

Abstract
Objective:
To determine whether white matter hyperintensity (WMH) progression rate is a better predictor of cognitive impairment risk than baseline WMH volume in healthy elderly individuals.
Method:
Ninety-eight cognitively intact elderly subjects were followed in the Oregon Brain Aging Study. Forty-nine had at least 3 brain MRIs and annual cognitive and neurologic assessments until diagnosed with persistent cognitive impairment (PCI). Brain, ventricular CSF (vCSF), intracranial volume (ICV), hippocampus, total WMH, periventricular (PV) WMH, and subcortical WMH volumes were measured. Cox proportional hazards survival analyses were used to assess cognitive impairment risk.
Results:
After adjusting for age, apolipoprotein E4 status, incident hypertension, ICV, entry Mini-Mental State Examination, baseline hippocampus, and both baseline vCSF volume and rate of vCSF volume change, increased progression of total WMH volume (hazard ratio [HR] 1.84, 95% confidence interval [CI] 1.3–2.7, p = 0.0007) and PV WMH volume (HR 1.94, 95% CI 1.3–3.1, p = 0.001) conferred higher risk of PCI, whereas baseline WMH volumes did not. Every 1 mL/y increase in PV WMH volume was associated with a 94% increased risk of PCI.
Conclusion:
Progression of total and periventricular (PV) white matter hyperintensity (WMH) volumes are better predictors of persistent cognitive impairment (PCI) than baseline WMH burden. Greater PV WMH burden progression is associated with the development of PCI, a potential precursor to Alzheimer or vascular dementia. Identification of factors that decrease WMH accumulation over time is needed to maintain cognitive health in our growing elderly population.
GLOSSARY
- AD
= Alzheimer disease;
- CDR
= Dementia Rating Scale;
- CI
= confidence interval;
- HR
= hazard ratio;
- HS
= hippocampal sclerosis;
- HTN
= hypertension;
- ICV
= intracranial volume;
- MCI
= mild cognitive impairment;
- MMSE
= Mini-Mental State Examination;
- NA
= not applicable;
- NS
= not significant;
- PCI
= persistent cognitive impairment;
- PV
= periventricular;
- SES
= socioeconomic status;
- TE
= echo time;
- TR
= repetition time;
- vCSF
= ventricular CSF;
- vol
= volume;
- WMH
= white matter hyperintensity.
White matter changes seen as white matter hyperintensities (WMHs) on T2-weighted MRI are commonly observed on brain imaging of elderly individuals1 and are associated with cognitive changes2–5 and conversion to mild cognitive impairment (MCI).6 It has been shown that such white matter change is likely to progress over time,7–9 with increased rate of progression in those with greater baseline WMH burden.7,8,10 A few longitudinal volumetric MRI studies have shown detrimental effects of total WMH progression on verbal IQ, memory, and executive function.11–13 One previous study showed more specific regional consequences of periventricular (PV) white matter progression on worsening executive function testing,14 whereas another volumetric study showed that progression of PV WMH is associated with onset of dementia in depressed elderly subjects.15 Most previous longitudinal studies calculated rates of WMH change from scans obtained from all subjects regardless of whether they had converted to cognitive impairment, thus increasing the chance of introducing confounding effects on CNS structural changes from non–WMH-related neurodegenerative disease (i.e., hippocampal or brain volume decline known to be associated with Alzheimer disease [AD]).
One recent study has shown that increased total baseline WMH burden is associated with onset of MCI.6 Few, if any, however, have studied the effects of WMH progression on the onset of mild cognitive symptoms, which in some cases precedes conversion to dementia, particularly in those with greater baseline and PV WMH burden.16 The objective of this study was to compare the regional effects of baseline WMH burden with rates of WMH volume change over time in cognitively intact elderly individuals on the risk of eventual cognitive impairment.
METHODS
Subjects.
Ninety-eight subjects aged 65 years or older underwent baseline brain MRI, ApoE allele testing, and detailed annual cognitive and neurologic assessments as part of an ongoing longitudinal study of brain aging and cognition (Oregon Brain Aging Study).17,18 Entry inclusion criteria included a score of 24 or greater on the Mini-Mental State Examination (MMSE)19 and a 0 on the Clinical Dementia Rating Scale (CDR).20 At entry, subjects were community-dwelling, functionally independent adults, free of comorbid conditions commonly associated with cognitive decline (e.g., stroke, heart disease, cancer, diabetes, neurologic disorders), and were not taking medications that affect cognition. Volunteers were solicited from retirement homes, senior citizens’ organizations, and public relation activities. Those who showed evidence of questionable dementia by CDR >0, had an MMSE score <24, or who had sought or planned to seek medical attention for memory problems were not enrolled. Each CDR was based on interviews with the participant and someone familiar with the participant who served as a collateral source, as well as examination by a neurologist. Elders who showed evidence of clinical depression were not enrolled. Participants who developed health problems were retained in the project. All subjects had total WMH volumes analyzed. In addition, WMH volumes were regionally defined as being subcortical or periventricular. Forty-nine subjects had 3 or more MRIs analyzed for total, PV, and subcortical WMH volumes corresponding with a cognitive and motor evaluation before conversion to persistent cognitive impairment (PCI). As defined previously, subjects were considered to have PCI if they had 2 consecutive semiannual CDR scores of 0.5 or greater and did not convert back to normal cognition (defined as having 2 or more consecutive CDR scores of 0 during the duration of their follow-up).21 For subjects who converted to PCI, neurologic and MRI data were used from the last visit before their conversion. All subjects signed written informed consent, and approval from the Institutional Review Board of Oregon Health & Science University was obtained. Information regarding subjects’ cardiac risk factors at enrollment was obtained from a detailed medical history form. Changes in subjects’ general medical conditions were obtained yearly through patient report, and from 1996 on, from a modified cumulative illness rating scale22 administered yearly. A subset of subjects agreed to brain autopsy at the time of their death. Neuropathologic diagnosis of AD and vascular dementia followed published criteria.23–26
MRI acquisition.
The general procedures have been described previously.27 Briefly, MRI scans were performed with a 1.5-T magnet. The protocol consists of slice thickness of 4 mm (no gap), 24-cm field of view with a 256 × 256 matrix (0.86 × 0.86-mm pixel size), and 0.5 repetitions per sequence. The brain was visualized in 2 planes using the following pulse sequences: 1) T1-weighted sagittal images centered in the midsagittal plane with the pituitary profile (including the infundibulum) and cerebellar vermis clearly delineated: repetition time [TR] = 600 msec, echo time [TE] = 20 msec-images; 2) multiecho sequence T2-weighted (TR = 2,800 msec, TE = 80 msec) and proton density (TR = 2,800 msec, TE = 32 msec) coronal images perpendicular to the sagittal plane. The coronal plane is determined by a line drawn from the lowest point of the splenium to the lowest point of the genu of the corpus callosum on the midsagittal image.
Image analysis.
Image analysts evaluated each scan independently and were blind to subjects’ cognitive or neurologic testing, demographic characteristics, and results from previous imaging. The image analysis software REGION is used to quantitatively assess regional brain volumes of interest.27,28 Briefly, recursive regression analysis of bifeature space based on relative tissue intensities was used to separate tissue types on each coronal image. Pixel areas were summed for all slices and converted to volumetric measures by multiplying by the slice thickness for each of the following regions of interest: PV WMH, subcortical WMH, total WMH (PV plus subcortical WMH), brain volume, ventricular cerebral spinal fluid (vCSF), and hippocampal volumes. Intracranial volume was determined by automatically regressing for brain tissue, CSF, and WMH collectively against bone, creating a boundary along the inner table of the skull. Additional boundaries were manually traced along the tentorium cerebelli and the superior border of the superior colliculus, the pons, and the fourth ventricle. The pituitary, vessels in the sphenoidal area, and any sinuses that may have been included by the automatic regression were also manually excluded. Hippocampal bodies were determined by manually outlining the structures with a cursor directly on the computer display, as previously described.28 Rates of change of outcomes were determined by calculating the slope obtained from the regression line created by all available data points from scans performed before the onset of conversion. The intraclass correlation coefficient as a measure of reliability of volume determination was ≥0.95 for all regions except for WMH volume, which was 0.85.
Quantification of WMH using REGION.
Using REGION′s sampling tools, the analyst selects representative, unambiguous pixels of WMH (as well as brain tissue, fluid, and bone) from the multiecho sequence display. A regression model including the proton density and T2 intensities and location of each pixel differentiates tissue types. WMH is distinguished from brain tissue and fluid based on higher signal on both the proton density and T2 images. Areas of high signal that immediately abut ventricular fluid as visualized on the coronal image are considered periventricular. WMH bounded by brain tissue on all sides is considered subcortical. Areas of infarct were determined separately from WMH volumes based on whether they had clean or sharp edges and were relatively dark on proton density images.
Statistical analysis.
Longitudinal clinical data and coded MRI data were exported from a dedicated database into the statistical program JMP (SAS Institute, Cary, NC). MRI regions and clinical outcomes of interest were examined as continuous variables. Change in MRI outcome measures over time was determined from the calculated slope of a regression line created from 3 or more time points for each subject. T tests for continuous variables and Fisher exact tests for categorical variables were used to determine differences in subject and MRI characteristics between those who converted to PCI and those who did not. Survival analyses (Cox proportional hazards) were used to determine associations between baseline brain volumes and rates of brain volume change over time with risk of PCI. Analyses were adjusted for age, intracranial volume, baseline MMSE, baseline hippocampal volume, and ApoE-4 status. When rate of volume change was included as a covariate, we also controlled for the baseline volume of that variable. Proportionality assumptions were examined through visual inspection of survival curves (−ln(−ln S(t))) as well as statistical assessments for all variables.29
RESULTS
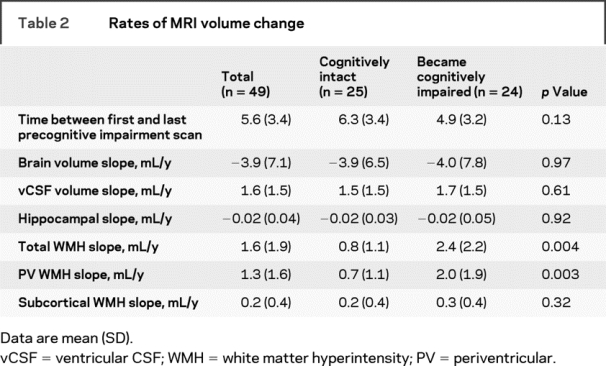
Ninety-eight participants were followed up to 15.8 years (average 9.5 years), during which time 53 subjects developed PCI. Two percent of subjects had a diagnosis of hypertension (HTN) at entry into the study. Over the total duration of follow-up, 32% had an HTN diagnosis. Subjects who eventually converted to PCI were more likely to have at least 1 ApoE-4 allele present (χ2 = 6.8, p = 0.009). Forty-nine subjects had 3 or more scans from which rates of volumetric change was calculated. These subjects did not differ in entry age, sex, socioeconomic status, education, ApoE-4 status, cerebrovascular risk factor status (presence of HTN, diabetes, TIA, stroke, or smoking history), or baseline MMSE from those without 3 or more scans. Baseline subject and MRI characteristics for subjects with longitudinal data are presented in table 1. Of the 49 subjects with multiple scans, 24 developed PCI. The average time between first and last precognitive impairment scan was 5.6 years. Longitudinal MRI characteristics are presented in table 2. Subjects who converted to PCI had greater progression of total and PV WMH burden over time.
Table 1 Baseline subject and MRI characteristics
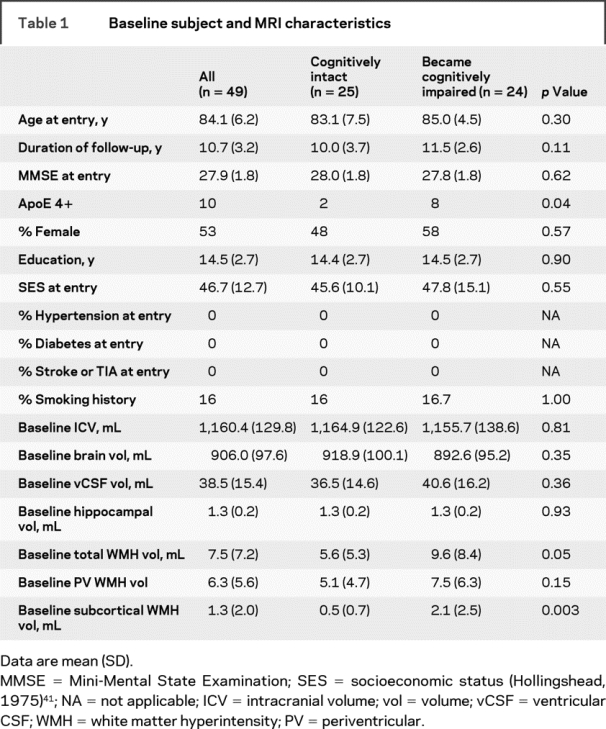
Table 2 Rates of MRI volume change
Baseline subject characteristics and risk of PCI.
Subject characteristics were individually entered into a Cox proportional hazards survival regression adjusted for age. Sex, education, socioeconomic status, a history of HTN, stroke or TIA, and smoking were not associated with cognitive decline. The absence of any ApoE-4 allele (hazard ratio [HR] 0.61, 95% confidence interval [CI] 0.45–0.84, p = 0.003) conferred a lower risk of PCI.
Baseline MRI volumes and risk of PCI (n = 98).
Baseline MRI volumes (cubic centimeters) were individually entered into a Cox proportional hazards survival regression adjusted for age, incident HTN, ICV, MMSE at entry, and ApoE-4 status. Because a larger baseline hippocampal volume conferred a lower risk of cognitive impairment (HR 0.11, 95% CI 0.02–0.73, p = 0.02), all other analyses were also adjusted for baseline hippocampal volume. Greater baseline PV WMH (HR 1.06, 95% CI 1.01–1.10, p = 0.02) and total WMH (HR 1.04, 95% CI 1.00–1.07, p = 0.03), and vCSF (HR 1.02, 95% CI 1.00–1.04, p = 0.04) conferred a higher risk of cognitive impairment. Brain volume at baseline was not associated with cognitive decline. After adjusting for baseline vCSF volume, higher PV WMH (HR 1.04, 95% CI 1.00–1.09, p = 0.078) remained a relatively weak predictor of PCI, whereas total WMH volume was no longer significant.
Rates of MRI volume change and risk of PCI (n = 49).
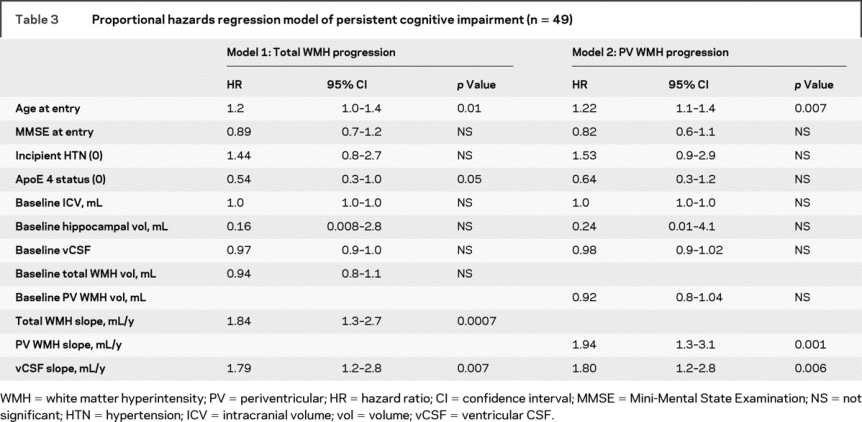
Rates of MRI volume change were individually entered into a Cox proportional hazards survival regression adjusted for age, incident HTN, ICV, MMSE at entry, baseline hippocampal and vCSF volume, ApoE-4 status, and baseline MRI volume for each region of interest. Because vCSF volume increase over time conferred a higher risk of PCI (HR 1.69, 95% CI 1.10–2.68, p = 0.02), all other analyses were also adjusted for vCSF slope. Greater rates of total WMH and PV WMH increase were associated with a higher risk of cognitive impairment (table 3). No relationship was observed between brain volume change or subcortical WMH volume change and cognitive decline.
Table 3 Proportional hazards regression model of persistent cognitive impairment (n = 49)
Brain pathology.
Of the 53 converters, 42 died during the follow-up period. Brain autopsy evaluations were available for 71% (30/42) of those subjects. There was no difference in age, sex, socioeconomic status, ApoE 4 status, baseline MMSE, or duration of follow-up between those with and without brain autopsy. Of those with pathology available, 70% (21/30) had a pathologic diagnosis of AD. Fifteen of these 21 AD subjects also had evidence of vascular disease, 1 had hippocampal sclerosis (HS), 1 had concurrent Lewy body disease, and 3 had a mixed AD/vascular dementia. Thirteen percent (4/30) had pathology consistent with vascular dementia, with 1 also having HS. Seven percent (2/30) had mixed pathology of HS, some AD pathology, and vascular disease, and 10 percent (3/30) had mild microvascular ischemic injury with moderate arteriolosclerosis.
DISCUSSION
Results from this study show that greater total and PV WMH burden predicts PCI in healthy elderly people. In addition, this is one of the first studies to demonstrate that progression of total and PV WMH volumes are more robust predictors of cognitive impairment than baseline WMH burden. In those with brain autopsy available, AD pathology was the most common diagnosis. However, evidence of cerebrovascular disease was seen in almost all cases. The high prevalence of vascular disease among other types of pathology has been reported by others.30–32 Because it is thought that AD pathology may precede the onset of cognitive symptoms by years,33 it is possible that total and PV WMH progression is an early imaging manifestation of neuronal degeneration in those who have underlying AD pathology and are destined to develop the disease. Alternatively, the effects of cerebrovascular disease may promote Aβ aggregation or plaque formation and cognitive decline in early or mild AD, as has been suggested by others.34–37 In this study, subcortical WMH progression was not associated with PCI. The total amount of subcortical WMH volume was small (<0.3% of brain), however, and likely reflective of the healthy nature of this cohort. It is possible that other subject populations with more cerebrovascular risk factors would have greater total subcortical WMH progression with subsequently increased impact on cognitive health. Alternatively, there may be a regional significance of PV WMH progression in conferring greater cerebrovascular injury or in being a specific indicator of neuronal vulnerability when compared with progression of subcortical lesions.
Limitations of the study include generalizing results to other populations, given that our subjects had few cerebrovascular risk factors at entry, and—perhaps related to this—the small volume of subcortical WMH. On the other hand, 48% had acquired at least 1 vascular risk factor (HTN, diabetes, stroke, or TIA) by the mean follow-up period of the cohort. This suggests that if vascular risk factors are important for subcortical WMH development, their effect results from sustained risk factor exposure for many years. A strength of this study is use of the designation of persistent cognitive decline as an endpoint which helped ensure that individuals had enduring meaningful change. We chose to use the term PCI rather than other definitions of mild cognitive decline (e.g., cognitive impairment nondemented, vascular cognitive impairment, or various forms of MCI) to capture the full spectrum of early and mild cognitive symptoms before the onset of dementia in those showing a temporal pattern of decline likely to be reflective of true cognitive change. A PCI diagnosis has the additional advantage of being relatively easily replicated by other population- and community-based studies in which detailed neuropsychological testing may not be readily available. Other strengths of this study include the relatively long follow-up period and availability of pathologic data on some of the subjects. In addition, rates of volumetric change were created from a minimum of 3 time points, a method that is likely to reduce the amount of variability inherent in longitudinal measures. MRI volumetric slopes were determined from the scan before conversion for those who became cognitively impaired, thus reducing potential confounding effects from neurodegenerative disease. It is possible that cognitive effects from progressive ischemic brain injury may be more apparent during this time period, before the onset of overt dementia. This hypothesis is supported by pathologic studies that show reduced cognitive repercussions of certain types of cerebrovascular lesions after the spectrum of AD pathology is taken into account.32,38,39 While prior studies have shown that concomitant vascular lesions do not affect rates of cognitive decline in those with AD,40 results from this study suggest that the total burden and rate of accumulation of such WMH lesions may have a more significant impact on the development of cognitive decline before a clinical diagnosis of AD.
Total and PV WMH progression in cognitively intact elderly individuals confers increased risk of eventual cognitive impairment in relatively healthy elderly individuals. Specifically, every 1-mL/year increase in PV WMH increases risk of PCI by 94%. Identifying those vulnerable to such progression may be important so that those at risk for cognitive decline can be targeted for early intervention and treatment trials.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by L.C. Silbert.
ACKNOWLEDGMENT
The authors thank D. Wasserman and M. Moore for longitudinal assessments and R. Guariglia for database management. The authors also thank C.S. Nelson and L.G. Perkins for their contribution in data collection and technical assistance for this study.
DISCLOSURE
L.C.S. receives research support from the NIH (K23 AG024826, P30 AG008017), receives reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients and for intraoperative neurophysiological monitoring, and is salaried to see patients at the Portland VA Medical Center. D.B.H. receives research funding from the NIH (P30 AG008017, AG029392); has received honorarium from the American Academy of Clinical Neuropsychology for a workshop presentation on “Preparing for Examination for ABPP/CN Board of Certification in Clinical Neuropsychology: ABCN Policies and Procedures”; receives royalties from Oxford University Press as coauthor of Neuropsychological Assessment, 4th Edition; and serves on the editorial board of the Journal of the International Neuropsychological Society. H.D. receives research support from the NIH (K01 AG023014, P30 AG008017) and serves on the Scientific Review Board of the National Alzheimer’s Coordinating Center. J.A.K. receives research support from the Department of Veterans Affairs (Merit Review grant) and the NIH (AG008017, AG024059, AG024978, AG026916, AG024826, AG10483, AG024904); directs a center that receives research support from the NIH, Elan Corporation, Intel Corporation, and previously Myriad Pharmaceuticals; receives reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients; is salaried to see patients at the Portland VA Medical Center; serves as an unpaid Chair for the Work Group on Technology and for the National Alzheimer’s Association and as an unpaid Commissioner for the Center for Aging Services and Technologies; receives an annual royalty from sales of the book Evidence-Based Dementia Practice; and serves on the editorial advisory board of the journal Alzheimer’s & Dementia.
Address correspondence and reprint requests to Dr. Lisa Silbert, Layton Aging and Alzheimer’s Disease Center, Department of Neurology, CR-131, Oregon Health & Science University, 3181 S.W. Sam Jackson Park Rd., Portland, OR 97239 silbertl@ohsu.edu
Supported in part by grants from the Department of Veterans Affairs and NIH (P30 AG 08017, M01 RR000334, and K23 AG 24826-01, K01AG023014), Paul B. Beeson Career Development Award in Aging, the Max Millis Fund for Neurological Research, and the Storms Family Fund at the Oregon Community Foundation.
Disclosure: Author disclosures are provided at the end of the article.
Received January 28, 2009. Accepted in final form April 2, 2009.
REFERENCES
- 1.Yue NC, Arnold AM, Longstreth WT Jr, et al. Sulcal, ventricular, and white matter changes at MR imaging in the aging brain: data from the cardiovascular health study. Radiology 1997;202:33–39. [DOI] [PubMed] [Google Scholar]
- 2.Baum KA, Schulte C, Girke W, Reischies FM, Felix R. Incidental white-matter foci on MRI in “healthy” subjects: evidence of subtle cognitive dysfunction. Neuroradiology 1996;38:755–760. [DOI] [PubMed] [Google Scholar]
- 3.DeCarli C, Murphy DG, Tranh M, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology 1995;45:2077–2084. [DOI] [PubMed] [Google Scholar]
- 4.Longstreth WT Jr, Manolio TA, Arnold A, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 1996;27:1274–1282. [DOI] [PubMed] [Google Scholar]
- 5.Adak S, Illouz K, Gorman W, et al. Predicting the rate of cognitive decline in aging and early Alzheimer disease. Neurology 2004;63:108–114. [DOI] [PubMed] [Google Scholar]
- 6.Smith EE, Egorova S, Blacker D, et al. Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Arch Neurol 2008;65:94–100. [DOI] [PubMed] [Google Scholar]
- 7.Sachdev P, Wen W, Chen X, Brodaty H. Progression of white matter hyperintensities in elderly individuals over 3 years. Neurology 2007;68:214–222. [DOI] [PubMed] [Google Scholar]
- 8.Silbert LC, Nelson C, Howieson DB, Moore MM, Kaye JA. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology 2008;71:108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt R, Fazekas F, Kapeller P, Schmidt H, Hartung HP. MRI white matter hyperintensities: three-year follow-up of the Austrian Stroke Prevention Study. Neurology 1999;53:132–139. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt R, Enzinger C, Ropele S, Schmidt H, Fazekas F; Austrian Stroke Prevention Study. Progression of cerebral white matter lesions: 6-year results of the Austrian Stroke Prevention Study. Lancet 2003;361:2046–2048. [DOI] [PubMed] [Google Scholar]
- 11.Garde E, Lykke Mortensen E, Rostrup E, Paulson OB. Decline in intelligence is associated with progression in white matter hyperintensity volume. J Neurol Neurosurg Psychiatry 2005;76:1289–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt R, Ropele S, Enzinger C, et al. White matter lesion progression, brain atrophy, and cognitive decline: the Austrian stroke prevention study. Ann Neurol 2005;58:610–616. [DOI] [PubMed] [Google Scholar]
- 13.Kramer JH, Mungas D, Reed BR, et al. Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology 2007;21:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Heuvel DM, ten Dam VH, de Craen AJ, et al. Measuring longitudinal white matter changes: comparison of a visual rating scale with a volumetric measurement. AJNR Am J Neuroradiol 2006;27:875–878. [PMC free article] [PubMed] [Google Scholar]
- 15.Steffens DC, Potter GG, McQuoid DR, et al. Longitudinal magnetic resonance imaging vascular changes, apolipoprotein E genotype, and development of dementia in the neurocognitive outcomes of depression in the elderly study. Am J Geriatr Psychiatry 2007;15:839–849. [DOI] [PubMed] [Google Scholar]
- 16.Debette S, Bombois S, Bruandet A, et al. Subcortical hyperintensities are associated with cognitive decline in patients with mild cognitive impairment. Stroke 2007;38:2924–2930. [DOI] [PubMed] [Google Scholar]
- 17.Howieson DB, Holm LA, Kaye JA, Oken BS, Howieson J. Neurologic function in the optimally healthy oldest old: neuropsychological evaluation. Neurology 1993;43:1882–1886. [DOI] [PubMed] [Google Scholar]
- 18.Kaye JA, Oken BS, Howieson DB, Howieson J, Holm LA, Dennison K. Neurologic evaluation of the optimally healthy oldest old. Arch Neurol 1994;51:1205–1211. [DOI] [PubMed] [Google Scholar]
- 19.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 20.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 21.Marquis S, Moore MM, Howieson DB, et al. Independent predictors of cognitive decline in healthy elderly persons. Arch Neurol 2002;59:601–606. [DOI] [PubMed] [Google Scholar]
- 22.Parmelee PA, Thuras PD, Katz IR, Lawton MP. Validation of the Cumulative Illness Rating Scale in a geriatric residential population. J Am Geriatr Soc 1995;43:130–137. [DOI] [PubMed] [Google Scholar]
- 23.Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R. Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology 1992;42:473–480. [DOI] [PubMed] [Google Scholar]
- 24.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997;56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 25.Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer’s disease: a primer for practicing pathologists. Arch Pathol Lab Med 1993;117:132–144. [PubMed] [Google Scholar]
- 26.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann NY Acad Sci 2002;977:9–23. [DOI] [PubMed] [Google Scholar]
- 27.Mueller EA, Moore MM, Kerr DC, et al. Brain volume preserved in healthy elderly through the eleventh decade. Neurology 1998;51:1555–1562. [DOI] [PubMed] [Google Scholar]
- 28.Kaye JA, Swihart T, Howieson D, et al. Volume loss of the hippocampus and temporal lobe in healthy elderly persons destined to develop dementia. Neurology 1997;48:1297–1304. [DOI] [PubMed] [Google Scholar]
- 29.Kleinbaum D, Klein M. Survival Analysis: A Self-Learning Text. New York: Springer; 2005. [Google Scholar]
- 30.Fernando MS, Ince PG; MRC Cognitive Function and Ageing Neuropathology Study Group. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci 2004;226:13–17. [DOI] [PubMed] [Google Scholar]
- 31.Neuropathology Group, Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 2001;357:169–175. [DOI] [PubMed] [Google Scholar]
- 32.Jellinger KA, Attems J. Prevalence and impact of cerebrovascular pathology in Alzheimer’s disease and parkinsonism. Acta Neurol Scand 2006;114:38–46. [DOI] [PubMed] [Google Scholar]
- 33.Morris JC, Storandt M, McKeel DW Jr, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996;46:707–719. [DOI] [PubMed] [Google Scholar]
- 34.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. J Neurol Sci 2007;257:80–87. [DOI] [PubMed] [Google Scholar]
- 35.Sadowski M, Pankiewicz J, Scholtzova H, et al. Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004;29:1257–1266. [DOI] [PubMed] [Google Scholar]
- 36.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997;277:813–817. [PubMed] [Google Scholar]
- 37.Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke 2003;34:335–337. [DOI] [PubMed] [Google Scholar]
- 38.Gold G, Giannakopoulos P, Herrmann FR, Bouras C, Kovari E. Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain 2007;130:2830–2836. [DOI] [PubMed] [Google Scholar]
- 39.Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol 2006;60:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JH, Olichney JM, Hansen LA, Hofstetter CR, Thal LJ. Small concomitant vascular lesions do not influence rates of cognitive decline in patients with Alzheimer disease. Arch Neurol 2000;57:1474–1479. [DOI] [PubMed] [Google Scholar]
- 41.Hollingshead A. Four Factor Index of Social Status. New Haven, CT: Yale University; 1975. [Google Scholar]