] [PubMed] [Google Scholar]
- 33.Pellecchia M, Sem DS, Wuthrich K. Nat Rev Drug Discov. 2002;1(3):211–219. [Google Scholar]
- 34.Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS. J Biol Chem. 2003;278(9):7645–7654. doi: 10.1074/jbc.M209751200. [DOI] [PubMed] [Google Scholar]
- 35.Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS, Wiesmann C. J Biol Chem. 2006;281(31):22312–22320. doi: 10.1074/jbc.M602901200. [DOI] [PubMed] [Google Scholar]
- 36.Runyon ST, Zhang Y, Appleton BA, Sazinsky SL, Wu P, Pan B, Wiesmann C, Skelton NJ, Sidhu SS. Protein Sci. 2007;16(11):2454–2471. doi: 10.1110/ps.073049407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schapira M, Totrov M, Abagyan R. J Mol Recognit. 1999;12(3):177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 38.AMBER 8. Scripps Research Institute; La Jolla, CA: 2004. [Google Scholar]
- 39.Wang J, Morin P, Wang W, Kollman PA. J Am Chem Soc. 2001;123(22):5221–5230. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Donini O, Reyes CM, Kollman PA. Annu Rev Biophys Biomol Struct. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 41.Loughlin WA, Tyndall JD, Glenn MP, Fairlie DP. Chem Rev. 2004;104(12):6085–6117. doi: 10.1021/cr040648k. [DOI] [PubMed] [Google Scholar]
- 42.Fuh G, Pisabarro MT, Li Y, Quan C, Lasky LA, Sidhu SS. J Biol Chem. 2000;275(28):21486–21491. doi: 10.1074/jbc.275.28.21486. [DOI] [PubMed] [Google Scholar]
- 43.Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H. J Mol Biol. 2004;343(3):703–718. doi: 10.1016/j.jmb.2004.08.064. [DOI] [PubMed] [Google Scholar]
- 44.Song E, Gao S, Tian R, Ma S, Huang H, Guo J, Li Y, Zhang L, Gao Y. Mol Cell Proteomics. 2006;5(8):1368–1381. doi: 10.1074/mcp.M600072-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 46.Dominguez C, Boelens R, Bonvin AM. J Am Chem Soc. 2003;125(7):1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. J Biomol NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 48.Koradi R, Billeter M, Wuthrich K. J Mol Graph. 1996;14(1):51–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 49.Jorgensen WL, Chandrasekhar J, Madura JD, Klein M. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 50.Orozco M, Luque FJ. Chem Rev. 2001;101(1):203–204. doi: 10.1021/cr000703z. [DOI] [PubMed] [Google Scholar]