

Abstract
Drug treatment of HIV type 1 (HIV-1) infection leads to a rapid initial decay of plasma virus followed by a slower second phase of decay. To investigate the role of HIV-1 retained on follicular dendritic cells (FDCs) in this process, we have developed and analyzed a mathematical model for HIV-1 dynamics in lymphoid tissue (LT) that includes FDCs. Analysis of clinical data using this model indicates that decay of HIV-1 during therapy may be influenced by release of FDC-associated virus. The biphasic character of viral decay can be explained by reversible multivalent binding of HIV-1 to receptors on FDCs, indicating that the second phase of decay is not necessarily caused by long-lived or latently infected cells. Furthermore, viral clearance and death of short-lived productively infected cells may be faster than previously estimated. The model, with reasonable parameter values, is consistent with kinetic measurements of viral RNA in plasma, viral RNA on FDCs, productively infected cells in LT, and CD4+ T cells in LT during therapy.
Treatment of HIV type 1 (HIV-1) infection with reverse transcriptase (RT) and protease inhibitors leads to decay of plasma virus (1–4), decay of virus associated with follicular dendritic cells (FDCs) (5–8), partial recovery of CD4+ T cells in blood (1, 9–11) and lymphoid tissue (LT) (12), and partial restoration of the FDC network (13). A feature of HIV-1 dynamics is biphasic decay of virus during therapy. At the start of therapy, plasma virus decays quickly, but by 2 wk, a second phase is reached and the rate of viral decay slows considerably (4, 14). Second-phase dynamics may be caused by one or more processes, including viral production by long-lived infected cells, activation of latently infected cells, and release of HIV-1 from viral reservoirs (3, 4).
The pool of HIV-1 on FDCs is a significant viral reservoir (15, 16). During the asymptomatic untreated stage of infection, the FDC network harbors ≈1011 copies of HIV-1 RNA (5, 17). This pool of virus, which composes a large fraction of the viral burden in an infected patient (17), may influence HIV-1 dynamics, given its large size and the observed loss of virus from FDCs during antiretroviral therapy (5–8). Here, to assess the influence of FDCs on HIV-1 dynamics, we use a mathematical model, which includes FDC-associated virus, to analyze quantitative kinetic measurements of viral and cellular dynamics in blood and LT (18). We consider measurements of plasma viral load (14), FDC-associated HIV-1 RNA (5), infected mononuclear cells in LT (5), and CD4+ T cells in LT (12) during treatment for eight patients. The model combines earlier models for HIV-1 dynamics (19) with a recent model for the reversible binding of HIV-1 to FDCs (20).
Model
Fig. 1 illustrates our model of HIV-1 dynamics during therapy with RT and protease inhibitors. We consider uninfected cells, short-lived productively infected cells, and long-lived chronically infected cells. We consider two types of free and bound viral particles: particles unaffected by therapy, which are potentially infectious, and therapy-modified particles, which are noninfectious because they lack functional gag and pol gene products caused by inhibition of HIV-1 protease. We assume the concentration of free virus in blood is the same as that in extracellular fluid throughout the body.
Figure 1.

HIV-1 and cell dynamics during antiretroviral therapy.
Cells.
Dynamics of cells are characterized by (4, 19, 21)
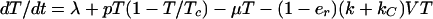 |
1 |
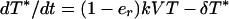 |
2 |
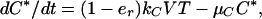 |
3 |
where T, T*, and C* are the numbers of uninfected, productively infected, and chronically infected cells, respectively, and V is the number of free potentially infectious viral particles (Fig. 1). Uninfected cells die with rate constant μ, are generated at constant rate λ, and proliferate according to a logistic law, in which p is the rate constant and Tc is the carrying capacity. [Only T appears in the logistic growth law, because T ≈ T + T* + C* (Table 1). The carrying capacity Tc is the target cell population at which proliferation is assumed to shut off because of limiting factors or homeostatic mechanisms.] Productively and chronically infected cells die at different rates, characterized by δ and μC. The overall rate at which cells are infected is given by (1 − er)(k + kC)VT, where k and kC characterize the rates of productive and chronic infection, respectively. The quantity er represents the efficacy of treatment with RT inhibitors. Before treatment, er = 0. For therapy with RT inhibitors that are 100% effective, analytical expressions can be derived from Eqs. 1–3 for T, T*, and C* as a function of treatment time t. These equations are available as supplemental material on the PNAS web site (www.pnas.org).
Table 1.
Baseline total body numbers of viral particles and cells
| Patient* | Free viral particles V0/109 | FDC-associated viral particles F0/1010 | Uninfected target cells T0/1010 | Productively infected cells T*0/108 | Chronically infected cells C*0/106 |
|---|---|---|---|---|---|
| 20485/10/3 | 0.41 | 4.6 | 9.9 | 1.1 | 3.0 |
| 20497/ 7/7 | 3.1 | 4.6 | 7.8 | 0.98 | 3.0 |
| 20490/ 2/1 | 7.1 | 5.6 | 5.9 | 6.8 | 1.1 |
| 20496/ 8/4 | 2.7 | 2.2 | 10 | 1.4 | 0.91 |
| 20491/ 5/6 | 0.44 | 7.0 | 11 | 3.5 | 1.2 |
| 20446/ 9/9 | 1.1 | 0.88 | 11 | 0.46 | 3.6 |
| 20449/ 3/5 | 0.25 | 12 | 13 | 1.8 | 180 |
| 20452/ 4/2 | 0.54 | 0.70 | 6.7 | 0.28 | 0.98 |
Free Virus.
Dynamics of free viral particles are characterized by (4, 19)
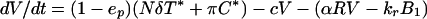 |
4 |
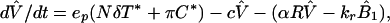 |
5 |
where V̂ is the number of free viral particles that are noninfectious because of therapy, R is the number of free receptors on FDC, and B1 and B̂1 are the numbers of potentially infectious and noninfectious viral particles on FDC that are bound to one receptor (Fig. 1). Productively infected cells produce viral particles at rate Nδ per cell, where N is the viral burst size, whereas chronically infected cells produce viral particles at rate π per cell. The quantity ep is the efficacy of protease inhibitors. Before treatment, ep = 0. The rate constant c characterizes clearance of free viral particles. The parameter α is an apparent rate constant for association of free viral particles with receptors on FDCs, and kr is the rate constant for dissociation of singly bound viral particles.
FDC-Associated Virus.
Dynamics of viral particles on FDCs are characterized by (20)
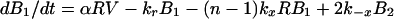 |
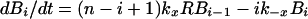 |
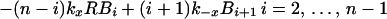 |
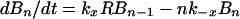 |
6 |
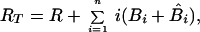 |
7 |
where Bi and B̂i represent the number of unmodified and therapy-modified viral particles, respectively, bound to i receptors (Fig. 1). A viral particle can bind up to n receptors. Eq. 6 governs the dynamics of potentially infectious viral particles; the governing equations for therapy-modified noninfectious viral particles are obtained from this equation by replacing V with V̂ and each Bi with B̂i. Eq. 7 is a conservation equation, in which RT represents the total number of FDC receptors. The forward and reverse crosslinking rate constants, kx and k−x, characterize reactions on the surface of FDCs. The total amount of virus bound to FDCs is given by the sum Σi=1n (Bi + B̂i).
These equations are based only on considerations of ligand-receptor binding (20). The model omits other processes that might influence the decay of FDC-associated virus, such as turnover of FDCs, internalization of HIV-1, shedding of HIV-1 on iccosomes (22), stripping of HIV-1 by cells that interact with FDCs, structural breakdown of virions, and recovery of the FDC network during therapy (13).
Parameter Estimates
Parameters were estimated for eight patients (Tables 1 and 2) who participated in the study of Notermans et al. (18) and for whom the level of plasma HIV-1 RNA (14), the number of infected mononuclear cells in LT (5), the amount of FDC-associated HIV-1 RNA (5), and the number CD4+ T cells in LT (12) were monitored during treatment. Four patients received triple therapy with a protease inhibitor (ritonavir) and two RT inhibitors (lamivudine and zidovudine), and the others received ritonavir monotherapy, followed by the triple combination after 3 wk. We assume drugs are 100% effective. Thus, for triple therapy patients, er = ep = 1 for t ≥ 0, and for ritonavir monotherapy patients, er = 0 and ep = 1 for t ≥ 0 (viral dynamics are unaffected by the shift to triple therapy at 3 wk; unpublished results). We also assume dynamics are in a steady state before treatment.
Table 2.
Rate constants and other parameters
| Parametera | Triple
therapy patientsb
|
Ritonavir
monotherapy patientsc
|
||||||
|---|---|---|---|---|---|---|---|---|
| 20485 | 20497 | 20490 | 20496 | 20491 | 20446 | 20449 | 20452 | |
| μC × 100 (d−1) | 0.35 | 0.71 | 0.38 | 0.38 | 2.4 | 0.97 | 3.5 | 0.36 |
| c (d−1) | 34 | 8.5 | 3.9 | 7.6 | 89 | 4.6 | 400 | 10 |
| δ (d−1) | ||||||||
| This studyd | 2.7 | 1.3 | 1.4 | 1.2 | —g | 1.2 | — | 0.55 |
| Notermans et al.e | 0.36 | 0.60 | 0.89 | 0.37 | 0.46 | 0.52 | 0.34 | 0.51 |
| Cavert et al.f | 0.11 | 1.1 | 1.5 | 1.2 | 2.6 | 0.73 | 0.37 | 0.24 |
| KxRT | 0.95 | 0.79 | 0.71 | 0.96 | 0.92 | 0.89 | 1.1 | 1.0 |
| p × 100 (d−1) | 1.0 | 0.75 | 3.4 | 1.1 | ND | ND | ND | ND |
| Tc × 10−11 | 2.9 | 2.8 | 1.4 | 2.0 | ND | ND | ND | ND |
| N | 49 | 210 | 30 | 120 | — | 87 | — | 360 |
| RT × 10−12 | 1.8 | 1.3 | 1.5 | 0.49 | 2.9 | 0.29 | 4.6 | 0.23 |
| k × 1012 (d−1) | 6.8 | 0.52 | 2.2 | 0.60 | — | 0.50 | — | 0.43 |
| kC× 1016 (d−1) | 2.9 | 0.89 | 0.10 | 0.13 | 5.7 | 3.1 | 1900 | 0.97 |
ND, not determined.
For all patients, n = 20, α = 1.5 × 10−10 d−1, k−x = kr = 8600 d−1, μ = 0.0038 d−1, and π = 0.
For these patients (ep = er = 1 for t ≥ 0), the values of p and Tc are determined simultaneously, with λ = 0 and δ given by the steady-state forms of Eqs. 1–3, from cellular data (5, 12). The values of c and KxRTthen are determined simultaneously, with δ fixed, from viral data (5, 14).
For these patients (ep = 1 and er = 0 for t ≥ 0), the values of c, δ, and KxRT are determined simultaneously, with T(t) = T0, from viral data (5, 14).
Confidence limits on δ, given in Tables 3 and 4, which are published as supplemental material, are broad in some cases; however, the lower limit is typically higher than the estimate of Notermans et al. (14). Estimates of c and KxRT are insensitive to the value of δ (unpublished results).
Estimates of δ with second-phase decay caused by long-lived infected cells (14).
Estimates of δ based on counts of infected mononuclear cells at d 0 and 2 of therapy (5).
Indeterminate. We find that any large value of δ is consistent with the data. Estimates of c, KxRT, and RT and calculations for these patients are based on δ → ∞.
Decay of Long-Lived Infected Cells.
To estimate μC, we use Eq. 3 and counts of infected mononuclear cells in LT at wks 3 and 24 of treatment (5). A count of infected cells at wk 3 was unavailable for patients 20490 and 20496. For these patients, we assume μC = 0.0038 d−1, which is approximately the slowest observed rate of decay (Table 2). Subsequent estimates of key parameters (e.g., δ) are essentially unchanged if we assume more rapid decay (unpublished results).
Baseline Total Body Numbers.
We determine the baseline total body numbers of free viral particles (V0), FDC-associated viral particles (F0), short-lived infected cells (T*0), and uninfected cells (T0) from baseline measurements (5, 12, 14). We use Eq. 3 to determine the baseline number of long-lived infected cells (C*0) from our estimate of μC (Table 2) and the count of infected mononuclear cells at wk 3 or 24 (5). Unit conversions to and from total body numbers are based on 700 g of LT (5, 12, 17), 15 liters of extracellular fluid (23, 24), and two copies of HIV-1 RNA per viral particle.
Recovery of Cells.
We analyze CD4+ cell recovery only for triple therapy patients; for these patients, viral and cellular dynamics are uncoupled. We assume a 6-mo half-life for uninfected cells: μ = 0.0038 d−1. This half-life is probably a lower limit (25, 26), but subsequent estimates of parameters (e.g., δ) change little if the half-life is longer (unpublished results). The relative contributions of cell generation in the thymus, characterized by λ, and proliferation, characterized by p and Tc, are uncertain. We consider the extremes: cell expansion is caused only by de novo generation, in which case p = 0, or cell expansion is caused only by proliferation, in which case λ = 0. For the case p = 0, we determine the value of λ that best simultaneously fits the counts of CD4+ T cells (12) and infected mononuclear cells (5) in LT. For the case λ = 0, we determine the values of p and Tc that best simultaneously fit these data. In the fitting procedure, we use Eqs. 1–3 to calculate the sums T + T* + C* and T* + C*, i.e., the total body numbers of (CD4+ T) cells and infected cells. The value of δ, which appears in Eq. 2, is determined as described below.
Decay of Short-Lived Infected Cells.
For triple therapy patients, we find δ from the steady-state forms of Eqs. 1–3: δ = [λ + pT0(1 − T0/Tc) − μT0 − μCC*0]/T*0. Thus, estimates of δ are derived from cellular data. Similar estimates are obtained for the cases p = 0 and λ = 0 (unpublished results). For the monotherapy patients, er = 0 during therapy, and cellular and viral dynamics are coupled (Eqs. 1–3). For this reason, we derive δ from viral data (see below). Estimates also can be obtained from viral data for triple therapy patients; these estimates are similar to those obtained when δ is derived from cellular data (unpublished results).
Production of Virus by Infected Cells.
To focus on the potential of FDCs to influence HIV-1 dynamics, we set π = 0, which ensures that long-lived infected cells do not contribute to viral dynamics. We find N, the burst size of productively infected cells, from the steady-state forms of Eqs. 4 and 6: N = cV0/(δT*0).
Decay of Free and FDC-Associated Virus.
The rate constants kr and k−x had been estimated earlier (20): k−x = kr ≈ 0.1 s−1. A reasonable value for αV0 is 0.3 d−1 if n = 20 (20). Typically, if n ≠ 20, the dimensionless crosslinking constant KxRT, where Kx ≡ kx/k−x, can be adjusted to yield kinetic behavior similar to that with n = 20 (20). The average V0 for patients that we consider is 2 × 109 (Table 1) (14). Thus, we specify n = 20 and α = 1.5 × 10−10 d−1 for all patients. The same qualitative results are obtained with a range of values for α (unpublished results). For treatment times of interest, we assume viral particles on FDCs are characterized by a single (mean) valence, although variation in n can influence the long-term dynamics of FDC-associated virus (20). For triple therapy patients, we determine the values of c and KxRT that best simultaneously fit measurements of plasma (14) and FDC-associated (5) virus. In this procedure, we numerically integrate Eqs. 4–7 and the set of equations for therapy-modified virus on FDC derived from Eq. 6. As part of the fitting procedure, the values of R, RT, and Bi for i = 1, … , n at t = 0 are determined by using the steady-state forms of Eqs. 4, 6, and 7, the baseline number of FDC-associated viral particles F0 (5), and the identity F0 = Σi=1nBi. These calculations are described in the supplemental material. For monotherapy patients, the fitting procedure is used to determine δ as well as c and KxRT. Here, the equations for viral dynamics are coupled with Eqs. 1–3. To simplify our analysis, we assume that T(t)≈T0 in Eqs. 2 and 3. To find k and kC, which appear in Eqs. 2 and 3, we use the steady-state forms of these equations: k = δT*0/(V0T0) and kC = μCC*0/(V0T0).
Results
We have developed a model for HIV-1 dynamics that includes FDC (Fig. 1; Eqs. 1–7) and used this model to analyze decay of plasma virus (14), decay of FDC-associated virus (5), decay of infected cells in LT (5), and recovery of CD4+ T cells in LT (12).
Release of Virus from FDCs Can Explain Biphasic Plasma Viral Decay.
Viral loads and best-fit theoretical time courses of viral decay are shown in Fig. 2 for two triple therapy patients. As is typical, the model is consistent with the data for these patients. Plots for all patients are available as supplemental material (Figs. 5 and 6). In previous analyses, the first and second phases of plasma viral decay have been attributed, respectively, to death of short- and long-lived infected cells (3, 4, 14). Here, time courses have been calculated on the basis that long-lived infected cells make no contribution to viral load (i.e., π = 0), but the model is still capable of matching the observed viral decay. Thus, production of virus by long-lived infected cells is not required to explain second-phase dynamics if release of virus from FDCs contributes to the plasma viral load.
Figure 2.

Decay of free and FDC-associated virus in triple therapy patients 20485 and 20497. (A and B) HIV-1 RNA per ml of plasma (points) (14) and the best-fit theoretical curve, found by calculating V + V̂ as a function of treatment time t. (C and D) HIV-1 RNA per g of LT (points) (5) and the best-fit theoretical curve, found by calculating Σi=1n(Bi + B̂i) as a function of treatment time t. Calculations are based on Eqs. 4-7 and parameter values in Tables 1 and 2.
Influence of FDCs on First- and Second-Phase Dynamics.
FDC may influence the first phase of plasma viral decay. We find that death of short-lived infected cells is faster than first-phase decay, as can be seen by comparing our estimates of δ with those determined earlier for the same patients by Notermans et al. (14) who used a model in which first-phase decay matches the rate of cell death (Table 2). Our higher estimates of δ are more consistent with direct counts of infected mononuclear cells in LT between d 0 and 2 of treatment (Table 2) (5). FDCs also may influence the second phase of plasma viral decay. Because the FDC reservoir is the only source of virus that we consider at treatment times much larger than the half-life of short-lived infected cells, the rate of second-phase decay corresponds to the net rate at which virus is lost from FDCs.
Rate of Viral Clearance.
Based on analysis of plasma viral decay, Perelson et al. (3) determined that 3 d−1 is a lower bound on the value of c, the rate constant for viral clearance. We typically estimate much larger values (Table 2), which are more consistent with recent direct measurements of c (27–29). Higher values for c are needed to explain the data because viral clearance is offset by release of virus from FDCs.
Persistence of Potentially Infectious Virus.
As illustrated in Fig. 3, potentially infectious virus may persist during combination therapy, even if therapy is 100% effective. The source of persistent potentially infectious virus is the FDC reservoir. This virus, which is present at the start of therapy, is not directly affected by antiviral drugs. Although therapy-modified virus competes with pretherapy virus for receptors on FDCs and displaces this potentially infectious virus to some extent, the amount of therapy-modified virus produced in triple therapy patients, NT*0, is insufficient to replace the virus on FDCs at the start of treatment, F0, i.e., NT*0 < F0 (Tables 1 and 2). Elsewhere (30), we show that when RT inhibitors are absent, more therapy-modified virus is produced, because of new infections generated by infectious virus from FDCs, and noninfectious virus displaces pretherapy virus to a greater extent than that shown in Fig. 3. Consistent with this finding, we find greater displacement of pretherapy virus on FDCs by therapy-modified virus for ritonavir monotherapy patients, as shown in the supplemental material (Fig. 7).
Figure 3.

Persistence of potentially infectious virus during therapy. The theoretical decay curves of Fig. 2 are replotted to show the total body numbers of potentially infectious (solid lines) and therapy-modified (dotted lines) viral particles that are (A and B) free in extracellular fluid and (C and D) associated with FDCs in LT.
Cellular Dynamics.
The model is consistent not only with measurements of viral dynamics but also measurements of cellular dynamics in LT. In Fig. 4, cell counts and two best-fit theoretical time courses are shown for each of two patients. Plots for all triple therapy patients are given in Fig. 8 of the supplemental material. One theoretical time course is based on target cell recovery caused only by proliferation and the other is based on recovery caused only by generation of new cells. The two time courses are similar, indicating that the relative contributions of proliferation and generation are indeterminate on the basis of this analysis alone.
Figure 4.

Decay of infected cells and recovery of target cells in triple therapy patients 20485 and 20497. (A and B) Infected mononuclear cells per g of LT (points) (5) and the best-fit theoretical decay curve, found by calculating T* + C* as a function of treatment time t. (C and D) CD4+ T cells per μg of LT (points) (12) and the best-fit theoretical recovery curve, found by calculating T + T* + C* as a function of treatment time t. Solid lines correspond to calculations with λ = 0 (all target cell expansion because of proliferation), which are based on Eqs. 1–3 and parameter values in Tables 1 and 2. Dotted lines correspond to calculations with p = 0 (all target cell expansion caused by de novo generation), which are based on Eqs. 1–3 and parameter values in Tables 1 and 2 with the following exceptions. For patient 20485, p = 0, λ = 7.2 × 108 d−1, and δ = 3.2 d−1. For patient 20497, p = 0, λ = 4.5 × 108 d−1, and δ = 1.5 d−1.
Discussion
Decay of HIV-1 during antiretroviral therapy is biphasic, with a slow second phase. Second-phase decay has been attributed to long-lived infected cells (4). By considering release of virus from FDCs (5), we have been able to reproduce biphasic viral dynamics without including production of virus by long-lived cells (Fig. 2). However, our analysis does not rule out a role for long-lived infected cells in viral dynamics: such cells and the FDC pool of virus both may contribute to the second-phase viral load. We find that a long-lived infected cell population, under the assumption of 100% effective drugs, is still required to explain the observed persistence of infected cells (Fig. 4). An alternative explanation for the persistence of infected cells is ongoing infection allowed by drugs that are less than 100% effective (31, 32).
Release of virus from FDCs can lead to biphasic plasma viral decay for two reasons. First, the amount of virus on FDCs is substantial (17) and its release (5, 20) can be expected to influence plasma measurements. For triple therapy patients, consistent with measurements of virus in cells and on FDCs (17), we predict that the initial viral load on FDCs, F0, is greater than the amount of virus produced by infected cells during treatment, NT*0 (Tables 1 and 2). Second, dissociation of virus from FDCs during treatment is biphasic because of reversible, multivalent binding of HIV-1 to FDCs (20). As viral particles dissociate from receptors on FDCs during first-phase decay, receptors are freed to interact with the particles that remain. These particles then attach, on average, to a greater number of receptors, causing the net rate of release to slow. A steady rate of dissociation, the second phase of decay, is reached when most receptors are free and the average number of bonds holding a viral particle on the cell surface changes little with time.
If the FDC network is a major source of second-phase virus, which is consistent with the available data (Fig. 2), FDCs may affect the long-term outcome of therapy. We predict that virus on FDCs is released continuously during treatment and that a significant fraction of the virus released at all times is virus present before therapy (Fig. 3). The infectivity of this virus depends on the length of time FDCs retain HIV-1 in an infectious state, which is unknown. However, protein antigens, which are quickly degraded in blood, remain intact on FDCs for months to years (33). Also, it has been shown in short-term experiments that HIV-1 on FDCs is infectious (34) and that target cells become infected when cocultured with FDC isolated from mice that were inoculated with HIV-1 42 d earlier (35) and even 9 mo earlier (G. F. Burton, personal communication).
In our calculations, plasma and FDC-associated virus decay in parallel (Fig. 3), and release of virus from FDCs limits the overall rate of viral decay. The influence of FDCs on first-phase decay is indicated by our parameter estimates (Table 2). We find that first-phase decay is slower than clearance of free virus, which is characterized by the rate constant c, and death of cells in the productive state of infection (models in which the eclipse phase of the viral life cycle is explicitly considered (36) show that δ is the death rate of cells in the posteclipse phase), which is characterized by the rate constant δ. In contrast, in earlier models (4, 14), the rate of first-phase decay matches δ. We obtain estimates for c and δ that are higher than expected on the basis of models without FDCs (3, 4, 14), because viral clearance and cell death are buffered by release of virus from FDCs. Direct measurements also indicate that c (27–29) and δ (5) (Table 2) are higher than earlier model-based estimates.
We have found that plasma viral decay is influenced by release of virus from FDCs and shown that this release may be the cause of slow second-phase decay (Fig. 2). This finding is significant because the virus seen in plasma, which is monitored to make clinical decisions, is of unknown origin. We now suggest that part of this virus is derived from the pool of virus on FDCs at the start of therapy and that plasma viral measurements do not necessarily reflect the current population of replicating virus, which may be masked by the release of stored virus. Furthermore, we suggest that blocking HIV-FDC interactions, which should significantly speed dissociation of FDC-associated virus (37), may increase the rate of viral decay during treatment and reduce the treatment time required to achieve a given level of virological control. Speeding viral decay in the face of less than perfect therapy will reduce the potential for development of drug resistance. The ability to speed viral decay also may affect the design of interrupted treatment strategies aimed at inducing immunological control of HIV infection. With faster decay, treatment periods can be shortened and compliance and drug side effects are less of an issue. Finally, speeding decay may aid in the identification of other persistent viral reservoirs, all of which must be considered in eradication strategies.
Our analysis leads to several predictions. Earlier studies (4, 14), in which second-phase decay is attributed to long-lived infected cells, suggest a sharp break between the two phases of viral decay. In contrast, we predict a gradual slowing between the first and second phases (Fig. 2), consistent with data from some studies (38). We also predict that a significant fraction of virus present during the second phase of therapy-induced decay will carry processed gag and pol gene products (Fig. 3). This finding is contrary to the predictions of earlier models (3), according to which virus present during second-phase decay is therapy modified if protease inhibitors are 100% effective. Finally, because we find that second-phase virus may include virus present before treatment (Fig. 3), we predict that virus arising after cessation of treatment will reflect some of the viral diversity present before treatment. A recent report indicates that viral reservoirs other than latently infected cells play a role in re-emergence of plasma viremia after discontinuation of antiretroviral therapy (39).
Supplementary Material
Acknowledgments
We thank A. T. Haase, J. E. Mittler, and J. M. Murray for helpful discussions. This work was performed in part under the auspices of the U. S. Department of Energy and was supported by National Institutes of Health Grants RR06555 and AI28433 (A.S.P.) and the German Cancer Research Center (N.I.S.).
Abbreviations
- HIV-1
HIV type 1
- FDC
follicular dendritic cell
- LT
lymphoid tissue
- RT
reverse transcriptase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190065897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190065897
References
- 1.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 2.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 3.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 4.Perelson A S, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho D D. Nature (London) 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 5.Cavert W, Notermans D W, Staskus K, Wietgrefe S W, Zupancic M, Gebhard K, Henry K, Zhang Z-Q, Mills R, McDade H, et al. Science. 1997;276:960–964. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 6.Wong J K, Günthard H F, Havlir D V, Zhang Z-Q, Haase A T, Ignacio C C, Kwok S, Emini E, Richman D D. Proc Natl Acad Sci USA. 1997;94:12574–12579. doi: 10.1073/pnas.94.23.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stellbrink H-J, van Lunzen J, Hufert F T, Fröschle G, Wolf-Vorbeck G, Zöllner B, Albrecht H, Greten H, Racz P, Tenner-Racz K. AIDS. 1997;11:1103–1110. doi: 10.1097/00002030-199709000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Tenner-Racz K, Stellbrink H-J, van Lunzen J, Schneider C, Jacobs J-P, Raschdorff B, Groβschupff G, Steinman R M, Racz P. J Exp Med. 1998;187:949–959. doi: 10.1084/jem.187.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Autran B, Carcelain G, Li T S, Blanc C, Mathez D, Tubiana R, Katlama C, Debré P, Leibowitch J. Science. 1997;277:112–116. doi: 10.1126/science.277.5322.112. [DOI] [PubMed] [Google Scholar]
- 10.Pakker N G, Notermans D W, de Boer R J, Roos M T L, de Wolf F, Hill A, Leonard J M, Danner S A, Miedema F, Schellekens P T A. Nat Med. 1998;4:208–214. doi: 10.1038/nm0298-208. [DOI] [PubMed] [Google Scholar]
- 11.Notermans D W, Pakker N G, Hamann D, Foudraine N A, Kauffmann R H, Meenhorst P L, Goudsmit J, Roos M T L, Schellekens P T A, Miedema F, et al. J Infect Dis. 1999;180:1050–1056. doi: 10.1086/315013. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z-Q, Notermans D W, Sedgewick G, Cavert W, Wietgrefe S, Zupancic M, Gebhard K, Henry K, Boies L, Chen Z, et al. Proc Natl Acad Sci USA. 1998;95:1154–1159. doi: 10.1073/pnas.95.3.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z-Q, Schuler T, Cavert W, Notermans D W, Gebhard K, Henry K, Havlir D V, Günthard H F, Wong J K, Little S, et al. Proc Natl Acad Sci USA. 1999;96:5169–5172. doi: 10.1073/pnas.96.9.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notermans D W, Goudsmit J, Danner S A, de Wolf F, Perelson A S, Mittler J. AIDS. 1998;12:1483–1490. doi: 10.1097/00002030-199812000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. Nature (London) 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 16.Pantaleo G, Graziosi C, Demarest J F, Cohen O J, Vaccarezza M, Gantt K, Muro-Cacho C, Fauci A S. Immunol Rev. 1994;140:105–130. doi: 10.1111/j.1600-065x.1994.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 17.Haase A T, Henry K, Zupancic M, Sedgewick G, Faust R A, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang Z-Q, et al. Science. 1996;274:985–989. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 18.Notermans D W, Jurriaans S, de Wolf F, Foudraine N A, de Jong J J, Cavert W, Schuwirth C M, Kauffmann R H, Meenhorst P L, McDade H, et al. AIDS. 1998;12:167–173. doi: 10.1097/00002030-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Perelson A S, Nelson P W. Soc Ind Appl Math Rev. 1999;41:3–44. [Google Scholar]
- 20.Hlavacek W S, Wofsy C, Perelson A S. Proc Natl Acad Sci USA. 1999;96:14681–14686. doi: 10.1073/pnas.96.26.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sachsenberg N, Perelson A S, Yerly S, Schockmel G A, Leduc D, Hirschel B, Perrin L. J Exp Med. 1998;187:1295–1303. doi: 10.1084/jem.187.8.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szakal A K, Kosco M H, Tew J G. J Immunol. 1988;140:341–353. [PubMed] [Google Scholar]
- 23.Niyongabo T, Melchior J C, Henzel D, Bouchaud O, Larouzé B. Nutrition. 1999;15:740–743. doi: 10.1016/s0899-9007(99)00146-x. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Wang Z, Gallagher D, Kotler D P, Ma K, Heymsfield S B. J Parenter Enteral Nutr. 1999;23:61–66. doi: 10.1177/014860719902300261. [DOI] [PubMed] [Google Scholar]
- 25.Michie C A, McLean A, Alcock C, Beverley P C L. Nature (London) 1992;360:264–265. doi: 10.1038/360264a0. [DOI] [PubMed] [Google Scholar]
- 26.McLean A R, Michie C A. Proc Natl Acad Sci USA. 1995;92:3707–3711. doi: 10.1073/pnas.92.9.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Dailey P J, He T, Gettie A, Bonhoeffer S, Perelson A S, Ho D D. J Virol. 1999;73:855–860. doi: 10.1128/jvi.73.1.855-860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L, Mittler J E, Markowitz M, Moore J P, Perelson A S, Ho D D. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 29.Igarashi T, Brown C, Azadegan A, Haigwood N, Dimitrov D, Martin M A, Shibata R. Nat Med. 1999;5:211–216. doi: 10.1038/5576. [DOI] [PubMed] [Google Scholar]
- 30.Hlavacek W S, Stilianakis N I, Perelson A S. Philos Trans R Soc London B. 2000;355:1051–1058. doi: 10.1098/rstb.2000.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, Talal A, Racz P, Perelson A S, Korber B T, et al. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 32.Furtado M R, Callaway D S, Phair J P, Kunstman K J, Stanton J L, Macken C A, Perelson A S, Wolinksy S M. N Engl J Med. 1999;340:1614–1622. doi: 10.1056/NEJM199905273402102. [DOI] [PubMed] [Google Scholar]
- 33.Mandel T E, Phipps R P, Abbot A, Tew J G. Immunol Rev. 1980;53:29–59. doi: 10.1111/j.1600-065x.1980.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 34.Heath S L, Tew J G, Tew J G, Szakal A K, Burton G F. Nature (London) 1995;377:740–744. doi: 10.1038/377740a0. [DOI] [PubMed] [Google Scholar]
- 35.Burton G F, Masuda A, Heath S L, Smith B A, Tew J G, Szakal A K. Immunol Rev. 1997;156:185–197. doi: 10.1111/j.1600-065x.1997.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 36.Mittler J E, Sulzer B, Neumann A U, Perelson A S. Math Biosci. 1998;152:143–163. doi: 10.1016/s0025-5564(98)10027-5. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein B, Wofsy C. Immunol Today. 1996;17:77–80. doi: 10.1016/0167-5699(96)80583-4. [DOI] [PubMed] [Google Scholar]
- 38.Grossman Z, Polis M, Feinberg M B, Grossman Z, Levi I, Jankelevich S, Yarchoan R, Boon J, de Wolf F, Lange J M A, et al. Nat Med. 1999;5:1099–1104. doi: 10.1038/13410. [DOI] [PubMed] [Google Scholar]
- 39.Chun T-W, Davey R T, Jr, Ostrowski M, Justement J S, Engel D, Mullins J I, Fauci A S. Nat Med. 2000;6:757–761. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.