

Abstract
Background:
Experimental studies indicate that dopaminergic neurons in the ventral periaqueductal gray matter (PAG) are involved in maintenance of wakefulness. Excessive daytime sleepiness (EDS) is a common manifestation of multiple system atrophy (MSA) and dementia with Lewy bodies (DLB) but involvement of these neurons has not yet been explored.
Methods:
We sought to determine whether there is loss of dopaminergic neurons in the ventral PAG in MSA and DLB. We studied the midbrain obtained at autopsy from 12 patients (9 male, 3 female, age 61 ± 3) with neuropathologically confirmed MSA, 12 patients (11 male, 1 female, age 79 ± 4) with diagnosis of DLB and limbic or neocortical Lewy body disease, and 12 controls (7 male, 5 female, ages 67 ± 4). Fifty-micron sections were immunostained for tyrosine hydroxylase (TH) or α-synuclein and costained with thionin. Cell counts were performed every 400 μm throughout the ventral PAG using stereologic techniques.
Results:
Compared to the total estimated cell numbers in controls (21,488 ± 8,324 cells), there was marked loss of TH neurons in the ventral PAG in both MSA (11,727 ± 5,984; p < 0.01) and DLB (5,163 ± 1,926; p < 0.001) cases. Cell loss was more marked in DLB than in MSA. There were characteristic α-synuclein inclusions in the ventral PAG in both MSA and DLB.
Conclusions:
There is loss of putative wake-active ventral periaqueductal gray matter dopaminergic neurons in both multiple system atrophy and dementia with Lewy bodies, which may contribute to excessive daytime sleepiness in these conditions.
GLOSSARY
- AD
= Alzheimer disease;
- B&B
= Braak and Braak;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- CPAP
= continuous positive airway pressure;
- DLB
= dementia with Lewy bodies;
- ECG
= electrocardiogram;
- EDS
= excessive daytime sleepiness;
- EOG
= electrooculography;
- ESS
= Epworth Sleepiness Scale;
- GCI
= glial cytoplasmic inclusion;
- LBD
= Lewy body disease;
- MSA
= multiple system atrophy;
- MSA-P
= MSA with predominant parkinsonism;
- MSA-C
= MSA with predominant cerebellar involvement;
- OSA
= obstructive sleep apnea;
- PAG
= periaqueductal gray matter;
- PSG
= polysomnogram;
- RBD
= REM sleep behavior disorder;
- TH
= tyrosine hydroxylase.
Recent rat studies have identified a group of dopaminergic neurons in the ventral periaqueductal gray matter (PAG) that appear to be critical for maintenance of wakefulness.1 These neurons are active during wakefulness, are connected with forebrain and brainstem areas involved in control of arousal, and their depletion results in increased total sleep time in rats.1 Excessive daytime sleepiness (EDS) is a common manifestation of multiple system atrophy (MSA)2–4 and Lewy body disorders, including dementia with Lewy bodies (DLB).5–7 In both MSA and Lewy body disorders, there is loss of several neuronal groups involved in maintenance of the waken state, including cholinergic neurons of the mesopontine tegmentum,8 noradrenergic neurons in the locus ceruleus,9 serotonergic neurons of the rostral raphe,9,10 and hypocretin/orexin neurons of the lateral hypothalamus.11–13 However, involvement of putative wake-active dopaminergic groups in the ventral PAG has not yet been explored in MSA or DLB. We sought to determine whether there was involvement of these neurons and accumulation of characteristic α-synuclein inclusions in the ventral PAG in cases with neuropathologically confirmed MSA or limbic or neocortical Lewy body disease (LBD).
METHODS
Subjects.
Brains were obtained at autopsy from 36 subjects (table). All patients had signed informed consent for autopsy according to the Institutional Review Board guidelines. Twelve cases (9 men, 3 women, age 61 ± 3 years) had clinical and neuropathologically proven MSA according to current criteria14,15; 12 cases (11 men, 1 woman, age 79 ± 4 years) had clinical diagnosis of DLB and neuropathologic neocortical or limbic stage Lewy body disease (LBD)16; and 12 control cases (7 men, 5 women, ages 67 ± 4 years) had no history of neurologic disease. As a group, the DLB cases were older (p < 0.01) than the control and MSA cases; there was no significant age difference between MSA and control cases. There was no significant difference in disease duration between the MSA (6 ± 1 year) and DLB (9 ± 2 years) cases.
Table Patient data
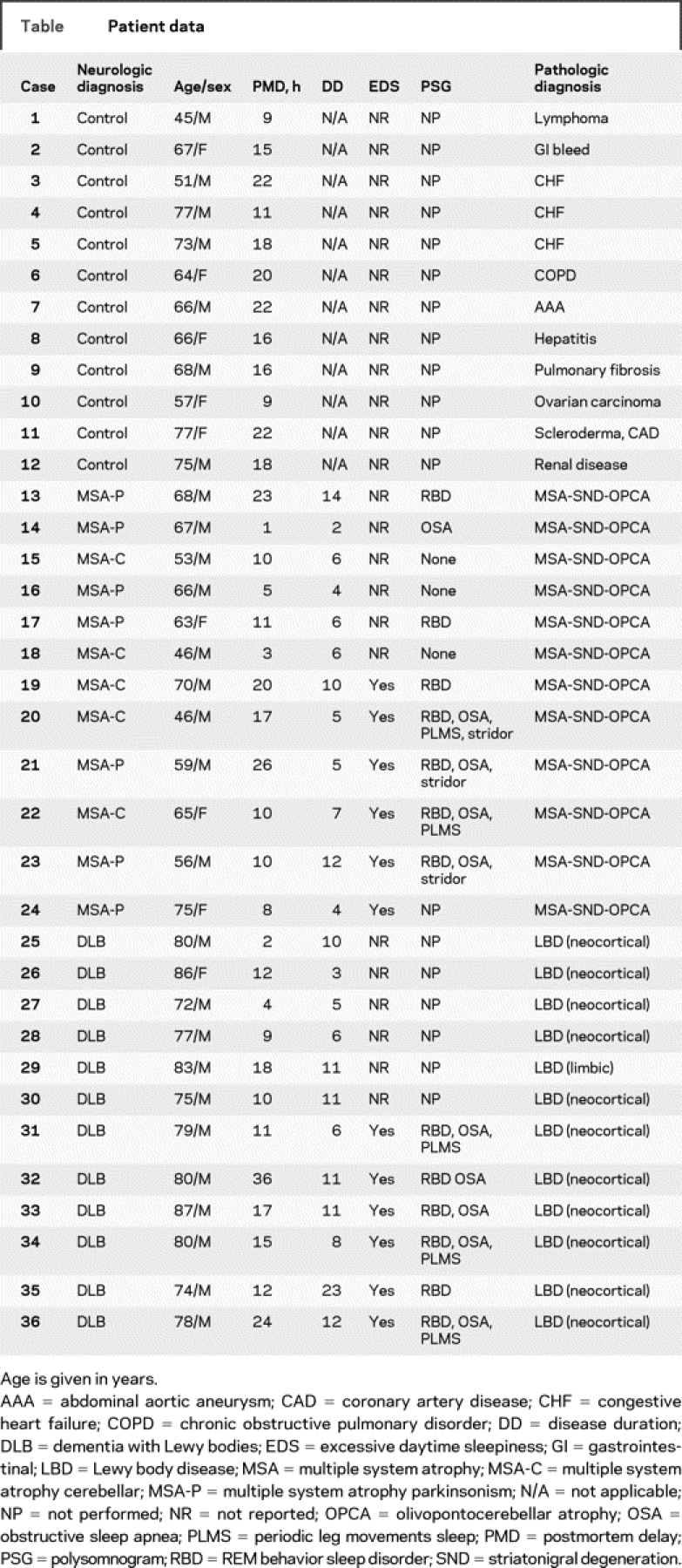
Of the 12 MSA cases, 7 had predominant parkinsonism (MSA-P) and 5 predominant cerebellar involvement (MSA-C) and all had severe autonomic failure, including orthostatic hypotension. All 12 cases with clinical diagnosis of DLB had dementia and parkinsonism; 10 of them had visual hallucinations, and 5 had orthostatic hypotension. Excessive daytime sleepiness was present, by history, in 6 of the 12 MSA (3 MSA-P and 3 MSA-C) and 6 of the 12 DLB cases. None of the DLB cases was on treatment with levodopa or dopamine agonists at the time of evaluation. Unfortunately, none of the MSA or DLB cases had undergone assessment with validated scales such as the Epworth Sleepiness Scale (ESS).17 Polysomnogram (PSG) had been performed in 4 of 6 patients with MSA and 6 of the 6 patients with DLB who had EDS. Polysomnography was performed according to current standards, using a digital polygraph (Cardinal Health/Viasys, NicoletOne) with 3 EEG leads (Fz-Cz, Cz-Oz, C3-A2 or C4-A1); 2 electrooculography (EOG); submental EMG; tibialis EMG, extensor or flexor digitorum EMG; electrocardiogram (ECG); rib cage and abdominal respiratory inductance plethysmography; nasal pressure transducer; oral/nasal Thermocouple; oxygen saturation via finger pulse oximetry; and sonography via decibel meter and body position (visual inspection). Scoring of all polysomnographic data were made according to the guidelines of The AASM Manual for the Scoring of Sleep and Associated Events; Rule, Terminology and Technical Specifications (American Academy of Sleep Medicine, Westchester, IL, 2007). Stridor was diagnosed via auditory identification and PSG documentation. Polysomnogram revealed REM sleep behavior disorder (RBD) in all MSA and DLB cases. Obstructive sleep apnea (OSA) was documented in the 4 patients with MSA and in 4 of the 6 patients with DLB who underwent PSG. Laryngeal stridor was present in 3 patients with MSA and none of the patients with DLB. Treatment with nocturnal continuous positive airway pressure (CPAP) resulted in improvement of EDS in these patients according to the medical record. However, the degree of improvement could not be assessed reliably given that EDS was not assessed using a standardized scale. By history, CPAP prevented laryngeal stridor in the MSA cases.
Postmortem delay was similar in control (17 ± 2 hours), MSA (18 ± 5 hours), and DLB (18 ± 4 hours) groups. The left half of the brain was examined for routine neuropathologic studies, including α-synuclein immunostaining. The neuropathologic diagnosis of MSA or LBD was made according to current consensus criteria.15,16 The presence of neurofibrillary tangles and senile plaques was classified in each case according to the stages of Alzheimer disease (AD) as described by Braak and Braak (B&B)18 and defined by the Consortium to Establish a Registry for Alzheimer's Disease (CERAD).19
Of the 12 MSA cases, 10 had severe involvement of the putamen and substantia nigra pars compacta and 6 had moderate to severe involvement of the pons, inferior olivary nucleus, and cerebellum. All cases had moderate to severe accumulation of glial cytoplasmic inclusions (GCIs) in these regions. One MSA case had mild to moderate and 2 had only rare associated Lewy body pathology. Eleven MSA cases had B&B staging 0–II, and 1 had stage III; none fulfilled CERAD criteria for AD. Of the DLB cases, 11 had neocortical and one had limbic stage LBD.16 Of the 11 neocortical stage LBD cases, 8 had B&B stage III AD pathology, 1 had CERAD criteria for possible AD; 1 had B&B stage 0–I and normal CERAD stage; and 1 case had B&B stage VI and CERAD criteria for definite AD. The single limbic stage LBD case had B&B stage 0–I and normal CERAD stage.
A block containing the midbrain from 33 to 47 mm rostral to the obex to include the whole extent of the PAG was separated in each case for the purposes of the present study. These blocks were immersion fixed in 5% formalin for 24 hours at 4°C and cryoprotected in buffered 30% sucrose for 5 to 7 days prior to processing. Serial 50-μm cryostat sections were obtained and every eighth section was processed for immunoreactivity for either tyrosine hydroxylase (TH, mouse monoclonal, 1:3,000, Immunostar, Hudson, WI) or α-synuclein (goat polyclonal, 1:400, Santa Cruz Biotechnologies, Santa Cruz, CA). Diaminobenzidine/glucose oxidase solution with nickel enhancement (SIGMA, St. Louis, MO) was used for the substrate reaction. Immunoreactive neurons were identified under bright-field illumination by a characteristic dark brown to black reaction product that densely fills the perikarya and their processes. Omission of the primary antibody or incubation with normal sera resulted in a lack of immunostaining. All sections were costained with thionin to identify surrounding structures and to determine whether loss of immunoreactivity reflected neuronal loss or lack of expression of the antigen. In selected cases, 5-μm paraffin-embedded sections were processed for immunofluorescence for α-synuclein (LB509 mouse monoclonal 1:50-dilution; Zymed, San Francisco, CA) with Alex fluor 488 fluorochrome (Molecular Probes, Eugene, OR); TH (rabbit polyclonal; 1:600, Affinity Bioreagents, Rockford, IL) with Alex fluor 568 fluorochrome (Molecular Probes, Eugene, OR), to assess colocalization of α-synuclein inclusions with TH-immunoreactive neurons in the ventral PAG.
Image analysis and quantitation.
For each control, MSA, or LBD case, we analyzed 14 sections, obtained 800 μm apart to span the length of the PAG according to the atlas of Paxinos and Huang.20 The sections were examined under bright-field microscopy using stereologic techniques. Cell counts were performed using a modified light microscope (Zeiss Axioimager A-1; Zeiss, Thornwood, NY) equipped with a motorized specimen stage for automated sampling (Ludl Electronics; Hawthorne, NY), CCD color video camera (Microfire; Optronics, Goleta, CA), and stereology software (Stereo Investigator, v9.2; MBF Bioscience, Williston, VT). Stereologic analysis of the total number of neurons well-defined by their strong immunoreactivity was performed using the optical fractionator method. All systematic unbiased random samples in each section were analyzed according to established criteria.21 The same investigator, blinded to the clinical and final neuropathologic diagnoses, performed all counts.
Statistical analysis.
Statistical analysis was performed using the software SPSS 14.0 for Windows. Multivariate analysis of variance was used to determine if age, gender, disease duration, postmortem delay, or history of EDS, OSA, or RBD had any effect on cell counts. We also analyzed the cell count comparing controls and the 2 patient groups using univariate analysis of variance. Post hoc analysis was then conducted using Dunnett's and Bonferroni's formulas. A p value of <0.05 was considered significant.
RESULTS
TH cell counts in the ventral PAG.
Tyrosine-hydroxylase cells were identified in the ventral PAG in all cases (figure 1). These cells were located slightly lateral to the midline and had a morphology and orientation similar to those described for the wake-active dopaminergic neurons in the ventral PAG in the rat,1 except in that they were only rarely seen in the area corresponding to the dorsal raphe in humans. Compared to the total estimated number of TH-immunoreactive cells in the ventral PAG in controls (21,488 ± 8,324 cells), there was marked loss of these cells in both MSA (11,727 ± 5,984, p = 0.001) and LBD cases (5,163 ± 1,926; p controls [p = 0.000]) (figure 2). The degree of cell loss was more marked in LBD than in MSA (p = 0.035; posthoc analysis, Bonferroni test) (figure 2I). Since only one of our LBD cases had limbic stage pathology, we could not compare the degree of cell loss with that in the neocortical stage LBD. On multivariate analysis, there was no significant effect of age, gender, disease duration, postmortem delay, disease phenotype (MSA-C vs MSA-P), or presence or absence of history of EDS (p = 0.507), OSA (p = 0.767), or RBD (p = 0.327) on the total estimated TH cell counts. There was no significant difference in the number of remaining non-TH-immunoreactive neurons in the ventral PAG among controls, MSA, or LBD cases. On multivariate analysis, age, gender, disease duration, postmortem delay, disease phenotype (MSA-C vs MSA-P), or presence or absence of history of EDS, OSA, or RBD did not have any significant effect on non-TH cell counts in the ventral PAG. Cell counts for each individual case are listed in table e-1 on the Neurology® Web site at www.neurology.org.

Figure 1 Distribution of TH-immunoreactive neurons in the ventral periaqueductal gray
Upper panel: Fifty-micrometer section of the human midbrain showing the distribution of tyrosine hydroxylase (TH) immunoreactive neurons in the ventral periaqueductal gray (PAG). Lower panel: TH-immunoreactive cells in the ventral PAG in a control (75-year-old man, 18-h postmortem delay), a 70-year-old man with multiple system atrophy (MSA) (postmortem delay 20 h), and an 80-year-old man with neocortical stage Lewy body disease (LBD) (postmortem delay, 15 h). Aq = aqueduct; bar = 500 μm upper panel; bar = 50 μm lower panels.
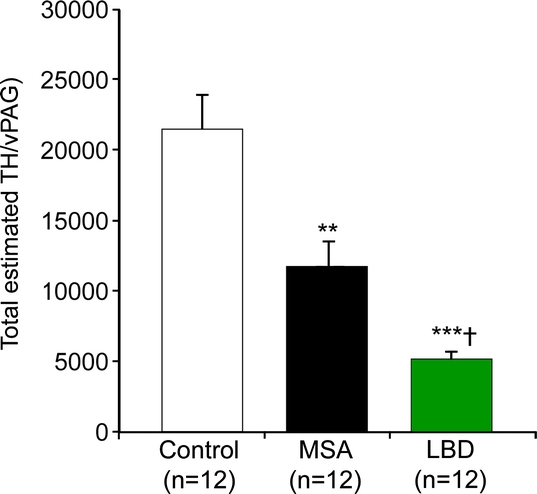
Figure 2 Total estimated numbers of TH-immunoreactive neurons in the vPAG in controls, patients with MSA, and patients with dementia with Lewy bodies and neocortical or limbic LBD
**p < 0.001, ***p = 0.000 compared to controls; †p < 0.05 compared to MSA. TH = tyrosine hydroxylase; vPAG = ventral periaqueductal gray; MSA = multiple system atrophy; LBD = Lewy body disease.
α-Synuclein-immunoreactive inclusions in the ventral PAG.
There were abundant GCIs in the ventral and other portions of the PAG in all MSA cases (figure 3) in the proximity of TH-immunoreactive cells (figure 4). In LBD cases, there were also Lewy bodies and neurites in the ventral and other portions of the PAG (figure 3); Lewy bodies were present in TH-immunoreactive cells (figure 4).

Figure 3 Distribution of α-synuclein-immunoreactive inclusions in the periaqueductal gray in a 59-year-old patient with MSA and a 72-year-old patient with neocortical stage LBD
Bar = 20 μm. MSA = multiple system atrophy; LBD = Lewy body disease.

Figure 4 Colocalization of α-synuclein inclusions and TH neurons in the ventral periaqueductal gray
Localization of α-synuclein immunofluorescent inclusions (A), tyrosine hydroxylase (TH) immunofluorescent neurons (B), or both (C) in the ventral periaqueductal gray in a 60-year-old man with no neurologic disease (control), a 57-year-old man with multiple system atrophy (MSA), and a 70-year-old man with dementia with Lewy bodies and neocortical stage Lewy body disease (LBD). Representative fields (60×) show α-synuclein-immunoreactive glial cytoplasmic inclusion in the vicinity of a TH cell in the MSA cases, and Lewy body in a TH cell in the LBD case.
DISCUSSION
Our findings indicate that there is loss of TH-immunoreactive neurons and accumulation of characteristic α-synuclein-immunoreactive inclusions in the ventral PAG in both MSA and LBD. Tyrosine hydroxylase is a marker of all catecholaminergic neurons and thus cannot differentiate dopaminergic from noradrenergic neurons. Although we did not process the tissue for markers of noradrenergic neurons, such as dopamine-β-hydroxylase, the distribution, morphology, and orientation of the TH cells studied was similar to those of the dopaminergic neurons described in the ventral PAG of the rat1 and probably represent a similar population. Elegant studies in the rat show that dopaminergic neurons in the ventral PAG have an important contributory role in maintenance of wakefulness.1 These neurons are selectively activated during wakefulness and have extensive reciprocal connections with areas involved in regulation of the sleep-wake cycle, including the ventrolateral preoptic area, lateral hypothalamic hypocretin/orexin neurons, cholinergic neurons of the laterodorsal tegmental nucleus, and locus ceruleus.1 Loss of these wake-active dopaminergic neurons as a result of local administration of 6-hydroxydopamine in the ventral PAG increased total day sleep time by approximately 20% in the rat.1 Therefore, our present findings might suggest that loss of TH-immunoreactive (presumably dopaminergic) neurons in the ventral PAG may contribute to EDS in MSA and DLB.
The degree of TH cell loss in the ventral PAG was significantly more marked in LBD than in MSA. Our patients with LBD were significantly older than our patients with MSA or controls, suggesting that age-related neuronal loss may have contributed to these differences. However, neither age nor disease duration was significantly associated with the degree of TH cell loss on multivariate analysis. Thus, like the case of serotonergic neurons in the dorsal raphe,10 the ventral PAG appears to be more vulnerable to TH cell loss in LBD than in MSA. Since most of our LBD cases were neocortical and only 1 had limbic stage LBD, extrapolation of our findings to other stages of LBD requires studies of a larger number of cases.
The presence of GCIs in the MSA and Lewy body pathology in DLB cases is consistent with evidence that the PAG is a target in neurodegenerative disorders such as Parkinson disease9,22 and AD.23 The PAG forms an interface between the forebrain and the lower brainstem and consists of separate columns that have specific connections and critical roles in coordinating behavioral, motor, autonomic, and pain-modulatory responses to a variety of stimuli.24 Many of these responses are mediated by projections from the PAG to the rostral ventrolateral medulla and medullary raphe, which are involved in both MSA and LBD.25 Our results thus indicate that the PAG is yet another target of disorders that systematically affect brainstem and forebrain areas involved in homeostasis. Given the different connections and functions of the different PAG columns, it is possible that there is a regional susceptibility to involvement in synucleinopathies, as occurs in the case of AD.23
We found no significant differences in the degree of dopaminergic cell loss in the ventral PAG in either MSA or DLB cases with or without history of EDS. However, these findings have to be interpreted with caution given several limitations in assessment EDS in our study. Since this was a retrospective analysis using data available from medical records, it is possible that many patients with MSA or LBD were not specifically asked about the presence of EDS and therefore the true prevalence of EDS in our patients may have been underestimated. EDS had not been assessed with standardized questionnaires in these patients, which makes it difficult to determine its severity or response to treatment. Most of our cases with history of EDS had coexisting conditions, particularly sleep apnea, that could at least partially account for this symptom. Finally, the number of cases analyzed may have been too small to show any clear differences. Therefore, the lack of a clear relationship between the degree of dopamine cell loss in the ventral PAG and EDS does not exclude a role of these cells in maintenance of wakefulness in humans.
However, other possibilities, not mutually exclusive, may also contribute to explain the lack of clear relationship between TH cell loss in the ventral PAG and EDS in our cases. The most likely explanation is that EDS in synucleinopathies is multifactorial. All our cases that presented with history of EDS had polysomnographic evidence of obstructive sleep apnea. This is consistent with the evidence that sleep apnea is an important cause of EDS in MSA2–4 and DLB.5–7 Furthermore, both MSA and LBD are associated with loss of other wake-active neuronal groups in the hypothalamus and brainstem, including cholinergic mesopontine,8 noradrenergic locus ceruleus,9 serotonergic raphe,9,10 and hypocretin/orexin lateral hypothalamic11–13 neurons.
Our findings indicate that, in both MSA and LBD, there is loss of putative wake-active dopaminergic neurons in the ventral PAG. Loss of these neurons may contribute to EDS in MSA and DLB. However, given the limitations of our study in assessing EDS and the presence of coexisting conditions, studies on larger groups of patients with well-defined EDS and no other contributory factors may help to further define the contribution of these neurons to maintenance of wakefulness in humans.
AUTHOR CONTRIBUTIONS
P. Sandroni, MD, PhD, conducted the statistical analysis.
DISCLOSURE
Dr. Benarroch serves as Section Editor, Clinical Implications of Neuroscience Research, for Neurology, and receives research support from the NIH [NS32352-P2]. Ms. Schmeichel receives research support from the NIH [NS32352-P2]. Ms. Dugger reports no disclosures. Dr. Sandroni serves on the scientific advisory boards of Cervelo Pharmaceuticals and Ono and receives research support from the NIH [NS32352-P2]. Dr. Parisi serves as Section Co-Editor, Neurology Clinical Pathological Conference, for Neurology; serves on the US Government Defense Health Board; receives royalties from Oxford, Principles & Practice of Neuropathology, 2nd ed, 2003; and receives research support from the NIH [NS32352-13]. Dr. Low receives research support from the NIH [NS32352-P2].
Supplementary Material
Address correspondence and reprint requests to Dr. E.E. Benarroch, Mayo Clinic, Department of Neurology, 811 Guggenheim Bldg., 200 First Street SW, Rochester, MN 55905 benarroch.eduardo@mayo.edu
Supplemental data at www.neurology.org
This study was supported by NIH grant NS32352-P2 and Mayo Funds.
Disclosure: Author disclosures are provided at the end of the article.
Received January 16, 2009. Accepted in final form April 2, 2009.
REFERENCES
- 1.Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci 2006;26:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iranzo A. Sleep and breathing in multiple system atrophy. Curr Treat Options Neurol 2007;9:347–353. [PubMed] [Google Scholar]
- 3.Ghorayeb I, Bioulac B, Tison F. Sleep disorders in multiple system atrophy. J Neural Transm 2005;112:1669–1675. [DOI] [PubMed] [Google Scholar]
- 4.Arnulf I. Excessive daytime sleepiness in parkinsonism. Sleep Med Rev 2005;9:185–200. [DOI] [PubMed] [Google Scholar]
- 5.Boddy F, Rowan EN, Lett D, O'Brien JT, McKeith IG, Burn DJ. Subjectively reported sleep quality and excessive daytime somnolence in Parkinson's disease with and without dementia, dementia with Lewy bodies and Alzheimer's disease. Int J Geriatr Psychiatry 2007;22:529–535. [DOI] [PubMed] [Google Scholar]
- 6.Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology 2004;62:181–187. [DOI] [PubMed] [Google Scholar]
- 7.Grace JB, Walker MP, McKeith IG. A comparison of sleep profiles in patients with dementia with Lewy bodies and Alzheimer's disease. Int J Geriatr Psychiatry 2000;15:1028–1033. [DOI] [PubMed] [Google Scholar]
- 8.Schmeichel AM, Buchhalter LC, Low PA, et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology 2008;70:368–373. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA. Pathology of Parkinson's disease: changes other than the nigrostriatal pathway. Mol Chem Neuropathol 1991;14:153–197. [DOI] [PubMed] [Google Scholar]
- 10.Benarroch EE, Schmeichel AM, Sandroni P, Parisi JE, Low PA. Rostral raphe involvement in Lewy body dementia and multiple system atrophy. Acta Neuropathol 2007;114:213–220. [DOI] [PubMed] [Google Scholar]
- 11.Benarroch EE, Schmeichel AM, Sandroni P, Low PA, Parisi JE. Involvement of hypocretin neurons in multiple system atrophy. Acta Neuropathol (Berl) 2007;113:75–80. [DOI] [PubMed] [Google Scholar]
- 12.Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain 2007;130:1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson's disease. Brain 2007;130:1577–1585. [DOI] [PubMed] [Google Scholar]
- 14.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 2007;33:615–620. [DOI] [PubMed] [Google Scholar]
- 16.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 17.Billiard M. [Excessive daytime sleepiness.] Rev Prat 2007;57:1555–1564. [PubMed] [Google Scholar]
- 18.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 19.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 20.Paxinos G, Huang XF. Atlas of the Human Brainstem. San Diego: Academic Press; 1995. [Google Scholar]
- 21.Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience 2005;130:813–831. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Rub U, Sandmann-Keil D, et al. Parkinson's disease: affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol 2000;99:489–495. [DOI] [PubMed] [Google Scholar]
- 23.Parvizi J, Van Hoesen GW, Damasio A. Selective pathological changes of the periaqueductal gray matter in Alzheimer's disease. Ann Neurol 2000;48:344–353. [PubMed] [Google Scholar]
- 24.Bandler R, Keay KA, Floyd N, Price J. Central circuits mediating patterned autonomic activity during active vs. passive emotional coping. Brain Res Bull 2000;53:95–104. [DOI] [PubMed] [Google Scholar]
- 25.Benarroch EE, Schmeichel AM, Low PA, Boeve BF, Sandroni P, Parisi JE. Involvement of medullary regions controlling sympathetic output in Lewy body disease. Brain 2005;128:338–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.